Introduction
Chromosome 13q deletion syndrome, a rare genetic
disorder, is characterized by partial deletions of one of the long
arms of chromosome 13, which leads to a number of human birth
defects. The clinical symptoms vary widely among patients and may
include, developmental delays (mental and growth retardation),
facial anomalies (microcephaly, hypertelorism, flattened nasal
bridge and micrognathia), and severe malformations in the distal
limbs, central nervous system (posterior encephalocele,
holoprosencephaly and neural tube defects), eyes (micro-ophthalmia
and retinoblastoma), heart (congenital heart defects), lungs,
kidneys, gastrointestinal tract and genitourinary tract
(penoscrotal transposition and hypospadias, ambiguous genitalia,
reduced anogenital distance, imperforate anus, bicornuate uterus
and imperforate anus with vaginal fistula or cloaca) (1–4).
Clinical characteristics and severity depend on the
size of the deleted region and the location on chromosome 13.
Chromosome 13 deletion syndrome was first described in 1969
(1), and efforts since then have
been made to identify the critical region involved in specific
anomalies using genotype-phenotype analysis (5,6).
However, due to the limited number of cases and the different
levels of penetrance, the causative genes have yet to be
determined. Previous studies have proposed the existence of the
following 3 groups of genotype-phenotypes based on the involvement
of the critical 13q32 band in the deletion (2,5): i)
proximal deletions with a non-deleted 13q32 band are observed
primarily in patients with mild mental retardation, growth delay
and inconstant retinoblastoma; ii) a 13q32 band deletion is
associated with severe congenital malformations; iii) a distal
deletion without a 13q32 deletion has been observed in patients
with severe mental retardation, no brain malformation or growth
delay. Previous studies involving the use of array comparative
genomic hybridization (CGH) coupled with fluorescence in
situ hybridization, reverse transcription-quantitative
polymerase chain reaction (qPCR) and multiple ligation-depended
probe amplification to determine the precise breakpoint of the
unbalanced chromosomal translocation have been conducted (3–5,7–11).
This analysis has provided information regarding the molecular
genotype-phenotype in chromosome 13 deletion syndrome.
In the present study, two patients diagnosed with
chromosome 13 deletion syndrome, which harbored 13q31.3q terminal
(qter) and 13q33.1qter deletions, respectively, were recruited for
genotype-phenotype analysis using array-CGH and qPCR.
Case report
The present study was approved by the institutional
review board of Shengjing Hospital affiliated to China Medical
University (Shenyang, China). Written informed consent regarding
participation and the publication of clinical information was
obtained from the parents of patients 1 and 2, and all clinical
investigations were conducted according to the principles expressed
in the Declaration of Helsinki.
Clinical descriptions
Patient 1 (gender, female; age, 14 months) was
diagnosed with anal atresia with rectoperineal fistula following
birth, and was referred to Shengjing Hospital for surgery in June
2009 due to recurrent constipation. Patient 1 was born at full-term
by Caesarean section, and was the first-born child to
non-consanguineous parents. Following birth, the patient was blue
and was diagnosed with a heart murmur as well as an imperforate
anus with navicular fossa fistula. At 1 month of age, the patient
suffered recurrent seizures. At 14 months-old, physical examination
identified marked growth retardation, with a height of 69 cm, which
was <3 standard deviations below the average for the patient's
age; a body weight of 7.8 kg and a head circumference of 44 cm. In
addition, facial dysmorphism was observed, including hypotelorism,
blepharophimosis and a broad nasal bridge with a flat philtrum. In
addition, psychomotor milestones, determined by developmental
quotient evaluation, were markedly delayed, as they were equivalent
to the level observed in a 5-month-old child. Angioma was observed
in the lateral side of the left foot and digits. A
systolic-diastolic phase heart murmur and a number of complex type
congenital heart defects were identified by echocardiography
including ventricular septal defect, double outlet right ventricle
(DORV) defects, single atrium (SA) defects, mixed type atrial
septal defect (ASD), persistent left superior vena cava (PLSVC)
defects and severe pulmonary stenosis (PS). Radiography analysis of
the fistula and anoplasty revealed a fistula with a 0.3-cm opening
at the navicular fossa (also known as the fossa of the vaginal
vestibule) located before the terminal rectum, which was 1.5 cm in
length, stopping 0.5-cm from the anterior wall of the rectal blind
end. Magnetic resonance imaging (MRI) of the brain revealed
cortical atrophy, agenesis of the corpus callosum, cerebral
ventricle dilation, a small cerebral cortex hippocampus, bilateral
otitis media and mastoiditis. The patient was treated with
anoplasty and the fistula was closed, however, the cardiac
anomalies were not corrected.
Patient 2 was a newborn (gender, female; age, 46 h),
born at full-term to a 39-year-old mother and 43-year-old father by
Caesarean section due to oligohydramnios. The parents were
non-consanguineous. At birth, patient 2 weighed 2,460 g (<3rd
centile), was 42 cm in length (<3rd centile) and demonstrated a
head circumference of 31.5 cm (<3rd centile). The patient
presented with poor feeding and vomiting following birth. Facial
dysmorphism was observed, including a round face with a small
forehead, hypotelorism and blepharophimosis. The patient's
heartbeat was strong with a regular rhythm, and no murmur was
observed. A nasogastric tube was passed into the stomach with great
difficulty, and an esophageal hiatus hernia and gastroesophageal
reflux were identified following upper gastrointestinal contrast
X-ray analysis. MRI results revealed limited damage to the cerebral
white matter, which was only observed in the posterior horn of the
left lateral ventricle, and suggested an optimistic prognosis.
Following treatment with gastrointestinal decompression, the
patient's vomiting was greatly relieved.
Cytogenetic analysis
Cytogenetic investigation using Giemsa-banding with
trypsin as the proteolytic enzyme (GTG banding) was performed on
metaphase spreads of peripheral blood lymphocytes using standard
procedures (12). The following
detailed procedures were followed: heparinized human whole blood
(0.5 ml) was cultured at 37°C for 70 h in 8 ml Gibco Chromosome
Medium (Thermo Fisher Scientific, Inc., Waltham, MA, USA). Cells
were arrested with colchicine (10 ug/ml) for 90 min. Chromosome
preparations were made by incubating the cell suspension in 0.075
mol/l potassium chloride at 37°C for 30 min, followed by a fixation
step in a freshly prepared mixture of 3:1 methanol:acetic acid at
−20°C for 30 min. GTG banding was performed by incubating the glass
slides in a 0.05% trypsin solution at 37°C for 15 sec, followed by
rinsing the slides in PBS buffer and staining in a 5% Giemsa stain
for 8 min at room temperature. The slides were rinsed with water
and air dried. Cytogenetic analysis was performed on GTG-banded
metaphase spreads collected from the patients and their parents at
a resolution of 400 bands according to standard lab procedures. A
total of 15 metaphases were analyzed for each individual
sample.
DNA isolation and array-CGH
analysis
Genomic DNA was isolated from the peripheral blood
samples using the DNeasy Blood & Tissue kit (Qiagen GmbH,
Hilden, Germany). DNA quality was confirmed by gel electrophoresis
with a 1.5% agarose gel, and the yield was confirmed by
spectrophotomery (NanoDrop ND-1,000; NanoDrop Technologies; Thermo
Fisher Scientific, Inc., Wilmington, DE, USA). Genomic DNA was
labeled using the Affymetrix Cytogenetics Reagent kit (Affymetrix,
Inc., Santa Clara, CA, USA) and the labeled DNA was loaded on to an
Affymetrix Cytogenetics Array Cytoscan 750K Chip (containing 7.5
million copy number markers; Affymetrix, Inc.), which was performed
by Gene Tech (Shanghai, China). The array was scanned and the data
were analyzed using the Affymetrix Chromosome Analysis Suite
(version 2.1; Affymetrix, Inc.).
qPCR validation
PCR was performed on lymphocyte DNA extracts using
the 7900HT Fast Real-Time PCR System (Applied Biosystems; Thermo
Fisher Scientific, Inc.). A total of 3 sequence-tagged sites on
chromosome 13 (D13S797 in 13q33.2, D13S628 in 13q31.1 and D13S258
in 13q21.33) were detected. The primers used are shown in Table I. The NCBI reference sequence of
D13S797 used to determine copy number alterations was NG_012694.1
(https://www.ncbi.nlm.nih.gov/nuccore/255958284) from
19563 nt to 19758 nt, the amplified product length is 196 bp. NCBI
reference sequence for D13S258 was AL356754.18 (https://www.ncbi.nlm.nih.gov/nuccore/AL356754) from
969 nt to 1239 nt, the amplified product length is 271 bp. NCBI
reference sequence for was AL160154.11 (https://www.ncbi.nlm.nih.gov/nucleotide/14160914);
from 1004 nt to 1243 nt, the amplified product length is 240 bp.
PCR reactions were prepared using the SYBR Premix Ex Taq II PCR
reagent kit (Takara Biotechnology Co., Ltd., Dalian, China)
according to the manufacturer's instructions. Amplification was
performed in a final reaction volume of 10 µl, containing 50 ng
genomic DNA, 0.4 µM PCR forward primer, 0.4 µM PCR reverse primer,
ROX Reference Dye II (1X) and SYBR Premix Ex Taq (Tli RNase H Plus;
1X). The thermal cycling conditions were as follows: initial
denaturation at 94°C for 30 sec, followed by 40 cycles of
denaturation at 95°C for 5 sec and annealing at 60°C for 35 sec.
Amplification levels were calculated using the
2−ΔΔCq method (13).
 | Table I.Primer sequences of STS markers used
for PCR analysis. |
Table I.
Primer sequences of STS markers used
for PCR analysis.
| STS | Forward primer | Reverse primer |
|---|
| D13S797 |
5′-GGTTTGCTGGCATCTGTATT-3′ |
5′-TGTCTGGAGGCTTTTCAGTC-3′ |
| D13S258 |
5′-ACCTGCCAAATTTTACCAGG-3′ |
5′-GACAGAGAGAGGGAATAAACC-3′ |
| D13S628 |
5′-CGCCACTTTTCTAAATGCC-3′ |
5′-GGAGTAACAAATAGCAAGGCT-3′ |
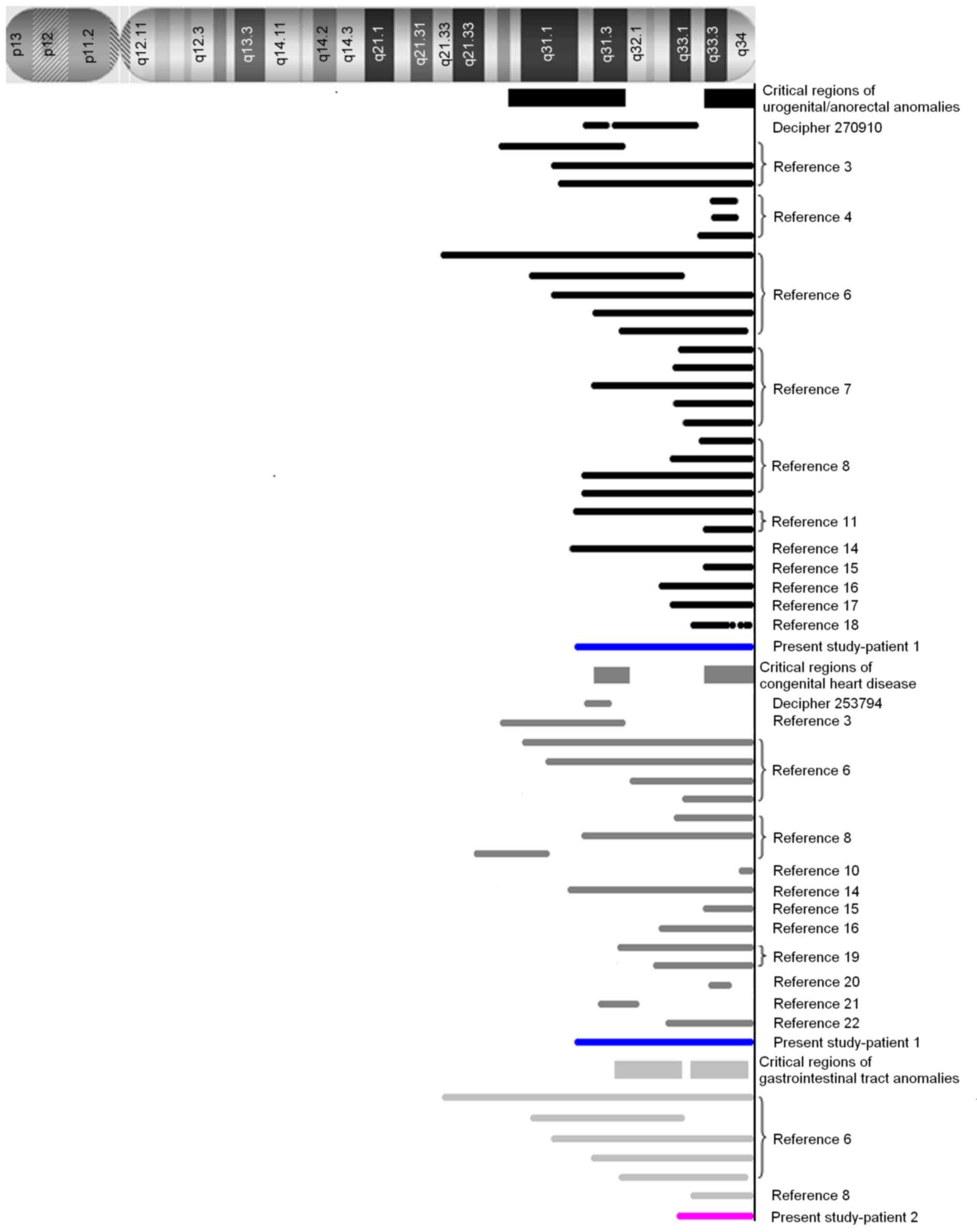
The array-CGH results are shown in Fig. 1 and Table II, and a summary of the regions on
chromosome 13 identified in the present and previous studies
(3,4,6–8,10,11,14–22)
are reviewed in Fig. 2.
Cytogenetic analysis of patient 1 revealed a karyotype of
46,XX,del(13)(pter→q31; data not
shown), and the array-CGH results revealed a distal 20.38 Mb
deletion in the 13q31.3-qter region, from 94,724,977 to 115,107,733
bp (terminal end). A total of 59 Refseq genes were detected in this
region of deletion, as determined by analysis by Affymetrix
Chromosome Analysis Suite software. The qPCR results verified the
presence of a deletion at D13S797 only, but not at D13S628 and
D13S258 (data not shown). The karyotype and qPCR results of the
parents of patient 1 were normal, demonstrating that the deletion
was not hereditary (data not shown).
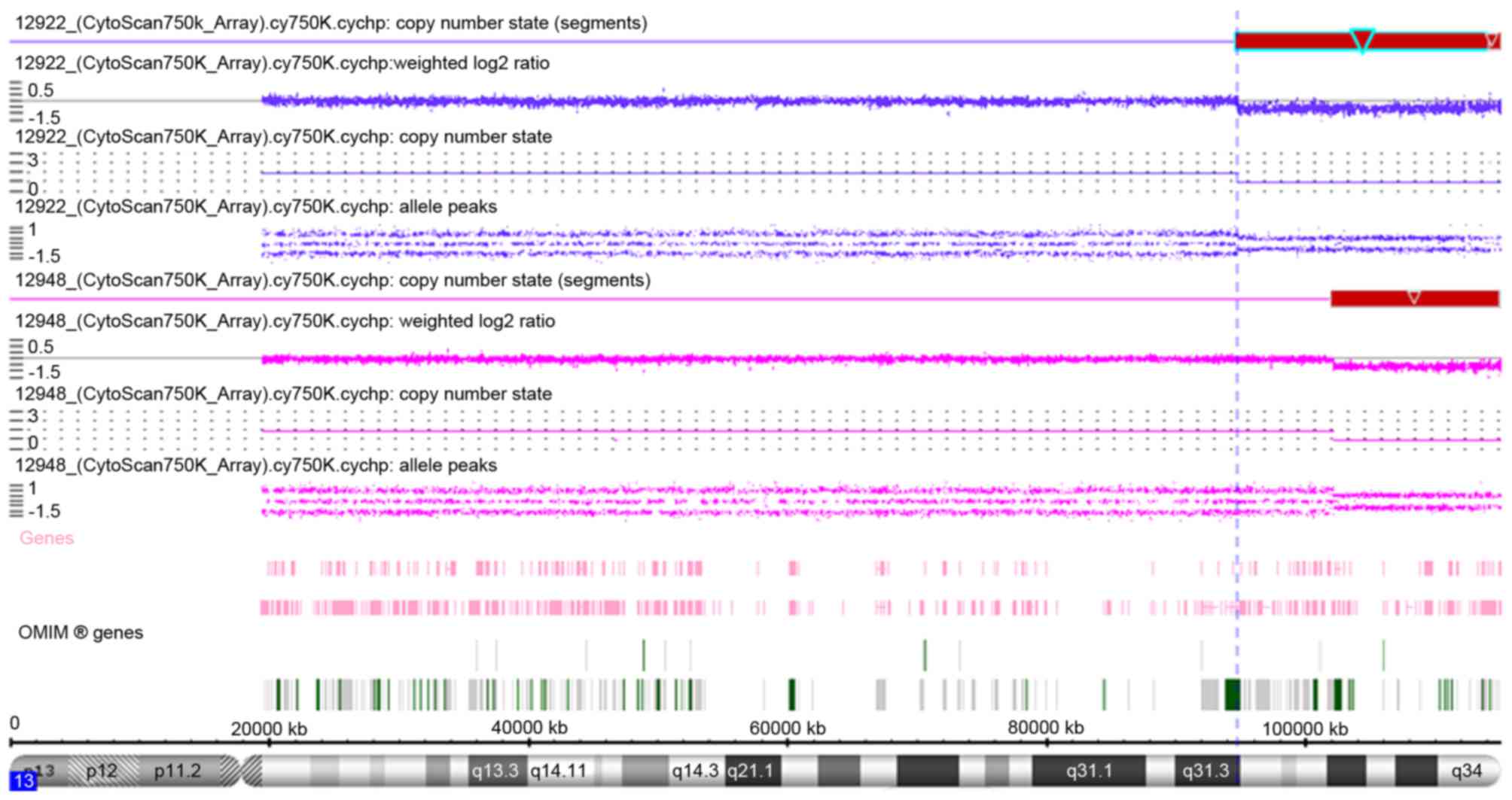 | Figure 1.Array-comparative genomic
hybridization results of patient 1 (12922; blue regions) and
patient 2 (12948; pink regions) as indicated by copy number state
using the Affymetrix Cytoscan 750K. The deleted region 13q31.3-qter
of patient 1 from 94,724,977 bp to 115,107,733 bp is indicated by
red box with light blue outline, with the darker blue line
indicating that copy number state is 1 (normal copy number state is
2). The deleted region 13q33.1-qter of patient 2 from 102,110,842
bp to 115,107,733 bp is indicated by red box, with the pink line
indicating that the copy number state is 1. OMIM, Online Mendelian
Inheritance in Man. |
| Table II.Amplified DNA copy numbers in the
13q31.3-q34 regions as determined using the Affymetrix Cytoscan
750K. |
Table II.
Amplified DNA copy numbers in the
13q31.3-q34 regions as determined using the Affymetrix Cytoscan
750K.
| A, Patient 1 |
|---|
|
|---|
| CN state | Type | Chromosome
band | Size (kbp) | Marker count | Confidence | Start (bp) | End (bp) |
|---|
|
| LOH | 13q33.1-q33.3 | 6,293.846 | 651 | 1 | 104,002,716 | 110,296,562 |
| 1 | Loss | 13q34-q34 | 861.336 | 320 | 0.9014088 | 114,246,397 | 115,107,733 |
| 1 | Loss | 13q31.3-q34 | 19,490.64 | 5,095 | 0.91336507 | 94,724,977 | 114,215,617 |
|
| B, Patient 2 |
|
| CN state | Type | Chromosome
band | Size (kbp) | Marker count | Confidence | Min (bp) | Max (bp) |
|
|
| LOH | 13q33.1-q34 | 12,987.398 | 1,170 | 1 | 102,108,307 | 115,095,705 |
| 1 | Loss | 13q31.3-q34 | 12,996.891 | 3,262 | 0.9176309 | 102,110,842 | 115,107,733 |
GTG banding analysis revealed that the karyotype of
patient 2 was 46,XX,del(13)(pter→q33:)(data not shown), and the
array-CGH results revealed a distal deletion of 12.99 Mb in the
13q33.1-qter region spanning 102,110,842 to 115,107,733 bp.
Analysis by Affymetrix Chromosome Analysis Suite software revealed
that a total of 34 Refseq genes are located in this region of
deletion. The qPCR results verified the deletion at positions
D13S797 and D13S628, but not D13S258 (data not shown). The
karyotype and qPCR results of the parents of patient 2 were normal
(data not shown), which suggested that this deletion was not
hereditary.
Discussion
Congenital heart disease, anorectal/genitourinary
and gastrointestinal tract malformations are the predominant
anomalies observed in 13q deletion syndrome, particularly as a part
of VACTERL association, a disorder characterized by vertebral
anomalies, anal atresia, cardiac defects, tracheoesophageal
fistula, renal anomalies and limb defects (14,23–25).
The aim of phenotype-genotype association analysis between these
anomalies and deleted regions of chromosome 13, is to identify a
limited number of candidate genes located in narrow regions of
deletion that may provide novel targets for molecular pathogenesis
research into the development of heart, anorectal/genitourinary and
gastrointestinal tract diseases.
Anorectal/genitourinary anomalies in 13q deletion
syndrome are generally rare and vary in manifestation and severity.
Typical cases observed in male patients involve anal atresia with
severe hypospadias and perineal fistula, whereas other patients may
present with distal hypospadias without anorectal anomalies. By
contrast, females are often diagnosed with anal atresia and vaginal
fistula (4,7). Human embryological studies have
demonstrated that the primitive urogenital sinus and anorectal
canal originate from the primitive cloaca, and there are number of
molecular events that govern the developmental processes underlying
cloacal septation, closure of the perineum and scrotum, urethral
tubularization and penoscrotal positioning (26–28).
The results obtained from patient 1 in the present study, together
with those obtained from patients with genitourinary/anorectal
anomalies in 13q deletion syndrome from previous studies (3,4,6–8,11,14–18),
have identified two critical regions, 13q33.3-qter and
13q22.1–31.3. In recent years, the non-morbid online Mendelian
Inheritance in Man (OMIM) gene, ephrin B2 (EFNB2) located in
13q33.3, has been recognized as a strong candidate gene for
hypospadias or anorectal anomalies in 13q deletion syndrome in a
number of studies (4,7–9,18).
Animal experiments have demonstrated that a partial
loss-of-function EFNB2 mutation in heterozygous male mice
induces severe hypospadias and incomplete cloacal septation, and
female mice exhibit similar defects in their external genitalia
(29). Molecular data has further
verified that the reverse signal, EFNB2 activation by EPH
receptor B2 (EPHB2) on EPHB2-expressing cells, is exhibited
in a dominant-negative manner in
Efnb2LacZ/+ mice with hypospadias,
which display a reduction in the effect of tyrosine phosphorylation
(29). The haploinsufficiency of
the EFNB2 gene product may provide a causative explanation
for the clinical results observed in patient 1 of the present
study. However, a limited number of studies have observed no
mutations in the EFNB2 gene among 331 patients with isolated
anorectal malformations (11), nor
in patients with persistent cloaca and associated kidney
malformations (30). This suggests
that mutations in the EFNB2 gene do not exclusively affect
the development of the anorectal and geniourinary tract. An
additional important region, 13q22.1–31.3, was identified in two
patients harboring an interstitial deletion (3), Database of Chromosomal Imbalance and
Phenotype in Humans using Ensembl Resources (DECIPHER) ID 270910]
(31). This region contains 27
OMIM genes, including the following 5 morbid genes: the
ceroid-lipofuscinosis, neuronal 5 gene, the endothelin receptor B
gene (EDNRB), the SLIT and NTRK like family member 1 gene,
the microRNA-17-92a-1 cluster host gene (MIR17HG) and the
glypican 6 gene (GPC6). Until recently, none of these genes
have been proven to be associated with urogenital/anorectal
anomalies, however, previous studies have indicated they may induce
a number of congenital malformations (32–39).
By contrast, homozygous or heterozygous mutations in EDNRB,
located in 13q22.3, are thought to give rise to 3 types of allelic
disease: Hirschsprung disease, albinism, black lock, cell migration
disorder syndrome (ABCD syndrome) and Waardenburg syndrome, which
are attributed to a defect in the migration of neural crest cells
(33–35). The occurrence of comorbid
Hirschsprung disease and hypospadisa/anorectal malformations has
been reported in a number of studies (40–42).
We hypothesize that aganglionic alterations that occur as a result
of EDNRB defects may be involved in the underlying
mechanisms of urogenital/anorectal anomalies. Therefore, the role
of the EDNRB gene in the development of the urogenital tract
requires further investigation to elucidate the molecular and
pathological effects of the EDNRB gene.
Congenital heart disease in 13q deletion syndrome is
more complex than in isolated cases, and include cases of Tetralogy
of Fallot combined with additional heart defects (6), at least 2 heart anomalies in one
patient (14,15,19),
or rare type complex heart anomalies (20,43).
In the present study, five of the six heart anomalies identified in
patient 1 were rare types, and included, DORV, SA defects,
mixed-type ASD, PLSVC and severe PS. These complex conditions
associated with cardiovascular anomalies in 13q deletion syndrome,
suggest that multiple genes may be involved in its pathogenesis. To
date, at least 2 critical regions, 13q31.3 and 13q33.3–13q34, have
been reviewed by the literature (3,6,8,10,14–16,19–22)
(DECIPHER ID 253794). In the 13q33.3–13q34 region, morbid OMIM
genes collagen type IV α1 chain (COL4A1) and COL4A2,
were identified as potential candidates for heart development in a
patient with an interstitial deletion of 13q33.3-q34 that presented
with DORV (20). Previous studies
have demonstrated that the collagen IV protein encoded by
COL4A1 and COL4A2 serves a vital role during early
cardiac development, and specifically in the development of the
atria and outflow tract in mouse and human cardiovascular
progenitor cells in fetal hearts (44,45).
EFNB2 located in 13q33.3, though not a morbid OMIM gene, is
an excellent candidate for congenital heart disease.
Efnb2−/− null homozygotes display early embryonic
lethality due to severe defects in cardiovascular development
(46,47). However, the majority of
Efnb2lacZ/lacZ homozygotes survive embryonic
development and are born live only to perish within the first day
due to cardiac abnormalities (48). These data suggest that insufficient
EFNB2 expression may serve an important role in the
pathogenesis of cardiovascular abnormalities. In an additional
region of 13q31.3, which was identified in two patients with a
microdeletion (DECIPHER ID 253794) (20,29),
2 OMIM morbid genes MIR17HG and GPC6 were present.
MIR17HG is associated with Feingold syndrome type 2
(37), which is an autosomal
dominant disorder characterized by variable combinations of
microcephaly, limb malformations, esophageal and duodenal atresia,
and learning disability/mental retardation. Cardiac and renal
malformations, vertebral anomalies and deafness have been described
in a minority of patients. GPC6 is associated with
omodysplasia 1 (38,39), a rare autosomal recessive skeletal
dysplasia characterized by severe congenital micromelia with
shortening and distal tapering of the humeri and femora producing a
club-like appearance. Patients with GPC6 mutations may
additionally present with cryptorchidism, hernias, congenital heart
defects and cognitive delay (38,39).
Heart development is a complex process, which involves atrial and
ventricular septation, giant vascular sprouting, branching and
tubularization. It has been hypothesized that multiple proteins and
factors may be required, with precise timing and spatial expression
patterns. Therefore, the haploid insufficiency of MIR17HG,
GPC6, EFNB2, COL4A1 and COL4A2 genes
may co-contribute to the complex heart anomalies observed in
patient 1 with a 13q31.3-qter deletion in the present study.
Gastrointestinal tract anomalies are rare in 13q
deletion syndrome, and primarily involve the esophagus and its
neighboring organs. They include the development of
tracheoesophageal fistula, esophageal atresia (4), pyloric stenosis (8) and esophageal hiatus hernia with
gastroesophageal reflux, and gastroesophageal reflux was observed
in patient 2 of the present study. Additional digestive anomalies
include common mesentery, pancreas anomalies, gall bladder
agenesis/hypoplasia and spleen hypoplasia/supernumerary spleen
(6). Two regions spanning
13q31.3-q33.1 and 13q33.2-q34 are thought to be involved, however,
it is not conclusive due to the limited number of cases. Among the
morbid OMIM genes in the 13q31.3-q33.1 region, only MIR17HG
in 13q31.3, which induces Feingold syndrome type 2, has been
associated with tracheoesophageal fistula and esophageal atresia
(37). However, in the 13q33.2-q34
region, no morbid OMIM gene may provide an explanation for these
anomalies. The non-morbid gene EFNB2 in 13q33.3, is thought
to be involved in intestinal epithelial architecture via the EPH
receptor B2-EFNB signaling pathway (49,50),
which may explain, in part, these gastrointestinal anomalies.
However, this does not explain the majority of reported cases, and
thus the precise regions require further investigation with more
detailed cytogenetic information and molecular data on the complex
symptoms of gastrointestinal anomalies.
When combining the information from patient 1 and 2
of the present study with the results of previous studies involving
patients with VACTERL syndrome, it is apparent that the
urogenital/anorectal anomalies, congenital heart disease and
gastrointestinal tract anomalies may involve common or overlapping
regions of deletion in chromosome 13q, and suggests they may share
a common molecular mechanism. Increasing numbers of microdeletions
are being identified in patients using the array-CGH technique, and
the morbid genes identified may therefore provide a greater
understanding of the molecular mechanisms underlying chromosome 13q
deletion syndrome. In addition, obtaining molecular data from
non-morbid OMIM genes in knockout animal models may reveal the
pathological processes during development.
Acknowledgements
The authors would like to thank the patients and
their parents, as well as all the associated physicians who
contributed to the analysis of patient samples and collection of
clinical data. The present study was funded by the Liaoning
Province Natural Science Foundation (grant no. 2013021021).
References
|
1
|
Allderdice PW, Davis JG, Miller OJ, Kliner
HP, Warburton D, Miller DA, Allen FH Jr, Abrams CA and McGilvray E:
The 13q-deletion syndrome. Am J Hum Genet. 21:499–512.
1969.PubMed/NCBI
|
|
2
|
Brown S, Gersen S, Anyane-Yeboa K and
Warburton D: Preliminary definition of a ‘critical region’ of
chromosome 13 in q32: Report of 14 cases with 13q deletions and
review of the literature. Am J Med Genet. 45:52–59. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Ballarati L, Rossi E, Bonati MT, Gimelli
S, Maraschio P, Finelli P, Giglio S, Lapi E, Bedeschi MF, Guerneri
S, et al: 13q Deletion and central nervous system anomalies:
Further insights from karyotype-phenotype analyses of 14 patients.
J Med Genet. 44:e602007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Walczak-Sztulpa J, Wisniewska M,
Latos-Bielenska A, Linné M, Kelbova C, Belitz B, Pfeiffer L,
Kalscheuer V, Erdogan F, Kuss AW, et al: Chromosome deletions in
13q33-34: Report of four patients and review of the literature. Am
J Med Genet A. 146A:337–342. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brown S, Russo J, Chitayat D and Warburton
D: The 13q-syndrome: The molecular definition of a critical
deletion region in band 13q32. Am J Hum Genet. 57:859–866.
1995.PubMed/NCBI
|
|
6
|
Quélin C, Bendavid C, Dubourg C, de la
Rochebrochard C, Lucas J, Henry C, Jaillard S, Loget P, Loeuillet
L, Lacombe D, et al: Twelve new patients with 13q deletion
syndrome: Genotype-phenotype analyses in progress. Eur J Med Genet.
52:41–46. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Garcia NM, Allgood J, Santos LJ, Lonergan
D, Batanian JR, Henkemeyer M, Bartsch O, Schultz RA, Zinn AR and
Baker LA: Deletion mapping of critical region for hypospadias,
penoscrotal transposition and imperforate anus on human chromosome
13. J Pediatr Urol. 2:233–242. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kirchhoff M, Bisgaard AM, Stoeva R,
Dimitrov B, Gillessen-Kaesbach G, Fryns JP, Rose H, Grozdanova L,
Ivanov I, Keymolen K, et al: Phenotype and 244k array-CGH
characterization of chromosome 13q deletions: An update of the
phenotypic map of 13q21.1-qter. Am J Med Genet Part A.
149A:894–905. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Shojaei A, Behjati F, Derakhshandeh-Peykar
P, Razzaghy-Azar M, Otukesh H, Kariminejad R, Dowlati MA,
Rashidi-Nezhad A and Tavakkoly-Bazzaz J: Partial trisomy 7q and
monosomy 13q in a child with disorder of sex development:
Phenotypic and genotypic findings. Gene. 517:137–145. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Yang YF, Ai Q, Huang C, Chen JL, Wang J,
Xie L, Zhang WZ, Yang JF and Tan ZP: A 1.1Mb deletion in distal 13q
deletion syndrome region with congenital heart defect and postaxial
polydactyly: Additional support for a CHD locus at distal 13q34
region. Gene. 528:51–54. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dworschak GC, Draaken M, Marcelis C, et
al: De novo 13q deletions in two patients with mild anorectal
malformations as part of VATER/VACTERL and VATER/VACTERL-like
association and analysis of EFNB2 in patients with anorectal
malformations. Am J Med Genet A. 161A:3035–3041. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
McKay RD: The mechanism of G and C banding
in mammalian metaphase chromosomes. Chromosoma. 44:1–14. 1973.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Walsh LE, Vance GH and Weaver DD: Distal
13q Deletion Syndrome and the VACTERL association: Case report,
literature review, and possible implications. Am J Med Gene.
98:137–144. 2001. View Article : Google Scholar
|
|
15
|
Kaylor J, Alfaro M, Ishwar A, Sailey C,
Sawyer J and Zarate YA: Molecular and cytogenetic evaluation of a
patient with ring chromosome 13 and discordant results. Cytogenet
Genome Res. 144:104–108. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Cain CC, Saul DO, Oehler E, Blakemore K
and Stetten G: Prenatal detection of a subtle unbalanced chromosome
rearrangement by karyotyping, FISH and array comparative genomic
hybridization. Fetal Diagn Ther. 24:286–290. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kataoka A, Hirakawa S, Iwamoto M, Sakumura
Y, Yoshinaga R and Ohba T: Prenatal diagnosis of a case of partial
monosomy/monosomy 13 mosaicism: 46,XX,r(13)(p11q33)/45,XX,-13
suspected by nuchal fold translucency increasing. Kurume Med J.
58:127–130. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Andresen JH, Aftimos S, Doherty E, Love DR
and Battin M: 13q33.2 deletion: A rare cause of ambiguous genitalia
in a male newborn with growth restriction. Acta Paediatr.
99:784–786. 2010.PubMed/NCBI
|
|
19
|
Mimaki M, Shiihara T, Watanabe M, Hirakata
K, Sakazume S, Ishiguro A, Shimojima K, Yamamoto T, Oka A and
Mizuguchi M: Holoprosencephaly with cerebellar vermis hypoplasia in
13q deletion syndrome: Critical region for cerebellar dysgenesis
within 13q32.2q34. Brain Dev. 37:714–718. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
McMahon CJ, Breathnach C, Betts DR,
Sharkey FH and Greally MT: De Novo interstitial deletion 13q33.3q34
in a male patient with double outlet right ventricle, microcephaly,
dysmorphic craniofacial findings, and motor and developmental
delay. Am J Med Genet A. 167A:1134–1141. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Valdes-Miranda JM, Soto-Alvarez JR,
Toral-Lopez J, González-Huerta L, Perez-Cabrera A, Gonzalez-Monfil
G, Messina-Bass O and Cuevas-Covarrubias S: A novel microdeletion
involving the 13q31.3-q32.1 region in a patient with normal
intelligence. Eur J Med Genet. 57:60–64. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Huang C, Yang YF, Yin N, Chen JL, Wang J,
Zhang H and Tan ZP: Congenital heart defect and mental retardation
in a patient with a 13q33.1–34 deletion. Gene. 498:308–310. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Czeizel A and Ludányi I: An aetiological
study of the VACTERL-association. Eur J Pediatr. 144:331–337. 1985.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
McMullen KP, Karnes PS, Moir CR and
Michels VV: Familial recurrence of tracheoesophageal fistula and
associated malformations. Am J Med Genet. 63:525–528. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Quan L and Smith DW: The VATER
association. Vertebral defects, anal atresia, T-E fistula with
esophageal atresia, radial and rnal dysplasia: A spectrum of
associated defects. J Pediatr. 82:104–107. 1973. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Rogers DS, Paidas CN, Morreale RF and
Huthcins GM: Septation of the anorectal and genitourinary tract in
the human embryo: Crucial role of the catenoidal shape of the
urorectal sulcus. Teratology. 66:144–152. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Hynes PJ and Fraher JP: The development of
the male genitourinary system. I. The origin of the urorectal
septum and the formation of the perineum. Br J Plast Surg.
57:27–36. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Baskin LS, Erol A, Jegatheesan P, Li Y,
Liu W and Cunha GR: Urethral seam formation and hypospadias. Cell
Tissue Res. 305:379–387. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Dravis C, Yokoyama N, Chumley MJ, Cowan
CA, Silvany RE, Shay J, Baker LA and Henkemeyer M: Bidirectional
signaling mediated by ephrin-B2 and EphB2 controls urorectal
development. Dev Biol. 271:272–290. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Jenkins D, Bitner-Glindzicz M, Thomasson
L, Malcolm S, Warne SA, Feather SA, Flanagan SE, Ellard S, Bingham
C, Santos L, et al: Mutational analyses of UPIIIA, SHH, EFNB2 and
HNF1beta in persistent cloaca and associated kidney malformations.
J Pediatr Urol. 3:2–9. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Firth HV, Richards SM, Bevan AP, Clayton
S, Corpas M, Rajan D, Van Vooren S, Moreau Y, Pettett RM and Carter
NP: DECIPHER: Database of chromosomal imbalance and phenotype in
humans using ensembl resources. Am J Hum Genet. 84:524–533. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Xin W, Mullen TE, Kiely R, Min J, Feng X,
Cao Y, O'Malley L, Shen Y, Chu-Shore C, Mole SE, et al: CLN5
mutations are frequent in juvenile and late-onset non-Finnish
patients with NCL. Neurology. 74:565–571. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Tanaka H, Moroi K, Iwai J, Takahashi H,
Ohnuma N, Hori S, Takimoto M, Nishiyama M, Masaki T, Yanagisawa M,
et al: Novel mutations of the endothelin B receptor gene in
patients with Hirschsprung's disease and their characterization. J
Biol Chem. 273:11378–11383. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Puffenberger EG, Hosoda K, Washington SS,
Nakao K, deWit D, Yanagisawa M and Chakravarti A: A missense
mutation of the endothelin-B receptor gene in multigenic
Hirschsprung's disease. Cell. 79:1257–1266. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Verheij JB, Kunze J, Osinga J, van Essen
AJ and Hofstra RM: ABCD syndrome is caused by a homozygous mutation
in the EDNRB gene. Am J Med Genet. 108:223–225. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Abelson JF, Kwan KY, O'Roak BJ, Baek DY,
Stillman AA, Morgan TM, Mathews CA, Pauls DL, Rasin MR, Gunel M, et
al: Sequence variants in SLITRK1 are associated with Tourette's
syndrome. Science. 310:317–320. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
de Pontual L, Yao E, Callier P, Faivre L,
Drouin V, Cariou S, Van Haeringen A, Geneviève D, Goldenberg A,
Oufadem M, et al: Germline deletion of the miR-17-92 cluster causes
skeletal and growth defects in humans. Nat Genet. 43:1026–1030.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Albano LM, Oliveira LA, Bertola DR, Mazzu
JF and Kim CA: Omodysplasia: The first reported Brazilian case.
Clinics (Sao Paulo). 62:531–534. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Elcioglu NH, Gustavson KH, Wilkie AO,
Yüksel-Apak M and Spranger JW: Recessive omodysplasia: Five new
cases and review of the literature. Pediat Radiol. 34:75–82. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Lukong CS, Mshelbwala PM, Anumah MA, Ameh
EA and Nmadu PT: Anorectal malformation coexisting with
Hirschsprung's disease: A report of two patients. Afr J Paediatr
Surg. 9:166–168. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Metts JC III, Kotkin L, Kasper S, Shyr Y,
Adams MC and Brock JW III: Genital malformations and coexistent
urinary tract or spinal anomalies in patients with imperforate
anus. J Urol. 158:1298–1300. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Raboei EH: Patients with anorectal
malformation and Hirschsprung's disease. Eur J Pediatr Surg.
19:325–327. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Jobanputra V, Wilson A, Shirazi M,
Feenstra H, Levy B, Anyane-Yeboa K and Warburton D: Partial
uniparental disomy with mosaic deletion 13q in an infant with
multiple congenital anomalies. Am J Med Genet A. 161A:2393–2395.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Hanson KP, Jung JP, Tran QA, Hsu SP, lida
R, Ajeti V, Campagnola PJ, Eliceiri KW, Squirrell JM, Lyons GE and
Ogle BM: Spatial and temporal analysis of extracellular matrix
proteins in the developing murine heart: A blueprint for
regeneration. Tissue Eng Part A. 19:1132–1143. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Schenke-Leyland K, Nsair A, Van Handel B,
Angelis E, Gluck JM, Votteler M, Goldhaber JI, Mikkola HK, Kahn M
and MacLellan WR: Recapitulation of the embryonic cardiovascular
progenitor cell niche. Biomaterials. 32:2748–2756. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Adams RH, Wilkinson GA, Weiss C, Diella F,
Gale NW, Deutsch U, Risau W and Klein R: Roles of ephrinB ligands
and EphB receptors in cardiovascular development: Demarcation of
arterial/venous domains, vascular morphogenesis, and sprouting
angiogenesis. Genes Dev. 13:295–306. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Wang HU, Chen ZF and Anderson DJ:
Molecular distinction and angiogenic interaction between embryonic
arteries and veins revealed by ephrin-B2 and its receptor Eph-B4.
Cell. 93:741–753. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cowan CA, Yokoyama N, Saxena A, Chumley
MJ, Silvany RE, Baker LA, Srivastava D and Henkemeyer M: Ephrin-B2
reverse signaling is required for axon pathfinding and cardiac
valve formation but not early vascular development. Dev Biol.
271:263–271. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Batlle E, Henderson JT, Beghtel H, van den
Born MM, Sancho E, Huls G, Meeldijk J, Robertson J, van de Wetering
M, Pawson T and Clevers H: Beta-catenin and TCF mediate cell
positioning in the intestinal epithelium by controlling the
expression of EphB/ephrinB. Cell. 111:251–263. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Batlle E, Bacani J, Begthel H, Jonkheer S,
Gregorieff A, van de Born M, Malats N, Sancho E, Boon E, Pawson T,
et al: EphB receptor activity suppresses colorectal cancer
progression. Nature. 435:1126–1130. 2005. View Article : Google Scholar : PubMed/NCBI
|