Introduction
Acute myeloid leukemia (AML) is one of the most
frequently occurring malignant diseases of the blood and patients
of all ages may present with symptoms. It has previously been
reported that AML in children accounts for 25% of pediatric
leukemia cases and affects ~180 patients annually in Japan
(1). A total of 19,000 cases of
AML are diagnosed each year, with ~10,000 of these in the United
States (2). Outcomes have improved
in younger patients, with a 40–50% 5-year overall survival rate
(3). However, the majority of AML
cases occur in adults, and in these cases the mortality rate
remains high. It has been demonstrated that only 10–20% of patients
aged >60 years survive to 5 years; 80% of patients are incurable
as a result of primary refractoriness, relapse or
treatment-associated mortality (4,5). AML
has several subtypes and treatment and prognosis varies among them.
AML is treated traditionally with chemotherapy and recent genetic
research has provided more personalized treatment options.
Clinicians can now predict which drug or drugs may work best for a
particular person, and how long that person is likely to survive.
Furthermore, numerous studies have reported that various genetic
abnormalities in the following genes: Nucleophosmin 1, runt related
transcription factor 1, Tet methylcytosine dioxygenase 2 and
isocitrate dehydrogenase [NADP(+)] 1 cytosolic, are associated with
the occurrence, progression and recurrence of AML and may therefore
be used to predict prognosis and guide future therapeutic research
(6–9). Döhner et al (4) summarized the frequency and clinical
significance of various important mutated genes. The primary first
line therapeutic for the treatment of AML is combined chemotherapy
with anthracycline and cytarabine (10). Further therapeutic options include
the hypomethylating agents decitabine and azacitidine) low-dose
cytarabine, investigational agents, and supportive care with
hydroxyurea and transfusions (11). Decitabine is a deoxynucleoside
analogue of cytidine that selectively inhibits DNA
methyltransferases. It is considered an effective and
well-tolerated alternative treatment to cytarabine or supportive
care in older patients with AML (12). To improve the efficacy and
structure of decitabine, the present study examined the mechanism
underlying the effects of decitabine and cytarabine on AML, via
microarray analysis. Although some progress has been made in
targeted therapy of AML, the diagnosis and treatment of it remain
challenging. The present study identified additional biomarkers
associated with the therapeutic effects of drugs, in order to
explore the corresponding mechanisms.
Materials and methods
Microarray data
The microarray datasets GSE40442 (13) and GSE40870 (13) were downloaded from the Gene
Expression Omnibus (GEO) database (www.ncbi.nlm.nih.gov/geo/). The expression profile of
GSE40442 contained 67 primary AML samples cultured with medium only
for 1 day, followed by 3 days treatment with decitabine (the case
group; n=17), cytarabine (the control group; n=16), dimethyl
sulfoxide (DMSO; n=17) or untreated (n=17). These data were
identified via the GPL5188 [HuEx-1_0-st] Affymetrix Human Exon 1.0
ST Array [probe set (exon) version] platform. The GSE40870 profile
presented the methylation data of AML cell samples treated with
decitabine (the case group; n=16), cytarabine (the control group;
n=16) or DMSO (n=16). Detection of the methylation data was
performed via GPL13534 Illumina HumanMethylation450 BeadChip
(HumanMethylation 450_15017482).
Data preprocessing
To create the expression profile, the original data
were converted into a recognizable format in R, and the affy
(14) package (bioconductor.org/ packages/release/bioc/html/affy.html) was used for
background correction and normalization, followed by conversion
from the probe symbol to the gene symbol with the biomaRt (15) package of R (bioconductor.org/packages/release/bioc/html/biomaRt.html).
The β-value of every methylated site in all samples was extracted
via GenomeStudio software version 2.0 (Illumina, Inc., San Diego,
CA, USA) to create a methylation profile.
Identification of differentially
expressed genes (DEGs) and differentially methylated sites
For GSE40442, the Linear Models for Microarray Data
(16) package of R
(bioconductor.org/packages/release/bioc/html/limma.html)
was used to identify the DEGs in AML cells treated with decitabine
compared with those treated with cytarabine. The DEGs were
identified according to the criteria P<0.05 and
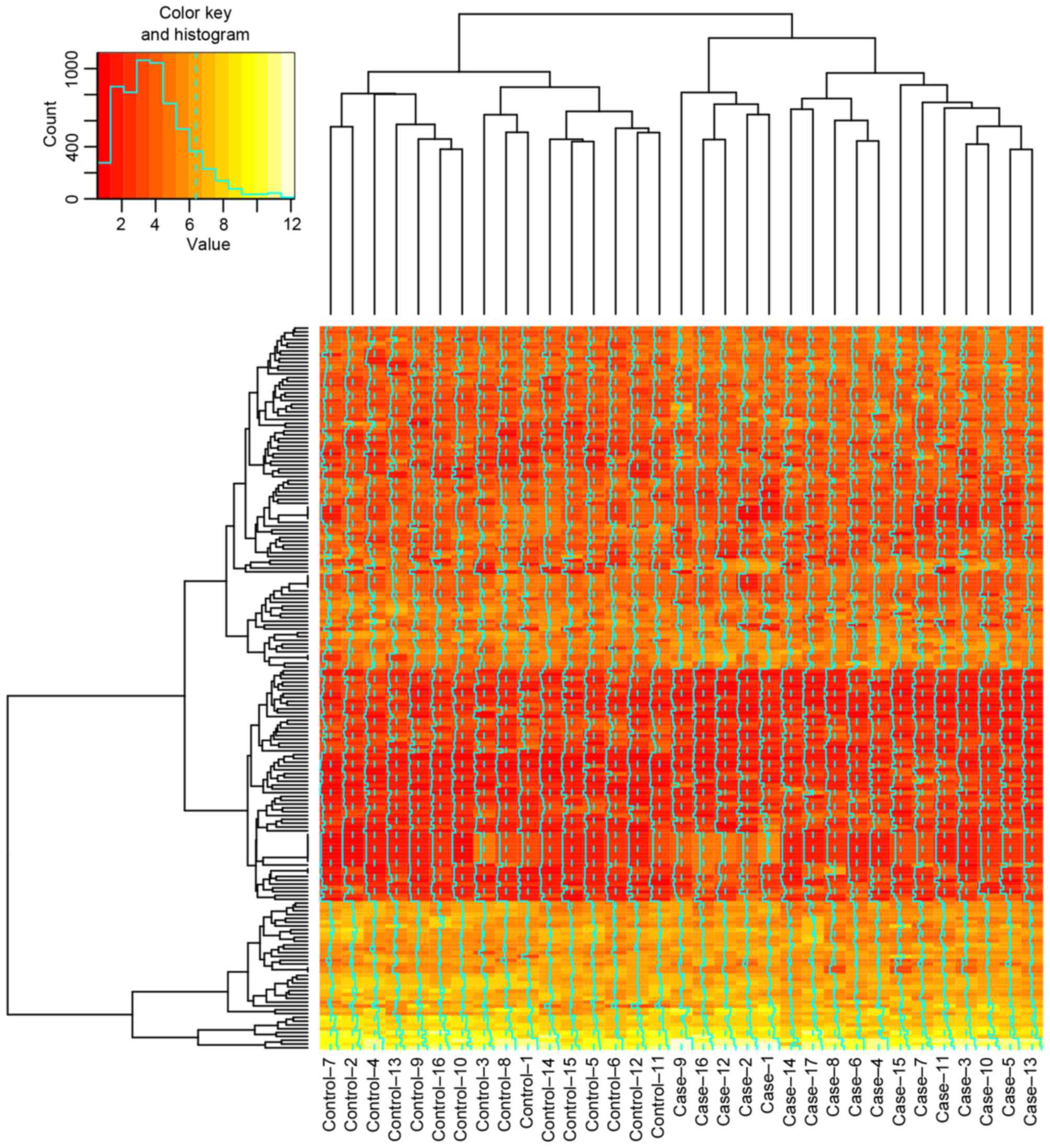
log(fold-change)>0.5. The heatmap of DEGs in every sample of the
control and the case group was constructed. For GSE40870, the
differentially methylated sites were identified in AML cells
treated with decitabine compared with cytarabine via the Illumina
Methylation Analyzer (17) package
of R (ima.r-forge.r-project.org/), and were screened out
with the criteria P<0.05 and log(fold-change)>0.2.
Functional enrichment analysis
Gene Ontology (GO) enrichment analysis of DEGs was
performed via the Database for Annotation, Visualization and
Integrated Discovery (david.abcc.ncifcrf.gov/) (18) with the threshold of P<0.05.
Screening of important genes and
methylated sites
Genes corresponding to the differentially methylated
sites were obtained by the annotation package of the methylation
microarray platform. The genes that exhibited an overlap compared
with DEGs were selected, and those that exhibited the opposite
trend in the methylation variation compared with their expression
were screened out.
Identification and analysis of
important transcription factor (TF)-gene pairs and establishment of
TF-gene regulated network
Methylation in the gene promoter region may affect
the binding of TFs to genes and result in the variation of gene
expression. Firstly, chromosomal locations of the methylation sites
were identified using the annotation package of the methylation
microarray platform. Following this, chromosomal locations of all
the known and predicted TF binding sites were downloaded from the
University of California Santa Cruz (UCSC) database (19) (genome.ucsc.edu/). The methylation sites were
considered to affect the binding of TFs and genes when the
chromosomal location of the methylation sites overlapped with the
region of the TF binding site. Furthermore, all the known and
predicted TF-gene pairs were downloaded from UCSC and the TF-gene
pairs were screened out. The TF-gene regulated network was
established via Cytoscape version 3.11 (www.cytoscape.org/).
Results
DEGs and differentially methylated
sites
A total of 190 DEGs (102 up- and 88 downregulated)
and 540 differentially methylated sites were identified in AML
cells treated with decitabine compared with cytarabine, and all the
identified differentially methylated sites were hypomethylated. The
top 30 DEGs and the top 30 differentially methylated sites are
presented in Tables I and II, respectively, and the heatmap of DEGs
is presented in Fig. 1.
 | Table I.Top 30 differentially expressed genes
in acute myeloid leukemia cells treated with decitabine compared
with those treated with cytarabine. |
Table I.
Top 30 differentially expressed genes
in acute myeloid leukemia cells treated with decitabine compared
with those treated with cytarabine.
| Gene | Log(fold-change) | P-value |
|---|
| PNMA5 | 1.008734 |
4.61×10−7 |
| COL14A1 | 0.981479 |
7.39×10−6 |
| LINC01344 | 0.688924 |
1.12×10−5 |
| PPP1R27 | 0.991804 |
1.52×10−5 |
| ACRC | 0.794564 |
1.99×10−5 |
| TKTL1 | 1.047284 |
2.91×10−5 |
| DAZL | 0.504424 | 0.000131 |
| RBMY3AP | −0.50665 | 0.000302 |
| MIR675 | 0.598142 | 0.000474 |
| MYBL2 | −0.62118 | 0.000611 |
| BNIP3P9 | 0.505733 | 0.00071 |
| HIST1H1C | 0.518146 | 0.000896 |
| TK1 | −0.63139 | 0.000925 |
| HIST1H1E | 0.612766 | 0.001018 |
| PCNA | −0.52607 | 0.001235 |
| TRAJ13 | −0.96523 | 0.001403 |
| RN7SKP60 | 0.640781 | 0.001425 |
| CDKN1A | −0.61749 | 0.0015 |
| HMGN5 | 0.575089 | 0.00238 |
| NFE4 | 1.069562 | 0.002412 |
| YPEL5P1 | 0.869379 | 0.002471 |
| FAM111B | −0.86319 | 0.002552 |
| HIGD1AP8 | 0.692903 | 0.002888 |
| OR2L3 | 0.913212 | 0.003109 |
| OR52P2P | −1.04122 | 0.003232 |
| MDM2 | −0.54666 | 0.003275 |
| GACAT2 | 0.692088 | 0.003317 |
| CCT4P2 | −0.72233 | 0.003713 |
| HIST1H1T | 0.761500 | 0.003745 |
| TMEM261P1 | −0.52041 | 0.003776 |
| Table II.Top 30 differentially methylated
sites in AML cells treated with decitabine compared with those
treated with cytarabine. |
Table II.
Top 30 differentially methylated
sites in AML cells treated with decitabine compared with those
treated with cytarabine.
| Methylation |
Log(fold-change) | P-value |
|---|
| cg22040989 | −0.46477 |
7.71×10−27 |
| cg19098118 | −0.32461 |
3.17×10−22 |
| cg14063817 | −0.34161 |
1.51×10−19 |
| cg17631454 | −0.33468 |
5.13×10−19 |
| cg02597199 | −0.32804 |
1.82×10−18 |
| cg08071595 | −0.35059 |
3.15×10−18 |
| cg27576136 | −0.20948 |
1.73×10−17 |
| cg09374462 | −0.25800 |
1.77×10−17 |
| cg12442125 | −0.35154 |
4.70×10−17 |
| cg05592278 | −0.31725 |
4.70×10−17 |
| cg27052900 | −0.24157 |
4.70×10−17 |
| cg22802167 | −0.26297 |
5.69×10−17 |
| cg08550094 | −0.35068 |
6.61×10−17 |
| cg21486341 | −0.20684 |
6.79×10−17 |
| cg08411833 | −0.27190 |
8.08×10−17 |
| cg17806847 | −0.25718 |
8.08×10−17 |
| cg03865944 | −0.20673 |
9.44×10−17 |
| cg03611733 | −0.21888 |
9.80×10−17 |
| cg12091641 | −0.31272 |
1.43×10−16 |
| cg17338368 | −0.24704 |
2.00×10−16 |
| cg23641672 | −0.21018 |
2.87×10−16 |
| cg23836413 | −0.20551 |
2.87×10−16 |
| cg05073880 | −0.22513 |
3.63×10−16 |
| cg12866103 | −0.29918 |
5.32×10−16 |
| cg03282689 | −0.25659 |
6.36×10−16 |
| cg07042346 | −0.24842 |
8.04×10−16 |
| cg09014775 | −0.21864 |
8.09×10−16 |
| cg05303739 | −0.20861 |
1.05×10−15 |
| cg11017535 | −0.24066 |
1.51×10−15 |
| cg13987334 | −0.23196 |
1.51×10−15 |
Enriched GO terms of the DEGs
A total of 36 enriched GO terms of DEGs, including
nucleosome, protein-DNA complex, and nucleosome, chromatin and
protein-DNA complex assemblies, were obtained and are presented in
Table III.
| Table III.Enriched GO terms of differentially
expressed genes. |
Table III.
Enriched GO terms of differentially
expressed genes.
| Category | GO ID | GO name | P-value |
|---|
| CC | 0000786 | Nucleosome |
1.03×10−7 |
| CC | 0032993 | Protein-DNA
complex |
6.69×10−7 |
| BP | 0006334 | Nucleosome
assembly |
8.70×10−7 |
| BP | 0031497 | Chromatin
assembly |
1.07×10−6 |
| BP | 0065004 | Protein-DNA complex
assembly |
1.40×10−6 |
| BP | 0034728 | Nucleosome
organization |
1.59×10−6 |
| BP | 0006323 | DNA packaging |
6.06×10−6 |
| CC | 0000785 | Chromatin |
7.65×10−6 |
| BP | 0006333 | Chromatin assembly
or disassembly |
9.73×10−6 |
| CC | 0005694 | Chromosome |
3.77×10−5 |
| CC | 0044427 | Chromosomal
part |
7.21×10−5 |
| BP | 0016584 | Nucleosome
positioning |
2.24×10−4 |
| BP | 0065003 | Macromolecular
complex assembly | 0.001008 |
| BP | 0034622 | Cellular
macromolecular complex assembly | 0.001458 |
| CC | 0031012 | Extracellular
matrix | 0.001562 |
| BP | 0043933 | Macromolecular
complex subunit organization | 0.001593 |
| BP | 0034621 | Cellular
macromolecular complex subunit organization | 0.002613 |
| BP | 0006259 | DNA metabolic
process | 0.003411 |
| BP | 0006325 | Chromatin
organization | 0.003467 |
| CC | 0005654 | Nucleoplasm | 0.004301 |
| CC | 0005578 | Proteinaceous
extracellular matrix | 0.006181 |
| BP | 0006260 | DNA
replication | 0.006457 |
| CC | 0044421 | Extracellular
region part | 0.007453 |
| BP | 0051276 | Chromosome
organization | 0.011355 |
| MF | 0003677 | DNA binding | 0.012103 |
| BP | 0006974 | Response to DNA
damage stimulus | 0.015069 |
| BP | 0030162 | Regulation of
proteolysis | 0.018269 |
| BP | 0033554 | Cellular response
to stress | 0.022660 |
| BP | 0006281 | DNA repair | 0.024911 |
| MF | 0005125 | Cytokine
activity | 0.024942 |
| BP | 0032026 | Response to
magnesium ion | 0.030924 |
| CC | 0031981 | Nuclear lumen | 0.035330 |
| BP | 0046685 | Response to
arsenic | 0.038507 |
| CC | 0000307 | Cyclin-dependent
protein kinase holoenzyme complex | 0.039727 |
| BP | 0051726 | Regulation of cell
cycle | 0.040353 |
| MF | 0004984 | Olfactory receptor
activity | 0.049749 |
Important genes and methylated
sites
A total of 240 genes were screened, in which 540
differentially methylated sites were identified. These 240 genes
were compared with the 190 DEGs, and the acid-repeat containing
protein (ACRC) exhibited an overlap. Furthermore,
ACRC corresponded to the methylated site of cg26924445 and
demonstrated the opposite trend in the methylation variation
compared with gene expression.
Important TF-gene pairs and the
TF-gene regulated network
A total of 60 TF-gene pairs and overlapped
methylated sites were screened out, including cg22475974-SET domain
bifurcated (SETDB)1, cg14063817-estrogen receptor
(ER)α A, cg22475974-ERα A,
cg05835309-SETDB1 and cg00334293-signal transducer and
activator of transcription-3. In addition, 140 pairs of TFs and
DEGs were identified, including CCAAT/enhancer binding protein β
(CEBPB)-cysteine rich secretory protein 3,
CEBPB-C-X-C motif chemokine ligand 2, CEBPB-fanconi
anemia complementation group I, CEBPB-histone cluster 1 H1
family member C and CEBPB-microRNA 378e. In addition, the 60
pairs of TFs and overlapped methylated sites contained 20 TFs and
51 methylated sites. The TF-gene regulated network was established
according to the 140 TF-gene pairs (Fig. 2). A total of 68 nodes and 140 pairs
were involved in the regulated network. Furthermore, the 68 nodes
contained 18 TFs (presented as triangles) and 50 genes (presented
as circles). The top 20 nodes according to connections with other
nodes in the network are presented in Table IV.
| Table IV.Top 20 most significant nodes
according to degree. |
Table IV.
Top 20 most significant nodes
according to degree.
| Node | Degree |
|---|
| TAF1 | 36 |
| CTCF | 25 |
| C-MYC | 14 |
| FOXA1 | 10 |
| RAD21 | 9 |
| CEBPB | 8 |
| PCNA | 8 |
| HEY1 | 6 |
| NRSF | 6 |
| FANCI | 6 |
| PU.1 | 5 |
| STAT3 | 5 |
| CXCL2 | 5 |
| THBS1 | 5 |
| HIST1H4H | 5 |
| NAMPT | 5 |
| RPL36AL | 5 |
| HIST1H1E | 5 |
| GABP | 4 |
| CCNE2 | 4 |
Discussion
Decitabine (Dacogen®;
5-aza-2′-deoxycytidine) has been extensively used for the treatment
of AML as an inhibitor of DNA methylation, which triggers
demethylation leading to consecutive reactivation of epigenetically
silenced tumor suppressor genes (20). When administered at low doses,
decitabine may reduce genomic DNA methylation as a consequence of
irreversible binding to DNA methyltransferases following
incorporation into newly synthesized DNA (21). Cytarabine inhibits DNA synthesis by
suppressing DNA polymerase activity; however, it additionally
inhibits the elongation of the polynucleotide chain and interferes
with the physiological function of DNA, which is important for the
treatment of hematological malignancies (22,23).
In the present study, all the identified differentially methylated
sites were hypomethylated in the primary AML samples treated with
low-dose decitabine compared with cytarabine, which is consistent
with differing underlying mechanisms of decitabine and cytarabine
in AML. The AML cell samples treated with decitabine differed from
those treated with cytarabine in the heatmap of DEGs.
In the present study, a total of 36 GO terms
enriched in DEGs were obtained. They were primarily associated with
the combination of protein and DNA, (protein-DNA complex and
protein-DNA complex assembly), chromosome conformation (chromatin
assembly or disassembly, chromosomal part and chromatin
organization), and biological processes associated with the
assembly of macromolecular complexes (nucleosome assembly,
protein-DNA complex assembly, macromolecular complex assembly and
cellular macromolecular complex assembly). It has previously been
demonstrated that DNA methylation is important in the biological
processes of genomic imprinting, X-chromosome inactivation,
suppression of transposable elements and carcinogenesis (24–28).
DNA methylation is considered a potent epigenetic modification and
may inhibit TF recruitment, resulting in suppression of
transcription (24,29), and closely associates with health
and disease in humans (30,31).
Furthermore, it has been previously reported that DNA methylation
is an epigenetic activity that affects the structure of
chromosomes, but not the sequence of genes (32–34).
Therefore, the aforementioned data demonstrated that decitabine may
affect AML via gene methylation.
Of the identified DEGs, ACRC was the only one
to additionally contain a differentially methylated site,
cg26924445, and demonstrated an opposite trend in methylation
variation compared with expression. Nestheide et al
(35) suggested that ACRC
is an important biomarker of Ewing sarcoma and concludes that
epigenetic dysregulation may contribute to the pathogenesis of
angiosarcoma, via analysis of expression and methylation profiles.
The results of the present study demonstrated that decitabine can
alter the methylation status of cg26924445, and that as in their
study, increasing expression of ACRC was conducive to
treating AML. Therefore, it was suspected that decitabine might
treat AML through altering the methylation status of cg26924445 in
ACRC. The results of the present study revealed that the
TATA-box binding protein associated factor 1 (TAF1) regulated the
most genes or TFs in the TF-gene regulated network. Therefore, TAF1
may act as a critical TF for decitabine treatment of AML, and may
be important in the differing underlying molecular mechanisms of
decitabine and cytarabine. Ben Abdelali et al (36) reported that the SET-NUP214
(TAF1/CAN) fusion gene is an important influencing factor in the
survival rate of AML. Therefore, TAF1 may be a potential novel
target gene in decitabine-treated AML. The CCCTC-binding factor
(CTCF) was another TF that regulated numerous genes or TFs.
Manodoro et al (37)
demonstrated that in AML, the methylation of CTCF binding sites may
result in loss of imprinting at 14q32. Furthermore, the present
study demonstrated that proliferating cell nuclear antigen
(PCNA) was the gene regulated by the greatest number of TFs,
and Buchi et al (38)
reported that the expression of PCNA was altered in AML
treated with decitabine or cytarabine.
In conclusion, the results of the present study
suggested that decitabine suppresses the function of certain
antitumor genes via methylation, in its role as a therapeutic agent
for the treatment of AML, and that this underlying mechanism of
action differs to that of cytarabine. The present study provides
information regarding potential drug targets, which may improve the
efficacy of decitabine in the treatment of AML.
Acknowledgements
The present study was supported by the Health Bureau
Science and Technology Foundation of Tianjin (grant no. 2012KZ063)
and the Municipal Science and Technology Commission of Tianjin
(grant no. 15ZLZLZF00440).
References
|
1
|
Taga T, Tomizawa D, Takahashi H and Adachi
S: Acute myeloid leukemia in children: Current status and future
directions. Pediatr Int. 58:71–80. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kadia TM, Ravandi F, Cortes J and
Kantarjian H: New drugs in acute myeloid leukemia. Ann Oncol.
27:770–778. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Döhner H, Estey EH, Amadori S, Appelbaum
FR, Büchner T, Burnett AK, Dombret H, Fenaux P, Grimwade D, Larson
RA, et al: Diagnosis and management of acute myeloid leukemia in
adults: Recommendations from an international expert panel, on
behalf of the European LeukemiaNet. Blood. 115:453–474. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Döhner H, Weisdorf DJ and Bloomfield CD:
Acute myeloid leukemia. N Engl J Med. 373:1136–1152. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Burnett A, Wetzler M and Löwenberg B:
Therapeutic advances in acute myeloid leukemia. J Clin Oncol.
29:487–494. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Marcucci G, Haferlach T and Döhner H:
Molecular genetics of adult acute myeloid leukemia: Prognostic and
therapeutic implications. J Clin Oncol. 29:475–486. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Meyer SC and Levine RL: Translational
implications of somatic genomics in acute myeloid leukaemia. Lancet
Oncol. 15:e382–e394. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Cancer Genome Atlas Research Network. Ley
TJ, Miller C, Ding L, Raphael BJ, Mungall AJ, Robertson A, Hoadley
K, Triche TJ Jr, Laird PW, et al: Genomic and epigenomic landscapes
of adult de novo acute myeloid leukemia. N Engl J Med.
368:2059–2074. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ding L, Ley TJ, Larson DE, Miller CA,
Koboldt DC, Welch JS, Ritchey JK, Young MA, Lamprecht T, McLellan
MD, et al: Clonal evolution in relapsed acute myeloid leukaemia
revealed by whole-genome sequencing. Nature. 481:506–510. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Roboz GJ: Current treatment of acute
myeloid leukemia. Curr Opin Oncol. 24:711–719. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang ES: Treating acute myeloid leukemia
in older adults. Hematology Am Soc Hematol Educ Program.
2014:14–20. 2014.PubMed/NCBI
|
|
12
|
Curran MP: Decitabine: A review of its use
in older patients with acute myeloid leukaemia. Drugs Aging.
30:447–458. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Klco JM, Spencer DH, Lamprecht TL,
Sarkaria SM, Wylie T, Magrini V, Hundal J, Walker J, Varghese N,
Erdmann-Gilmore P, et al: Genomic impact of transient low-dose
decitabine treatment on primary AML cells. Blood. 121:1633–1643.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: affy-analysis of Affymetrix GeneChip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Durinck S, Spellman PT, Birney E and Huber
W: Mapping identifiers for the integration of genomic datasets with
the R/Bioconductor package biomaRt. Nat Protoc. 4:1184–1191. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Smyth GK: Linear models and empirical
bayes methods for assessing differential expression in microarray
experiments. Stat Appl Genet Mol Biol. 3:Article32004. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang D, Yan L, Hu Q, Sucheston LE, Higgins
MJ, Ambrosone CB, Johnson CS, Smiraglia DJ and Liu S: IMA: An R
package for high-throughput analysis of Illumina's 450K Infinium
methylation data. Bioinformatics. 28:729–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sherman BT, da Huang W, Tan Q, Guo Y, Bour
S, Liu D, Stephens R, Baseler MW, Lane HC and Lempicki RA: DAVID
Knowledgebase: A gene-centered database integrating heterogeneous
gene annotation resources to facilitate high-throughput gene
functional analysis. BMC Bioinformatics. 8:4262007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kent WJ, Sugnet CW, Furey TS, Roskin KM,
Pringle TH, Zahler AM and Haussler D: The human genome browser at
UCSC. Genome Res. 12:996–1006. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Daskalakis M, Blagitko-Dorfs N and
Hackanson B: Decitabine. Small Molecules in Oncology Berlin
Heidelberg: Springer; pp. 131–157. 2010, View Article : Google Scholar
|
|
21
|
Flotho C, Claus R, Batz C, Schneider M,
Sandrock I, Ihde S, Plass C, Niemeyer CM and Lübbert M: The DNA
methyltransferase inhibitors azacitidine, decitabine and zebularine
exert differential effects on cancer gene expression in acute
myeloid leukemia cells. Leukemia. 23:1019–1028. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Löwenberg B: Sense and nonsense of
high-dose cytarabine for acute myeloid leukemia. Blood. 121:26–28.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Momparler RL: Optimization of cytarabine
(ARA-C) therapy for acute myeloid leukemia. Exp Hematol Oncol.
2:202013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hu S, Wan J, Su Y, Song Q, Zeng Y, Nguyen
HN, Shin J, Cox E, Rho HS, Woodard C, et al: DNA methylation
presents distinct binding sites for human transcription factors.
Elife. 2:e007262013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Egger G, Liang G, Aparicio A and Jones PA:
Epigenetics in human disease and prospects for epigenetic therapy.
Nature. 429:457–463. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Robertson KD: DNA methylation and human
disease. Nat Rev Genet. 6:597–610. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Feinberg AP: Phenotypic plasticity and the
epigenetics of human disease. Nature. 447:433–440. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Reik W: Stability and flexibility of
epigenetic gene regulation in mammalian development. Nature.
447:425–432. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Weaver IC, Hellstrom IC, Brown SE, Andrews
SD, Dymov S, Diorio J, Zhang TY, Szyf M and Meaney MJ: The
methylated-DNA binding protein MBD2 enhances NGFI-A
(egr-1)-mediated transcriptional activation of the glucocorticoid
receptor. Philos Trans R Soc Lond B Biol Sci. 369:pii: 20130513.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Robertson KD and Wolffe AP: DNA
methylation in health and disease. Nat Rev Genet. 1:11–19. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hu Y, Liu F, Lin IY, Gao GF and Zhu B:
Dissemination of the mcr-1 colistin resistance gene. Lancet Infect
Dis. 16:146–147. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lewis JD, Meehan RR, Henzel WJ,
Maurer-Fogy I, Jeppesen P, Klein F and Bird A: Purification,
sequence, and cellular localization of a novel chromosomal protein
that binds to methylated DNA. Cell. 69:905–914. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Weber M, Davies JJ, Wittig D, Oakeley EJ,
Haase M, Lam WL and Schübeler D: Chromosome-wide and
promoter-specific analyses identify sites of differential DNA
methylation in normal and transformed human cells. Nat Genet.
37:853–862. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
34
|
Cokus SJ, Feng S, Zhang X, Chen Z,
Merriman B, Haudenschild CD, Pradhan S, Nelson SF, Pellegrini M and
Jacobsen SE: Shotgun bisulphite sequencing of the Arabidopsis
genome reveals DNA methylation patterning. Nature. 452:215–219.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Nestheide S, Bridge JA, Barnes M, Frayer R
and Sumegi J: Pharmacologic inhibition of epigenetic modification
reveals targets of aberrant promoter methylation in Ewing sarcoma.
Pediatr Blood Cancer. 60:1437–1446. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ben Abdelali R, Roggy A, Leguay T, Cieslak
A, Renneville A, Touzart A, Banos A, Randriamalala E, Caillot D,
Lioure B, et al: SET-NUP214 is a recurrent γδ lineage-specific
fusion transcript associated with corticosteroid/chemotherapy
resistance in adult T-ALL. Blood. 123:1860–1863. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Manodoro F, Marzec J, Chaplin T,
Miraki-Moud F, Moravcsik E, Jovanovic JV, Wang J, Iqbal S, Taussig
D, Grimwade D, et al: Loss of imprinting at the 14q32 domain is
associated with microRNA overexpression in acute promyelocytic
leukemia. Blood. 123:2066–2074. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Buchi F, Spinelli E, Masala E, Gozzini A,
Sanna A, Bosi A, Ferrari G and Santini V: Proteomic analysis
identifies differentially expressed proteins in AML1/ETO acute
myeloid leukemia cells treated with DNMT inhibitors azacitidine and
decitabine. Leuk Res. 36:607–618. 2012. View Article : Google Scholar : PubMed/NCBI
|