Introduction
Syndactyly is one of the most common congenital limb
malformations and is associated with webbing of adjacent
fingers/toes (1). It is a
heterogeneous hereditary disease, with a prevalence of between 3
and 10 in every 10,000 births; however, a higher prevalence of
between 10 and 40 in every 10,000 births has been reported
(1,2). In addition, it is twice as common in
males, indicating that this disease may demonstrate incomplete
penetrance (3). Syndactyly exists
either as an isolated non-syndromic anomaly or as part of >300
syndromic abnormalities. Non-syndromic syndactyly may be cutaneous
or bony and unilateral or bilateral (4). To date, at least nine types (I–IX) of
phenotypically diverse non-syndromic syndactyly have been
identified. Five primary types (type I–V) are inherited as an
autosomal dominant trait with variable disease phenotypes and
incomplete penetrance (4); while
the remaining types and subtypes may demonstrate autosomal
recessive or X-linked hereditary patterns of inheritance (3). The phenotype of a specific patient
appears to be dependent on a specific gene, as well as their
genetic background and the subsequent signaling pathways involved
in limb formation (3).
Non-syndromic syndactyly is genetic in origin (5). At present, at least 11 disease loci
and eight disease-associated genes have been identified, including
the homeobox D13 gene (HOXD13), the fibulin 1 gene, the gap
junction protein alpha 1 gene, the limb development membrane
protein 1 gene, the LDL receptor related protein 4 gene, the
gremlin 1 gene, the formin 1 gene and the fibroblast growth factor
16 gene (1,6). Mutations in the HOXD13 gene
are responsible for the development of syndactyly type II-a and
syndactyly type V (7,8). The typical clinical features of
syndactyly type I-c include bilateral cutaneous or bony fusion of
the third and fourth fingers and occasionally of the third to fifth
fingers, with normal feet (2).
The aim of the present study was to identify the
genetic basis of syndactyly type I-c in four generations of a
single Chinese family.
Materials and methods
Participators and clinical data
The present study enrolled a single Chinese Han
family (15 members), and clinical data from physical examinations
and peripheral blood samples were collected from 11 members
(generation:family member: II:1, male, 64 years old; II:3, male, 54
years old; II:4, female, 51 years old; III:1, female, 41 years old;
III:2, female, 39 years old; III:3, male, 39 years old; III:4,
female, 38 years old; III:6, female, 35 years old; IV:1, female, 16
years old; IV:2, female, 8 years old; IV:3, male, 15 years old) of
the four-generation family enrolled at the Third Xiangya Hospital
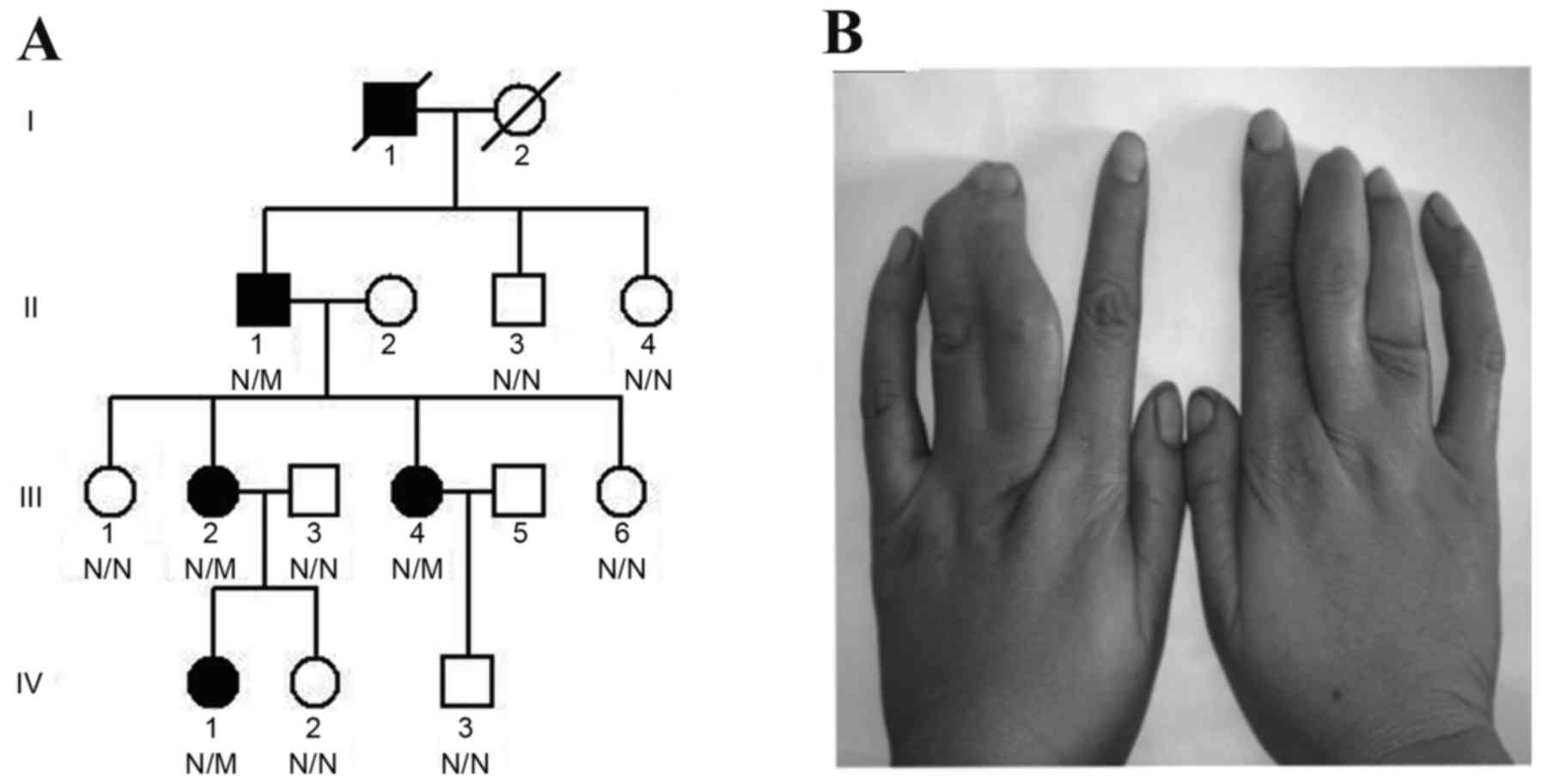
of Central South University in April 2013 (Changsha, China;
Fig. 1A); 2 family members were
deceased when the present study was performed. Physical
examinations and radiological examinations were conducted in the 4
affected patients (Table I), and
all 4 patients presented with cutaneous syndactyly of the third and
fourth fingers (Fig. 1B), without
foot involvement. Blood samples were collected from 100
ethnically-matched unrelated normal controls (gender, 50 males, 50
females; age, 39.24±8.31 years) during June and October in 2013.
They were recruited from the health examination center of the Third
Xiangya Hospital of Central South University (Changsha, China) and
examined by two professional surgeons according to the
classification system described previously (9). All participants included in the
present study provided written informed consent. The present study
was performed in compliance with the Declaration of Helsinki, and
was approved by the Institutional Review Board of the Third Xiangya
Hospital of Central South University.
 | Table I.Clinical and genetic data of four
patients from the same family harboring the HOXD13
c.917G>A (p.R306Q) mutation. |
Table I.
Clinical and genetic data of four
patients from the same family harboring the HOXD13
c.917G>A (p.R306Q) mutation.
| Factor |
| Generation,
member |
|
|---|
| Characteristic | II, 1 | III, 2 | III, 4 | IV, 1 |
| Sex | Male | Female | Female | Female |
| Age (years) | 64 | 39 | 38 | 16 |
| Genotype | Heterozygote | Heterozygote | Heterozygote | Heterozygote |
| Syndactyly | Yes | Yes | Yes | Yes |
| Affected fingers | 3rd and 4th | 3rd and 4th | 3rd and 4th | 3rd and 4th |
| Bilateral
affected | Yes | Yes | Yes | Yes |
| Cutaneous fusion | Yes | Yes | Yes | Yes |
| Bony fusion | No | No | No | No |
| Toes affected | No | No | No | No |
| Polydactyly | No | No | No | No |
| Brachydactyly | No | No | No | No |
Exome capture
Genomic DNA (gDNA) was extracted from peripheral
blood samples and used to extract the exome library using the
phenol-chloroform extraction method (phenol, Beijing Solarbio
Science & Technology Co., Ltd., Beijing, China; chloroform,
Tianjin Fuyu Fine Chemical Co., Ltd., Tianjin, China) as described
previously (10). Genomic DNA from
the proband (Fig. 1A: Generation
IV, member 1) was randomly sheared using the Illumina Paired-End
Sample Prep kit (Illumina, Inc., San Diego, CA, USA) and hybridized
using SureSelect biotinylated RNA library and SureSelect
hybridization buffers (Agilent Technologies, Inc., Santa Clara, CA,
USA), as well as Optimized gDNA prep and library prep kits (Agilent
Technologies, Inc.) to a concentration of 147 ng/mg at 45°C for
enrichment; all experimental operations were performed according to
the manufacturer's instructions for each kit. The enriched
exome-targeting library was sequenced using the Illumina HiSeq 2000
platform (Illumina, Inc.) to generate 90-bp paired-end reads, as
described previously (11). A mean
exome coverage of 69.64x was obtained, thus allowing for the
examination of selected regions at a sufficient depth to accurately
match 99.42% of the targeted exome (12).
Exome sequencing and variant
analysis
Human reference genomic sequences were obtained from
the University of California Santa Cruz genome browser database
(version hg19; build 37.1; http://genome.ucsc.edu/). Sequences obtained from the
proband were aligned using the short oligonucleotide analysis
package (SOAP) aligner software (soap2.21), and single nucleotide
polymorphisms (SNPs) were identified using SOAPsnp software
(version 1.03) (13). Small
insertions or deletions in the coding sequence and splicing sites
were detected as previously described (11,12),
and candidate SNPs were isolated. Variants were compared against
several public databases including the Single Nucleotide
Polymorphism database (dbSNP; build, 137; www.ncbi.nlm.nih.gov/projects/SNP/), the 1000 Genomes
Project (http://www.1000genomes.org/), the
International HapMap Project (http://hapmap.ncbi.nlm.nih.gov/) and the YanHuang
project (http://yh.genomics.org.cn/). Online
tools including Sorting Intolerant from Tolerant (SIFT) prediction
(http://sift.jcvi.org/), HumVar-trained
Polymorphism Phenotyping version 2, (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2/),
and MutationTaster prediction (http://www.mutationtaster.org/) algorithms were used
to estimate whether a variant may affect protein function (14,15).
The SIFT prediction scoring system ranges from 0 to 1. Amino acid
substitution is predicted as damaging (may have an important effect
on the function of the protein) if the score is ≤0.05, and
tolerated (may have no effect on the function of the protein) if
the score is >0.05. For PolyPhen-2, the predicted results
include benign, possibly damaging and probably damaging. The
PolyPhen-2 prediction scoring system ranges from 0 to 1. The closer
score is to 1, the more influence the variant has on the function
of the protein, and vice-versa. The predicted results of
MutationTaster include disease causing (have an important effect on
the function of the protein), disease causing automatic (have an
important effect on the function of the protein in several public
databases), polymorphism (may be a tolerant SNP) and polymorphism
automatic (may be a tolerant SNP in several public databases). The
probability of the prediction was measured by a scoring system
ranging from 0 to 1; the closer the score was to 1, the higher the
probability of the prediction, and vice-versa (14,15).
Known mutations and genes responsible for syndactyly, nonsense
mutations, read-through mutations and splicing mutations, or
mutations predicted to be damaging were prioritized in identifying
the pathogenic mutation. The National Center for Biotechnology
Information-Basic Local Alignment Search Tool (NCBI-BLAST;
http://blast.ncbi.nlm.nih.gov/Blast.cgi) was used to
examine sequence conservation among different species.
Mutation validation
Polymerase chain reaction primers were designed
using Primer 3 online primer design software (version 0.4.0;
http://frodo.wi.mit.edu/primer3). Sanger
sequencing was performed to validate the potential
disease-associated variants using the ABI 3500 sequencer (Applied
Biosystems; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
according to the standard procedures described previously (16,17).
The sequences of primers used to determine the causative variation
in the HOXD13 gene were as follows: Forward,
5′-GGGAGGAAGAAGAGAGTGCC-3′ and reverse,
5′-TCTAAGCTGTCTGTGGCCAA-3′.
Results
Exome sequencing of the proband (Fig. 1A: Generation IV, member 1) in the
syndactyly type I-c-affected family was first performed. A total of
5.92 billion bases of 90-bp paired-end read sequences for the
patient were generated, and 106,958 genetic variants, including
14,830 non-synonymous alterations, were identified in the coding
regions or the splicing sites. A prioritization scheme was applied
to identify the pathogenic mutation, using similar methods
described in a previous study (18). Given that the frequency of
syndactyly type I is 0.03% (2),
known variants identified in the dbSNP (build, 137), the 1000
Genomes Project, the International HapMap project and the YanHuang
project with a minor allele frequency value of >0.50% in each
database, were excluded. Using the aforementioned criteria, the
number of candidate genes were reduced by >92.99%. Variants not
annotated in public databases were prioritized for further
analysis.
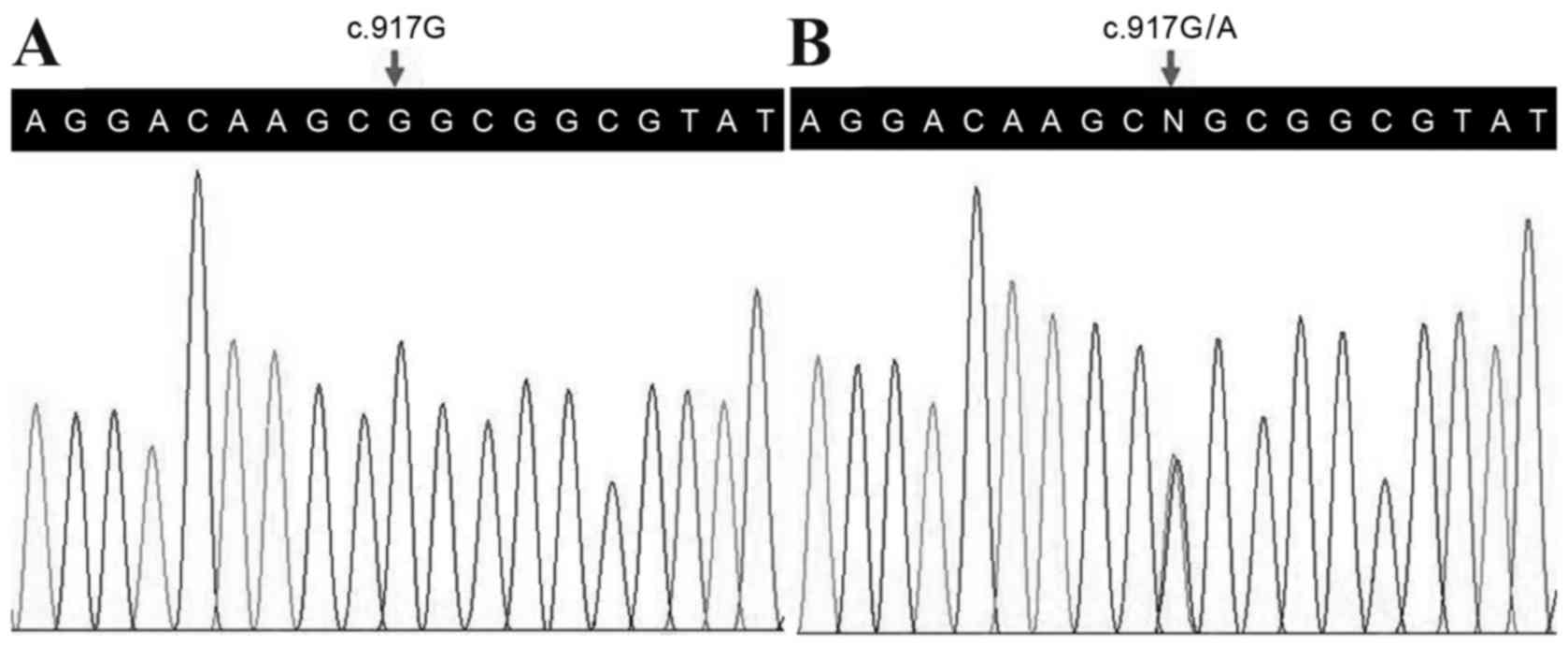
Following validation by Sanger sequencing, a
heterozygous mutation in the HOXD13 gene (c.917G>A,
p.R306Q) was observed in the proband (Fig. 2), however, no other variants in the
known disease-causing genes for syndactyly were identified by exome
sequencing. The mutation co-segregated among the affected family
members, and was absent in the unaffected family members, as well
as in the 100 normal controls. This suggests that the c.917G>A
mutation in HOXD13 may have been the pathogenic mutation. In
further support of this notion, this mutation was also absent in an
in-house database from the BGI-Shenzhen (Shenzhen, China) with
2,375 ethnically-matched controls. The results indicate that the
c.917G>A (p.R306Q) variant of the HOXD13 gene may be the
disease-associated mutation in this family. The c.917G>A
mutation is located in exon 2, which encodes the homeodomain in the
C-terminal region (a highly-conserved DNA binding motif) that is
responsible for binding to consensus DNA sequences (19). The mutation alters the amino acid
sequence at position 306 from Arg to Gln at residue 31 of this
homeodomain. This position was observed to be highly conserved
among different species (Fig. 3),
indicating its structural and functional importance. This mutation
was predicted to be disease-associated and affect protein function
using the MutationTaster and SIFT prediction algorithms, and
potentially damaging using the PolyPhen-2 algorithm (score,
1.00).
Discussion
Syndactyly displays marked genetic heterogeneity and
high clinical variability, and is characterized by limb
malformation (1). Syndactyly type
I-c is the rarest type among the four subtypes of syndactyly type I
(I-a, I-b, I-c and I-d), which is the most common form of
non-syndromic syndactyly (2). Two
mutations (p.R306Q and p.R306 G) in the HOXD13 homeodomain
have been identified in two independent Chinese families diagnosed
with syndactyly type I-c in a previous study (20). However, inconsistencies between the
clinical phenotypes and diagnostic criteria in selected patients
from the two families were observed, including a mild
synpolydactyly phenotype, involvement of the feet, clinodactyly and
fusion of the second and third fingers, which questions the
accuracy of the clinical diagnosis (20). The present study included a Chinese
Han family consisting of members diagnosed with typical syndactyly
type I-c that presented with bilateral cutaneous webbing of the
third and fourth fingers, normal toes, and an absence of additional
abnormalities. The results, together with those obtained from the
different prediction programs, identified the HOXD13
c.917G>A (p.R306Q) variant as the potential pathogenic mutation
for this family. Given that G>A or C>T transitions within CpG
dinucleotides are reportedly responsible for one-third of single
base-pair mutations in gene coding regions in human genetic
diseases (21), the c.917G>A
mutation may be caused by cytosine methylation followed by
deamination to thymine (C>T) on the complementary strand.
To the best of our knowledge, this is the first
study to present a family diagnosed with syndactyly type I-c, with
affected members harboring the HOXD13 p.R306Q mutation with
complete penetrance. The two independent Chinese families with
syndactyly reported previously (20), may have been diagnosed with unknown
complex types of non-syndromic syndactyly or syndactyly type I-c
variant types with incomplete penetrance and atypical phenotypes.
As two different amino acid substitutions from different families
have been identified at position 306, this position may be a
mutation hotspot. In addition, in a previous study, common mutation
screening was unable to exclude the genes involved in finger/toe
development, which was excluded by exome sequencing used in the
present study (20).
The HOXD13 gene is a member of the HOX
gene family, and encodes a transcription factor that serves a
crucial role in limb development (22). HOXD13 is composed of 2 exons
and encodes a 343-amino acid polypeptide sequence (23). At least 36 different HOXD13
mutations, including 17 missense/nonsense mutations, 8 repeats, 6
small deletions, 2 splicing mutations, a small insertion, a gross
deletion, as well as a gross insertion/duplication mutation, have
been described in the Human Gene Mutation Database (http://www.hgmd.cf.ac.uk/ac/index.php).
The majority of these mutations are observed in patients with limb
malformations, including syndactyly, synpolydactyly and
brachydactyly (24). Mouse models
with different genetic deficiencies in the HOXD13 gene have
been described previously in the literature. The majority of these
mouse models mimic some aspects of human synpolydactyly phenotypes,
further supporting the importance of the HOXD13 gene in limb
development (25–27).
In conclusion, the results of the present study
indicate that the c.917G>A (p.R306Q) mutation in the
HOXD13 gene may be the genetic basis of syndactyly type I-c
in a single Chinese family, which increases the phenotypic spectrum
of the HOXD13 gene. Whole exome sequencing provides a
cost-effective and expedited approach for identifying
disease-associated genes, and excludes the effects of background
genes in disorders displaying high clinical heterogeneity. The
results of the present study may provide novel insights into the
genetic diagnosis of syndactyly, and may have implications for
genetic counseling and the clinical management of this
condition.
Acknowledgements
The authors would like to thank the participating
members and investigators for their cooperation and efforts in
collecting clinical and genetic information and DNA specimens. The
present study was supported by grants from the National Key
Research and Development Program of China (grant no.
2016YFC1306604), the National Natural Science Foundation of China
(grant no. 81670216), the Natural Science Foundation of Hunan
Province (grant nos. 2015JJ4088 and 2016JJ2166), the Grant for the
Foster Key Subject of the Third Xiangya Hospital Clinical
Laboratory Diagnostics (H.D.), Zhishan Lead Project of the Third
Xiangya Hospital (grant no. 20150301) and the Fundamental Research
Funds for the Central Universities of Central South University
(grant no. 2015zzts318).
References
|
1
|
Malik S: Syndactyly: Phenotypes, genetics
and current classification. Eur J Hum Genet. 20:817–824. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Malik S, Schott J, Ali SW, Oeffner F,
Amin-ud-Din M, Ahmad W, Grzeschik KH and Koch MC: Evidence for
clinical and genetic heterogeneity of syndactyly type I: The
phenotype of second and third toe syndactyly maps to chromosome
3p21.31. Eur J Hum Genet. 13:1268–1274. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jordan D, Hindocha S, Dhital M, Saleh M
and Khan W: The epidemiology, genetics and future management of
syndactyly. Open Orthop J. 6:14–27. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sobreira NL, Cernach MC, Brunoni D and
Perez AB: Complex toe syndactyly with characteristic facial
phenotype: A new syndrome? Am J Med Genet A. 146A:1725–1728. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Fujii S, Yabe K, Kimura Y, Ito Y, Rokukawa
M, Furukawa M, Ito K, Matsuura M and Kiguchi M: Syndactyly lethal:
New mutation with multiple malformations occurring in Sprague
Dawley rats. Congenit Anom (Kyoto). 49:262–268. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jamsheer A, Zemojtel T, Kolanczyk M,
Stricker S, Hecht J, Krawitz P, Doelken SC, Glazar R, Socha M and
Mundlos S: Whole exome sequencing identifies FGF16 nonsense
mutations as the cause of X-linked recessive metacarpal 4/5 fusion.
J Med Genet. 50:579–584. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Zhou X, Zheng C, He B, Zhu Z, Li P, He X,
Zhu S, Yang C, Lao Z, Zhu Q and Liu X: A novel mutation outside
homeodomain of HOXD13 causes synpolydactyly in a Chinese family.
Bone. 57:237–241. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhao X, Sun M, Zhao J, Leyva JA, Zhu H,
Yang W, Zeng X, Ao Y, Liu Q, Liu G, et al: Mutations in HOXD13
underlie syndactyly type V and a novel brachydactyly-syndactyly
syndrome. Am J Hum Genet. 80:361–371. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deng H and Tan T: Advances in the
molecular genetics of non-syndromic syndactyly. Curr Genomics.
16:183–193. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Guo Y, Yuan L, Yi J, Xiao J, Xu H, Lv H,
Xiong W, Zheng W, Guan L, Zhang J, et al: Identification of a GJA3
mutation in a Chinese family with congenital nuclear cataract using
exome sequencing. Indian J Biochem Biophys. 50:253–258.
2013.PubMed/NCBI
|
|
11
|
Wang JL, Cao L, Li XH, Hu ZM, Li JD, Zhang
JG, Liang Y San-A, Li N, Chen SQ, et al: Identification of PRRT2 as
the causative gene of paroxysmal kinesigenic dyskinesias. Brain.
134:3490–3498. 2011. View Article : Google Scholar :
|
|
12
|
Shi Y, Li Y, Zhang D, Zhang H, Li Y, Lu F,
Liu X, He F, Gong B, Cai L, et al: Exome sequencing identifies
ZNF644 mutations in high myopia. PLoS Genet. 7:e10020842011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li R, Li Y, Kristiansen K and Wang J:
SOAP: Short oligonucleotide alignment program. Bioinformatics.
24:713–714. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu J and Ng PC: SIFT Indel: Predictions
for the functional effects of amino acid insertions/deletions in
proteins. PLoS One. 8:e779402013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Adzhubei I, Jordan DM and Sunyaev SR:
Predicting functional effect of human missense mutations using
PolyPhen-2. Curr Protoc Hum Genet Chapter. 7:Unit 7.20. 2013.
View Article : Google Scholar
|
|
16
|
Lei J, Deng X, Zhang J, Su L, Xu H, Liang
H, Huang X, Song Z and Deng H: Mutation screening of the HDC gene
in Chinese Han patients with Tourette syndrome. Am J Med Genet B
Neuropsychiatr Genet. 159B:72–76. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yuan L, Song Z, Xu H, Gu S, Zhu A, Gong L,
Zhao Y and Deng H: EIF4G1 Ala502Val and Arg1205His variants in
Chinese patients with Parkinson disease. Neurosci Lett. 543:69–71.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Guo Y, Yuan J, Liang H, Xiao J, Xu H, Yuan
L, Gao K, Wu B, Tang Y, Li X and Deng H: Identification of a novel
COL4A5 mutation in a Chinese family with X-linked Alport syndrome
using exome sequencing. Mol Biol Rep. 41:3631–3635. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Brison N, Tylzanowski P and Debeer P: Limb
skeletal malformations-what the HOX is going on? Eur J Med Genet.
55:1–7. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dai L, Liu D, Song M, Xu X, Xiong G, Yang
K, Zhang K, Meng H, Guo H and Bai Y: Mutations in the homeodomain
of HOXD13 cause syndactyly type 1-c in two Chinese families. PLoS
One. 9:e961922014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Cooper DN and Youssoufian H: The CpG
dinucleotide and human genetic disease. Hum Genet. 78:151–155.
1988. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Kurban M, Wajid M, Petukhova L, Shimomura
Y and Christiano AM: A nonsense mutation in the HOXD13 gene
underlies synpolydactyly with incomplete penetrance. J Hum Genet.
56:701–706. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Akarsu AN, Stoilov I, Yilmaz E, Sayli BS
and Sarfarazi M: Genomic structure of HOXD13 gene: A nine
polyalanine duplication causes synpolydactyly in two unrelated
families. Hum Mol Genet. 5:945–952. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jamsheer A, Sowinska A, Kaczmarek L and
Latos-Bielenska A: Isolated brachydactyly type E caused by a HOXD13
nonsense mutation: A case report. BMC Med Genet. 13:42012.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bruneau S, Johnson KR, Yamamoto M, Kuroiwa
A and Duboule D: The mouse Hoxd13(spdh) mutation, a polyalanine
expansion similar to human type II synpolydactyly (SPD), disrupts
the function but not the expression of other Hoxd genes. Dev Biol.
237:345–353. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zakany J and Duboule D: Synpolydactyly in
mice with a targeted deficiency in the HoxD complex. Nature.
384:69–71. 1996. View
Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dollé P, Dierich A, LeMeur M, Schimmang T,
Schuhbaur B, Chambon P and Duboule D: Disruption of the Hoxd-13
gene induces localized heterochrony leading to mice with neotenic
limbs. Cell. 75:431–441. 1993. View Article : Google Scholar : PubMed/NCBI
|