Introduction
Acute respiratory distress syndrome (ARDS), the most
severe form of acute lung injury (ALI), is the leading cause of
acute respiratory failure, and has a mortality rate of ~40%
worldwide (1), with a variety of
detrimental clinical disorders, including hypoxemia, respiratory
distress and pulmonary edema (2,3).
Although the morbidity and mortality rates associated with ARDS in
patients has decreased due to advances in protective ventilation
(4) and fluid conservative
supportive treatments (5), those
surviving suffer from significant physical impairments (6). Therefore, improved comprehension of
the molecular mechanism of ARDS is urgently required.
In the majority of cases, ARDS is induced by
inflammatory pulmonary diseases or bacterial sepsis, and
Gram-negative bacteria are common culprits (7). The endotoxin of Gram-negative
bacteria, lipopolysaccharide (LPS), has been reported to be
important in eliciting lung inflammation by inducing
proinflammatory cytokines, including tumor necrosis factor (TNF)-α,
interleukin (IL)-6, IL-1β and IL-8 (8), which increase the infiltration of
inflammatory cells to the lungs in the development of ALI.
Pro-inflammatory gene expression can be inhibited by the activation
of peroxisome proliferator-activated receptor (PPAR)-α (9), which is a member of the
ligand-activated transcription factors involved in the nuclear
hormone receptor superfamily (10,11).
Transforming growth factor-β (TGF-β) signaling has
been reported to be important in development and disease. TGF-β is
known to be a major inducer of epithelial to mesenchymal transition
via the small mothers against decapentaplegic (Smad)-dependent or
Smad-independent pathways (12,13).
Previous studies have reported that, through the stimulation of
fibroblast proliferation, TGF-β1 is involved in ARDS, which leads
to the development of pulmonary fibrosis. Pretreatment with
rosiglitazone, a ligand of PPAR-γ, can protect against ALI by
repressing the activation of nuclear factor-κB and inhibiting TGF-β
signaling (14). However, whether
PPAR-α is involved in TGF-β signaling, and whether PPAR-α is
involved for the recovery of lung function following ALI, remain to
be fully elucidated. The aim of the present study was to
investigate the protective effects of PPAR-α in LPS-induced ALI
in vivo and in vitro, and examine the underlying
mechanisms involving the PPAR-α and TGF-β signaling pathway.
Materials and methods
Patient selection
The present study was approved by the Ethics
Committee of Zigong First People's Hospital. A total of 18 patients
(including 8 females and 10 males, ages 55–75) with ARDS caused by
sepsis were enrolled between June 2010 and October 2013. All
peripheral blood samples were collected with written informed
consent. The clinical characteristics of ARDS were summarized and
disease severity was determined using the Acute Physiology and
Chronic Health Evaluation (APACHE) II, the Murray Lung Injury Score
(LIS) and the Simplified Acute Physiology Score (SAPS) II (15). The characteristics of patients with
ARDS were as follows: i) PaO2/FiO2 ≤300; ii)
presence of bilateral pulmonary infiltrates on frontal chest
radiograph; iii) no clinical evidence of left atrial hypertension;
iv) requirement for positive pressure ventilation via an
endotracheal tube; v) composite of oxygenation, compliance,
positive end expiratory pressure and the appearance of chest
radiograph (16). Blood was
collected using an indwelling arterial catheter at the time of ICU
admission (baseline) into sterilized, silicone-coated glass tubes,
and at 3 and 7 days subsequently. Healthy blood donors were used as
controls (also including 8 females and 10 males, ages from 55–75).
Serum samples (4 ml) were obtained by centrifugation at 2,500 × g
for 10 min at room temperature and frozen at −70°C until use. The
buffy coat cell layer was carefully aspirated and buffy coat cells
were suspended. Concentrations of TGF-β1 and PPAR-α were determined
using commercial, standardized enzyme-linked immunosorbent assay
(ELISA) kits. The TGF-β1 ELISA kit (cat. no. ADI-900-155) was from
Enzo Life Sciences, Inc. (Farmingdale, NY, USA) and the PPAR-α
ELISA kit (cat. no. 40,196) was from Active Motif, Inc. (Carlsbad,
CA, USA). The lower detection limit for TGF-β1 was 0.10 pg/ml and
for PPAR-α was 0.30 pg/ml.
Cell lines and cell culture
Human lung fibroblast (HLFs; IMR-90 cells) and
Mus musculus monocyte/macrophage RAW 264.7 cells were
purchased from American Type Culture Collection (Manassas, VA, USA)
and cultured in Eagle's Minimum Essential Medium with 10% fetal
bovine serum (FBS; Invitrogen; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA). The RAW 264.7 macrophages were cultured in
Dulbecco's modified Eagle's medium containing 10% FBS. The cells
were cultured in a humidified incubator at 37°C in 5%
CO2.
Reagents
LPS (isolated from Escherichia coli) and
TGF-β1 (used at a dose of 10 µg/ml) were purchased from
Sigma-Aldrich; Merck Millipore (Darmstadt, Germany). Mouse
monoclonal antibodies against TGF-β1 were from Abcam (cat. no.
PB190503; Cambridge, MA, USA) and rabbit monoclonal antibody
against PPARα was from Cell Signaling Technology, Inc. (cat. no.
2443S; Beverly, MA, USA). HRP-conjugated anti-mouse and anti-rabbit
secondary antibodies were from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA). Specific small interfering short hairpin (sh)RNA
targeting TGF-β1 and PPAR-α and the negative control shRNA were all
from Sigma-Aldrich; Merck Millipore. Following allowing cellular
attachment to the plates, 2×105 cells were treated with
relative shRNAs using Lipofectamine 2000 (Invitrogen; Thermo Fisher
Scientific, Inc.) RNA and protein were collected 72 h following
treatment.
Induction of ARDS in mice
All experiments involving mice were performed in
strict accordance with Animal Care and Use guidelines from Beijing
Charles River Laboratory Animal Centre institutional committee
(Beijing, China). A total of 30 (15 male and 15 female) mice (8–10
weeks old; 19–22 g) were purchased from Beijing Charles River
Laboratory Animal Centre, and kept under standard conditions at
room temperature (24°C) with a 12 h day/night cycle under specific
pathogen-free conditions. At 3 h following intratracheal
instillation of LPS (4 mg/kg) (17), the mice were administered with an
intravenous injection in the tail vein of 10% chloral hydrate (3.5
ml/kg) for anesthetization at 4, 12 and 24 h. The mice were divided
into the following groups: Vector group, injected with the same
volume of pyrogen-free PBS; LPS group, stimulated with LPS; PPARα
group, pre-treated with PPAPα prior to stimulation with LPS.
Following the successful induction of ALI, the mice were
administered with a tail vein injection of adeno-associated viruses
(AAV) carrying PPAR-α short hairpin RNA (shPPAR-α group) or with a
scrambled (SCR) sequence as a control. Primary murine alveolar
epithelial cells were isolated, as described previously (18).
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA was extracted from the cultured cells
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
For analysis of the expression of messenger RNA (mRNA), cDNA
synthesis was performed by reserve transcription using the
Transcriptor First Stand cDNA synthesis kit (Roche Diagnostics,
Basel, Switzerland). The qPCR was performed in duplicate with a
QuantiTect SYBR Green PCR kit (Qiagen, Inc., Valencia, CA, USA) on
an Applied Biosystems 7500 Real-Time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.), according to
manufacturer's protocol. The primers were as follows: TGF-β1,
forward 5′-CCACCTGCAAGACCATCGAC-3′ and reverse:
5′-CTGGCGAGCCTTAGTTTGGAC-3; PPARγ, forward
5′-GAGATCATCTACACGATGCTGGC-3′ and reverse
5′-CGCAGGCTTTTGAGGAACTC-3′; and GAPDH, forward
5′-GAGAAGTATGACAACAGCCTC-3′ and reverse 5′-ATGGACTGTGGTCATGAGTC-3′.
All primers were purchased from Thermo Fisher Scientific, Inc. The
PCR amplification was performed at 95°C for 1 min, followed by 35
cycles of 95°C for 15 sec, 60°C for 15 sec, and 72°C for 30 sec.
The expression levels of genes were normalized against that of
GAPDH and relative fold changes in mRNA expression were calculated
using the formula 2−ΔΔCq (19).
Western blot analysis
Whole cell lysates were collected and protein from
the cultured cells was extracted using RIPA lysis buffer on ice,
following centrifugation at 4°C, 10,000 × g for 15 min, the
supernatants were collected. The BCA protein assay kit (Thermo,
USA) was used to determine the protein concentration, and 30 µg
from each sample was mixed with 4X SDS loading buffer and heated at
100°C for 10 min. Protein samples were then separated by 10%
SDS-polyacrylamide gel electrophoresis and transferred onto a PVDF
membrane (EMD Millipore, Billerica, MA, USA). A mixture of 5%
non-fat milk in Tris-buffered saline Tween-20 (TBST) was used to
block the nonspecific proteins for 1 h at room temperature. The
membrane blots were then probed with primary antibodies at 4°C
overnight, as follows: PPAR-α (1:500), TGF-β1 (1:1,000), TNF-α
(1:1,000), and β-actin (1:2,000). The membranes were washed five
times with TBST for 5 min, followed by incubation with horseradish
peroxidase-conjugated secondary antibody (Santa Cruz Biotechnology,
Inc.) for 1 h at room temperature. An enhanced chemiluminescent
system (GE Healthcare Life Sciences, NJ, USA) was used to visualize
the protein antigen. The signals were recorded using X-ray film
(Kodak, Rochester, NY, USA). Images were captured, representative
of three repeats.
ELISA
The levels of PPAR-α and TGF-β1 in the
bronchoalveolar lavage fluid (BALF), serum and cell culture
supernatants were determined using sandwich ELISA, according to the
manufacturer's protocol.
Statistical analysis
All statistical analyses were performed using SPSS
18.0 statistical software (SPSS, Inc., Chicago, IL, USA). Data were
analyzed by comparing the mean ± standard deviation from three
experiments using Student's t-test P<0.05 was considered to
indicate a statistically significant difference. One-way analysis
of variance with a Bonferoni correction was used for statistical
analysis, followed by a Fisher's exact test, as necessary.
Results
PPAR-α is effective at suppressing
TGF-β in HLF cells and RAW 264.7 cells
In order to investigate whether PPAR-α was involved
in TGF-β signaling, the present study treated HLFs with PPAR-α and,
48 h following treatment, the cells were harvested. The mRNA and
nuclear proteins were isolated, followed by the examination of
TGF-β1 using RT-qPCR and western blot analyses. It was found that
PPAR-α inhibited the expression of TGF-β1 in HLF cells at the mRNA
level (Fig. 1A) and protein level
(Fig. 1B); the suppression of
TNF-α by PPAR-α was used as a positive control. To further confirm
these findings, RAW 264.7 cells were used and a similar experiment
was performed. As shown in Fig. 1C and
D, the expression of TGF-β1 was suppressed by the enforced
expression of PPAR-α.
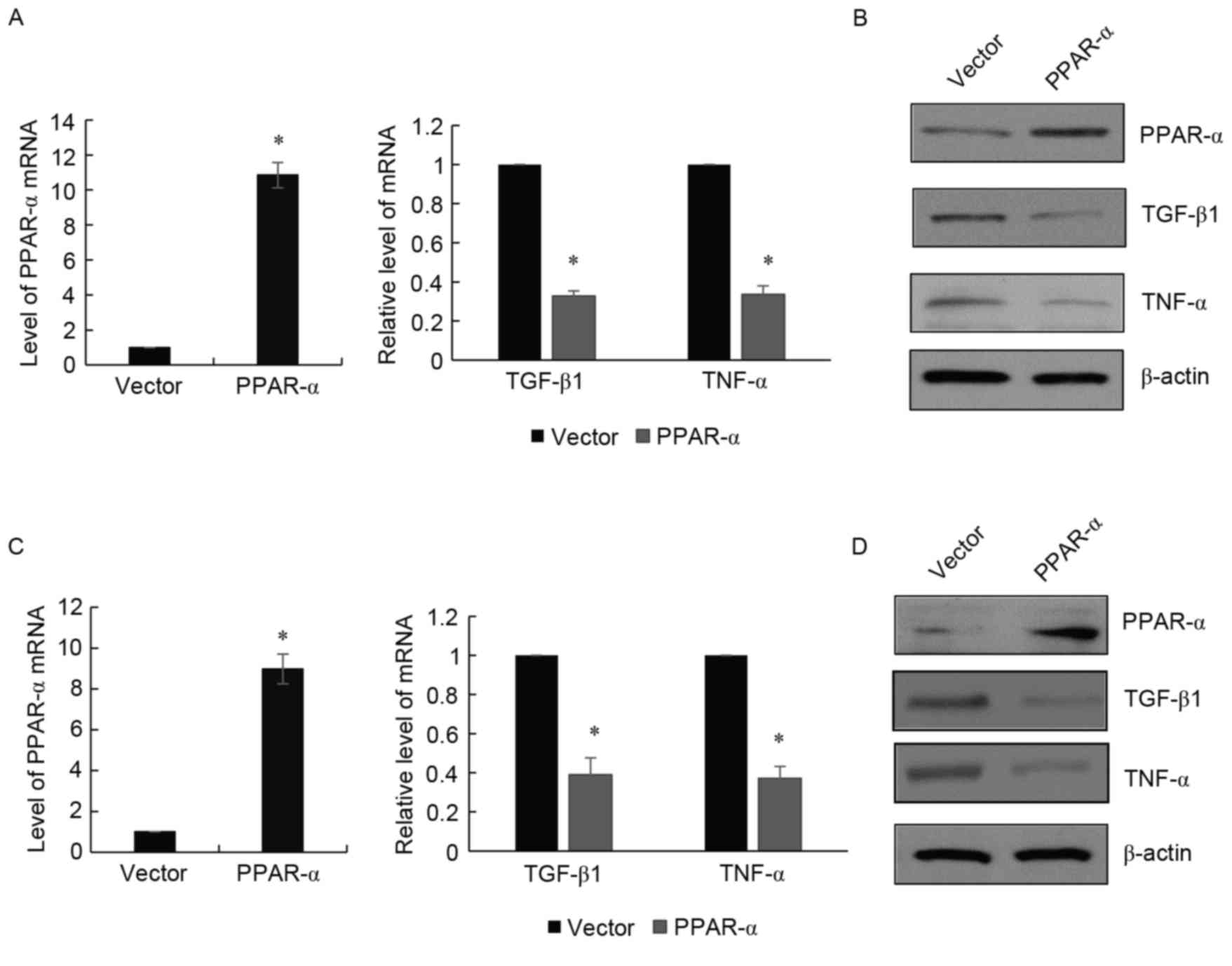 | Figure 1.PPAR-α is effective in suppressing the
activity of TGF-β in IMR-90 cells and RAW 264.7 cells. (A) RT-qPCR
analysis was used to detect the mRNA levels of TGF-β1, TNF-α and
PPAR-α in IMR-90 cells treated with PPAR-α, compared with the
vector group. Data are presented as the mean ± standard deviation
of three independent experiments. *P<0.05. GAPDH was used as an
intrinsic control. (B) Western blot analysis was used to detect the
protein levels of TGF-β1, TNF-α and PPAR-α in IMR-90 cells treated
with PPAR-α, compared with the vector group. β-actin was used as a
control. (C) RT-qPCR analysis was used to detect levels of TGF-β1,
TNF-α and PPAR-α in RAW 264.7 cells treated with PPAR-α, compared
with the vector group. (D) Western blot analysis was used to detect
levels of TGF-β1, TNF-α and PPAR-α in RAW 264.7 cells treated with
PPAR-α, compared with the vector group. RT-qPCR, reverse
transcription-quantitative polymerase chain reaction; TGF-β,
transforming growth factor-β; TNF-α, transforming growth factor-α;
PPAR-α, peroxisome proliferator-activated receptor-α. |
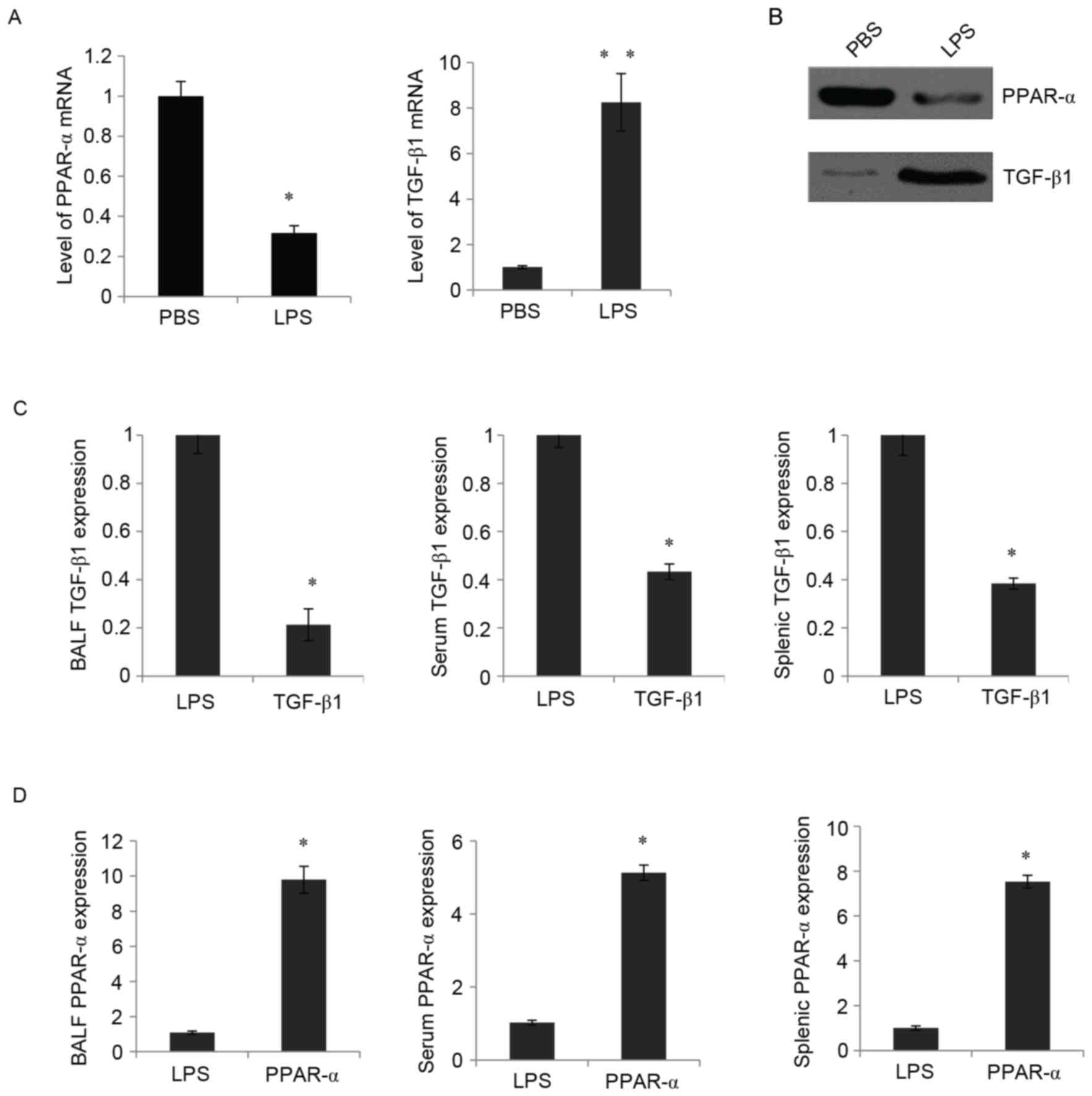
PPAR-α treatment downregulates the
expression of TGF-β1 in the mouse model of LPS-induced ALI
To further investigate the roles of PPAR-α and
TGF-β1 in ALI, LPS-induced ALI mice were used, and the expression
levels of PPAR-α and TGF-β1 were examined using RT-qPCR and western
blot analyses. Following LPS treatment, in the murine lung, a
decrease in the mRNA expression of PPAR-α and an increase in the
mRNA expression of TGF-β1 were observed using RT-qPCR analysis
(Fig. 2A). Similarly, the western
blot analysis showed that the protein expression PPAR-α was
decreased and that of TGF-β1 was significantly increased by
treatment with LPS (Fig. 2B).
PPAR-α treatment prior to stimulation with LPS resulted in a
decrease in the mRNA expression levels of TGF-β1 in the BALF,
peripheral blood and splenocytes (Fig.
2C). Consistently, the present study found that the expression
level of PPAR-α in the BALF was significantly elevated by treatment
with the PPAR-α expression vector (Fig. 2D, left panel). The expression
levels of PPAR-α in the peripheral blood and splenocytes were also
upregulated (Fig. 2D, middle and
right panels). These findings demonstrated that the enforced
expression of PPAR-α downregulated the expression of TGF-β1 in
LPS-induced ALI mice.
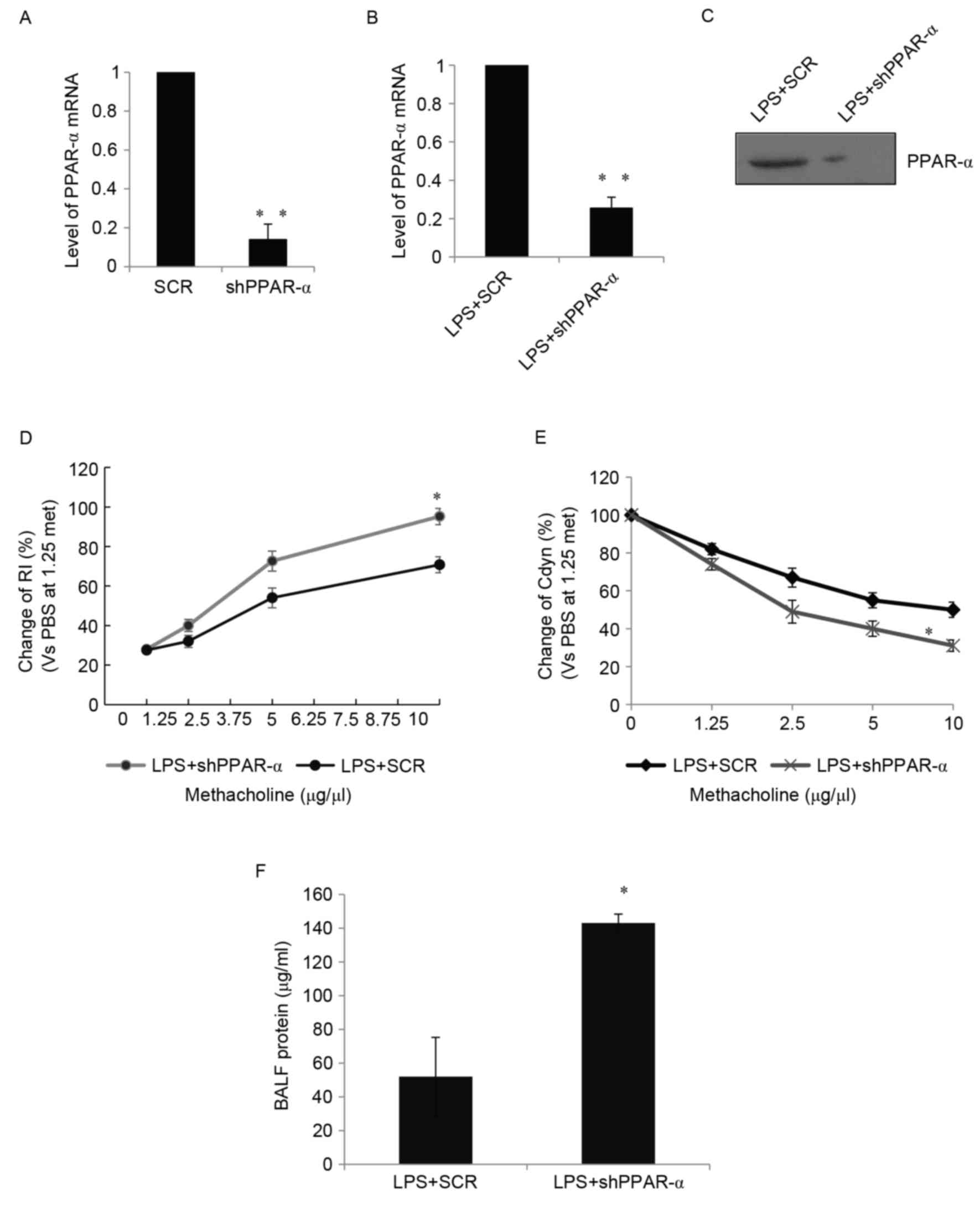
Activation of PPAR-α is essential for
recovery of lung function following ALI
The question of whether PPAR-α was involved in the
recovery of lung function following ALI remained; therefore, to
evaluate the role of PPAR-α in the recovery of lung function, the
present study developed a virus carrying the shPPAR-α or SCR
sequence as a control. It was found that the RAW 264.7 cells, which
were transduced with shPPAR-α, exhibited significantly decreased
levels of PPAR-α (Fig. 3A).
Following injecting of the virus expressing either shPPAR-α or SCR
into the mouse following LPS treatment, the effects of PPAR-α on
lung recovery were examined. Significantly decreased levels of
PPAR-α in the mouse lung were detected using RT-qPCR analysis
(Fig. 3B) and western blot
analysis (Fig. 3C). Impaired lung
function has been considered to cause a dose-dependent increase in
lung resistance index (RI) and decrease in dynamic compliance
(Cdyn) in response to a cholinergic stimulus (methacholine) and a
significant increase in BALF protein (11). The analysis of pressure and flow
waveforms indicated that the knockdown of PPAR-α resulted in a
marked increase in RI (Fig. 3D), a
notable decrease in Cdyn (Fig. 3E)
and a significant increase in BALF protein (Fig. 3F). These data suggested that PPAR-α
was essential for the recovery of lung function following ALI.
Suppression of TGF-β by PPAR-α
improves recovery of lung function following ALI
In order to further evaluate the association between
TGF-β with PPAR-α in the recovery of lung function following ALI,
TGF-β1 was inhibited in the mouse lung following LPS treatment by
injecting AAV expressing shTGF-β1 (or SCR as a control).
TGF-β1-knockdown efficiency was first confirmed in the mouse lung.
It was found that the application of these viruses significantly
decreased the levels of TGF-β1 (Fig.
4A), resulting in a significant decrease in RI (Fig. 4B), a significant increase in Cdyn
(Fig. 4C) and a significant
decrease in BALF protein (Fig.
4D). These data suggested that shTGF-β1 was essential for the
recovery of lung function following ALI. The results showed that
the knockdown of PPAR-α and knockdown of TGF-β1 in the mouse lung
partially reduced the shPPAR-α-impaired recovery of lung function,
as detected by RI (Fig. 4E), Cdyn
(Fig. 4F) and BALF protein
(Fig. 4G). Together, these
experiments suggested that PPAR-α was essential for the recovery of
lung function following ALI, possibly through the suppression of
TGF-β.
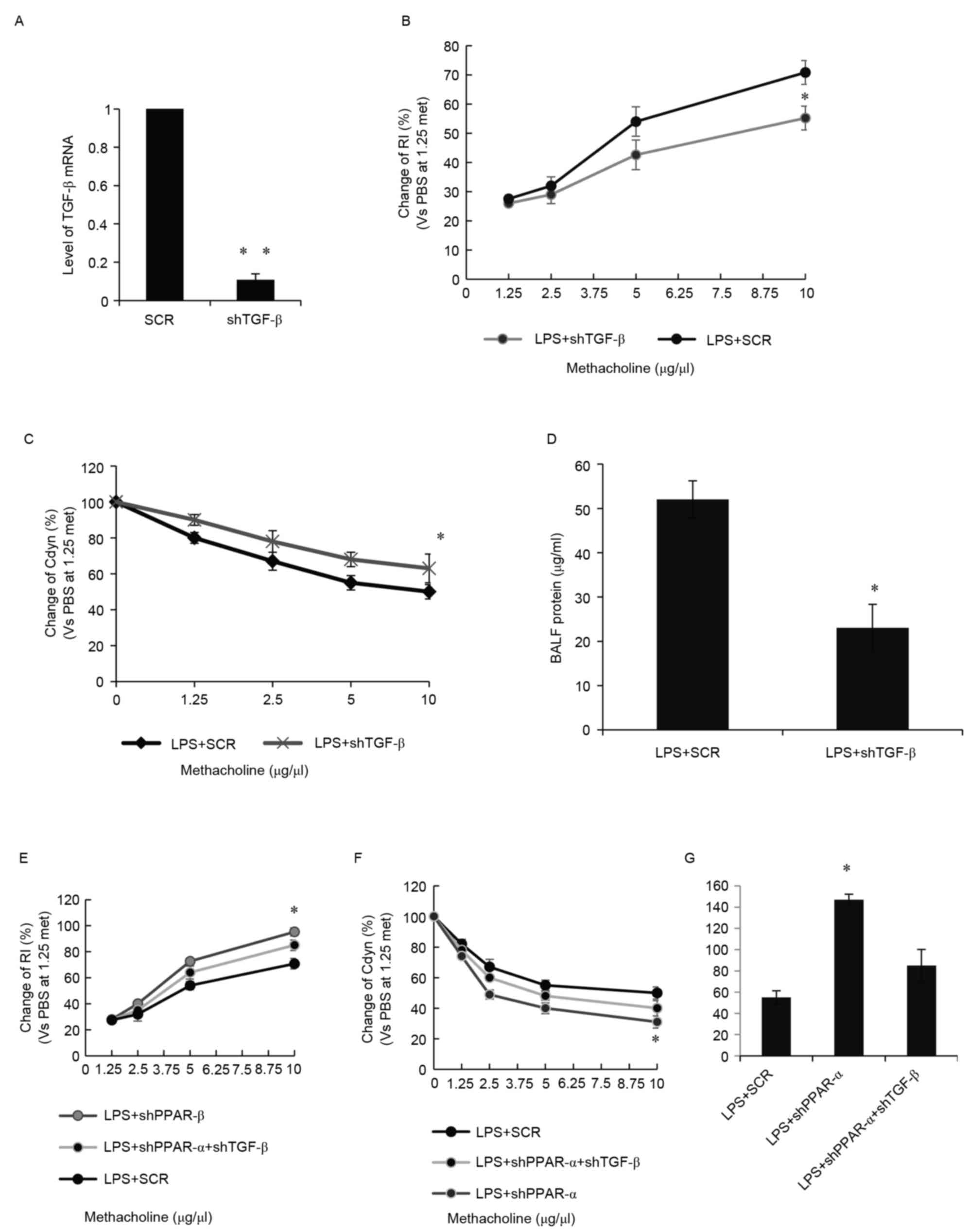 | Figure 4.Suppression of TGF-β1 by PPAR-α
improves recovery of lung function following ALI. (A) Effect of
TGF-β1 knockdown was confirmed in the mouse lung by injecting AAV
expressing either shRNA for TGF-β1 or SCR following LPS treatment.
Reverse transcription-quantitative polymerase chain reaction
analysis was performed. (B) Effects of TGF-β1 knockdown on lung
recovery were determined according to RI in response to a high dose
methacholine. (C) Effects of TGF-β1 knockdown on lung recovery were
determined using Cdyn in response to high dose methacholine. (D)
Total protein in BALF was measured using the bovine serum albumin
standard curve to record the absorbance at 595 nm. (E) Mice were
treated with SCR, shPPAR-α or shPPAR-α plus shTGF-β1 following LPS
treatment. RI in response to high dose methacholine was determined.
(F) Cdyn in response to high dose methacholine were determined. (G)
Total protein in BALF was determined to measure recovery of lung
function following ALI. *P<0.05; **P<0.01. TGF-β,
transforming growth factor-β; LPS, lipopolysaccharide; ALI, acute
lung injury; AAV, adeno-associated viruses; sh, short hairpin RNA;
BALF, bronchoalveolar lavage fluid; SCR, scrambled sequence; RI,
resistance index; Cdyn, dynamic compliance. |
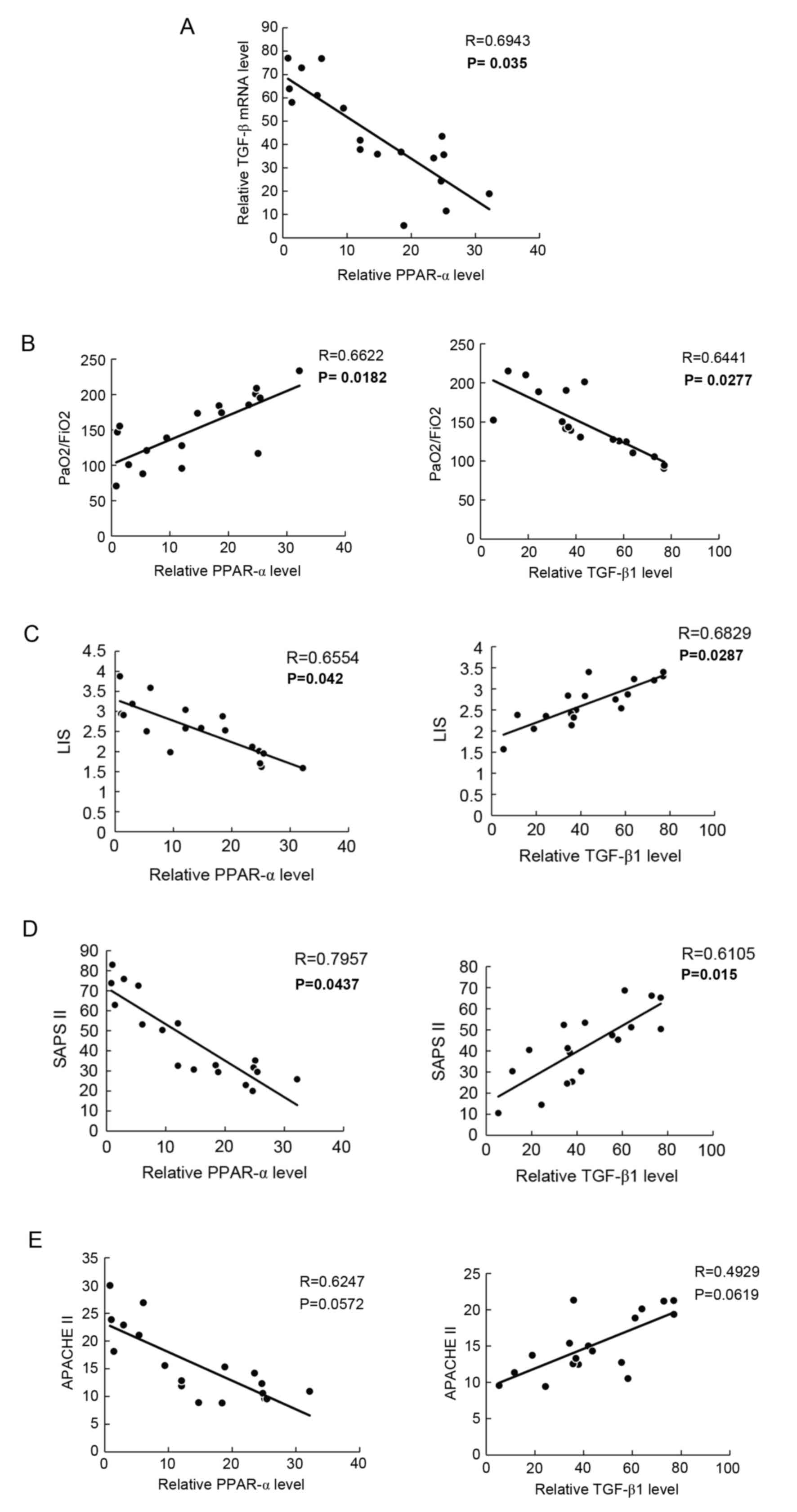
Serum levels of PPAR-α are negatively
correlated with TGF-β1 in patients with ARDS
To further investigate the clinical significance of
the above-mentioned findings, the present study detected the
serological expression levels of PPAR-α and TGF-β1, which revealed
that PPAR-α was inversely correlated with TGF-β1 in patients with
ARDS (Fig. 5A; P<0.05). In
addition, the correlation with disease activity in patients with
ARDS was analyzed. It was found that serum PPAR-α was positively
associated with the ratio of PaO2/FiO2,
whereas the serum level TGF-β1 was negatively associated with
PaO2/FiO2 (Fig.
5B; P<0.05).
Using the LIS (Fig.
5C; P<0.05) and SAPS II (Fig.
5D; P<0.05) scoring methods to determine disease activity,
it was revealed that the serum expression level of PPAR-α was
inversely correlated with these indices, whereas the serum
expression level of TGF-β1 was consistent with these indices. No
significant correlation was observed between the APACHE II scoring
method and PPAR-α or TGF-β1 serum levels (Fig. 5E; P>0.05). On the whole, these
results suggested that PPAR-α was negatively correlated with TGF-β1
and involved in the pathogenesis of ARDS.
Discussion
One of the primary findings of the present study was
that PPAR-α was effective in suppressing TGF-β1 in HLF cells and
RAW 264.7 cells. The present study further analyzed the levels of
TGF-β1 and PPAR-α in the mouse lung following LPS treatment. In the
murine lung, there was a decrease in the mRNA expression of PPAR-α
and an increase in the mRNA expression of TGF-β1. Treatment with
PPAR-α prior to stimulation with LPS resulted in a dose-dependent
decrease in the expression levels of TGF-β1 in BALF, peripheral
blood and splenocytes. The above data indicated that PPAR-α
treatment downregulated the expression of TGF-β1 in the LPS-induced
ALI model in vitro and in vivo. Taken together, the
present study demonstrated that the enforced expression of PPAR-α
ameliorated the development of ALI using the LPS-induced model.
Following the report that TNF-α is one of the primary cytokines
involved in the response to LPS, the present study revealed that,
in addition to TNF-α, TGF-β1 was another key regulator in
LPS-induced ALI.
In order to further elucidate the molecular
mechanism underlying post-ALI lung recovery, the present study
further focused on whether the suppression of TGF-β1 by PPAR-α was
involved in the recovery of lung function following ALI. The
knockdown of PPAR-α and knockdown of TGF-β1 in the mouse lung
partially reduced the shPPAR-α impaired recovery of lung function,
and it was concluded that the activation of PPAR-α and suppression
of TGF-β1 were essential for the recovery of lung function. The
clinical significance of PPAR-α being inversely correlated with
TGF-β1 in patients with ARDS was consistent with this mechanism.
Taken together, the results of the present study provided evidence
supporting critical role of PPAR-α in the suppression of TGF-β1 in
lung recovery, and revealed a novel mechanism controlling post-ALI
lung recovery.
References
|
1
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Taeusch HW: Treatment of acute (Adult)
respiratory distress syndrome. The holy grail of surfactant
therapy. Biol Neonate. 77:(Suppl 1). S2–S8. 2000. View Article : Google Scholar
|
|
4
|
Acute Respiratory Distress Syndrome
Network. Brower RG, Matthay MA, Morris A, Schoenfeld D, Thompson BT
and Wheeler A: Ventilation with lower tidal volumes as compared
with traditional tidal volumes for acute lung injury and the acute
respiratory distress syndrome. N Engl J Med. 342:1301–1308. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Reisinger MW, Moss M and Clark BJ:
National Heart, Lung, and Blood Institute Acute Respiratory
Distress Syndrome Network Investigators: Brief versus full alcohol
use disorders identification test in national heart, lung, and
blood institute acute respiratory distress syndrome network
clinical trials. Crit Care Med. 43:e382–e385. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thompson BT and Matthay MA: The Berlin
definition of ARDS versus pathological evidence of diffuse alveolar
damage. Am J Respir Crit Care Med. 187:675–677. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Martin MA and Silverman HJ: Gram-negative
sepsis and the adult respiratory distress syndrome. Clin Infect
Dis. 14:1213–1228. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Bhatia M and Moochhala S: Role of
inflammatory mediators in the pathophysiology of acute respiratory
distress syndrome. J Pathol. 202:145–156. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Chen C, Xu S, Wang WX, Ding YM, Yu KH,
Wang B and Chen XY: Rosiglitazone attenuates the severity of sodium
taurocholate-induced acute pancreatitis and pancreatitis-associated
lung injury. Arch Med Res. 40:79–88. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Schaefer MB, Pose A, Ott J, Hecker M,
Behnk A, Schulz R, Weissmann N, Günther A, Seeger W and Mayer K:
Peroxisome proliferator-activated receptor-alpha reduces
inflammation and vascular leakage in a murine model of acute lung
injury. Eur Respir J. 32:1344–1353. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Delerive P, De Bosscher K, Besnard S,
Vanden Berghe W, Peters JM, Gonzalez FJ, Fruchart JC, Tedgui A,
Haegeman G and Staels B: Peroxisome proliferator-activated receptor
alpha negatively regulates the vascular inflammatory gene response
by negative cross-talk with transcription factors NF-kappaB and
AP-1. J Biol Chem. 274:32048–32054. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Derynck R and Zhang YE: Smad-dependent and
Smad-independent pathways in TGF-beta family signalling. Nature.
425:577–584. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rastaldi MP: Epithelial-mesenchymal
transition and its implications for the development of renal
tubulointerstitial fibrosis. J Nephrol. 19:407–412. 2006.PubMed/NCBI
|
|
14
|
Wu M, Melichian DS, Chang E,
Warner-Blankenship M, Ghosh AK and Varga J: Rosiglitazone abrogates
bleomycin-induced scleroderma and blocks profibrotic responses
through peroxisome proliferator-activated receptor-gamma. Am J
Pathol. 174:519–533. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bernard GR, Artigas A, Brigham KL, Carlet
J, Falke K, Hudson L, Lamy M, Legall JR, Morris A and Spragg R: The
American-European Consensus Conference on ARDS. Definitions,
mechanisms, relevant outcomes, and clinical trial coordination. Am
J Respir Crit Care Med. 149:818–824. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Murray JF, Matthay MA, Luce JM and Flick
MR: An expanded definition of the adult respiratory distress
syndrome. Am Rev Respir Dis. 138:720–723. 1988. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang H, Xu L, Zhao J, Wang D, Guo R, Wang
J, Gong W, Liu T, Zhang Y and Dong L: Regulatory mechanism of
pyrrolidine dithiocarbamate is mediated by nuclear factor-κB and
inhibits neutrophil accumulation in ARDS mice. Exp Ther Med.
8:614–622. 2014.PubMed/NCBI
|
|
18
|
Rastaldi MP: Epithelial-mesenchymal
transition and its implications for the development of renal
tubulointerstitial fibrosis. J Nephrol. 19:407–412. 2006.PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|