Introduction
Atherosclerosis is the predominant pathological
cause of coronary artery disease, stroke and peripheral vascular
disease. Endothelial dysfunction is considered to be the primary
step of atherosclerosis, and various stimuli may induce this
(1). Synthesis of adhesion
molecules and chemokines by dysfunctional endothelial cells results
in their cellular junctions becoming leakier; these steps induce
early atherosclerotic lesions by mediating transient/rolling-type
and firm adhesions of leucocytes and macrophages, as well as
facilitating monocyte infiltration into the injured atheroma area
(2). Leucocyte adhesion to
vascular endothelial cells is a critical step in transendothelial
migration. The infiltration of monocytes and other immune cells
such as neutrophils, is mainly governed by interactions between
chemokines or adhesion molecules and their associated receptors,
such as vascular cell adhesion molecule-1 (VCAM-1) to α4β1
integrin, intercellular adhesion molecule-1 (ICAM-1) to
αLβ2-integrin, and galectin-3 interaction with integrin β1, β3 and
VCAM-1, either directly or indirectly (3).
Galectin-3 has numerous cellular locations,
including the nucleus, the cell surface, and the extracellular
environment. The location of galectin-3 depends on its ability to
recognize extracellular matrix (ECM) components; particularly
laminin, elastin and fibronectin, and these interactions facilitate
cell-ECM attachment and transendothelial migration. Furthermore,
this lectin may bridge neutrophilic and monocytic connections with
the endothelium, either directly or indirectly (4). The release of reactive species and
proteolytic enzymes, stimulated by the adhesion of neutrophils and
monocytes, may subsequently induce endothelial erosion, aggravate
endothelial cell dysfunction, and result in amplified vascular
permeability and leucocyte accumulation in the atherosclerotic
area. The predominant adhesion molecules expressed on the surface
of neutrophils are β2-integrins; these serve a critical role in the
recruitment and transmigration of neutrophils and monocytes
(5–7). This is followed by endothelial cell
surface-galectin-3 binding, which strengthens the adhesion
(4). Galectin-3 has also been
considered to promote cell-cell adhesion through a cross-linking of
the Mac-2 binding protein, which is a member of the macrophage
scavenger receptor cysteine-rich domain superfamily (8,9).
Galactin-3 may also have a similar effect to
monocyte chemoattractant protein-1 (10), whereby extracellular galectin-3 may
initiate alternative macrophage activation via interaction with
cluster of differentiation (CD) 98 on macrophages. Furthermore,
galectin-3 may serve as an advanced glycosylation end product
clearance receptor, and a mediator of macrophage-led endocytosis of
modified low-density lipoprotein (LDL), thus facilitating foam cell
formation and subsequent amplification of inflammation (11,12).
Cell surface and extracellular galectin-3 may therefore accelerate
or decelerate atherosclerotic initiation and progression, by
inducing the accumulation and activation of leucocytes.
Modified citrus pectin (MCP) is a complex
polysaccharide obtained from the peel and pulp of citrus fruits.
MCP powder is produced from citrus pectin using pH- and
temperature-based processes to break it into shorter, non-branched,
galactose-rich carbohydrate chains. This shorter polysaccharide
enables MCP to access and bind tightly to galectin-3 by adhering to
the carbohydrate recognition domain, thereby modulating the
bioactivity of galectin-3 (13).
Previous research indicated that either galectin-3 knockout or oral
administration with MCP may reduce the atherosclerotic area in
apolipoprotein E (apoE)-deficient mice; however, the exact
mechanism remains unclear (14–16).
The present study explored the effects of MCP on the initiation of
atherosclerosis, and it was demonstrated that MCP may reduce the
size of the atherosclerotic lesion by inhibiting leucocyte adhesion
to endothelial cells.
Materials and methods
Preparation of animals
Male apoE−/− mice (age, 8 weeks, 20±2 g)
bred from a C57BL/6J background were obtained from Jackson
Laboratory (Peking University Health Science Center, Beijing,
China). Mice (n=16) were fed a high-fat, cholesterol-rich
atherogenic diet containing 21% fat, 19.5% casein and 1.25%
cholesterol for 4 weeks and during the course of the experiment,
all mice were allowed ad libitum access to food and water. They
were maintained at 20–24°C with 45–55% humidity with a 12-h
light-dark cycle. The mice were divided into two groups: MCP (n=8)
and model (n=8) groups. A total of 1% MCP (Centrax International
Corporation, San Francisco, USA) was added into the drinking water
of the mice in the MCP group, for 4 weeks. All animal protocols
were approved by the Animal Ethics Committee of the Beijing
Institute of Geriatrics (Beijing, China).
Quantification of atherosclerotic
lesions in the aortic root
Mice were sacrificed and hearts were sectioned
throughout the aortic root; serial paraffinic sections of the heart
were taken every 10 µm, according to the identified methods
(12). Aortic roots were dissected
and fixed overnight in 4% polymerized formaldehyde, paraffin
embedded, and subsequently sectioned into 5 µm slices. Every sixth
section was stained with a modified Movat pentachrome stain
(SS0236; Xinhua Luyuan Science and Technology Ltd, Beijing, China),
Sirius Red (DC0041; Leagene Biotechnology Co., Ltd., Beijing,
China), to evaluate plaque areas and collagen. Briefly, the
sections were stained with alcian blue, Verheoff's hematoxylin
solution and then differentiated in 2% aqueous ferric chloride and
subsequently stained in crocein scarlet-acid fuchsin and again
differentiated in 5% aqueous phosphotungstic acid to achieve movat
staining. The slides were stained with saturated picric acid Sirius
red and alcoholic hematoxylin progressively, to observe lesional
collagen content. All the stained slides were checked and captured
microscopically by an upright microscope (Carl Zeiss AG,
Oberkochen, Germany). Atherosclerotic lesions were analyzed using
Image-Pro® Plus-6 software (Media Cybernetics, Inc.,
Rockville, MD, USA).
Immunohistochemistry
Regular sections from the excised aortic root were
used for immunohistochemistry, to identify macrophages and smooth
muscle cells (SMCs), according to a previously reported method
(12). Briefly, sections were
incubated with polyclonal antibodies at 37°C for 90 min or at 4°C
overnight, and then labeled with horseradish peroxidase- or
tetramethylrhodamine-conjugated anti-rabbit immunoglobulin G
(catalog no. PV9000; OriGene Technologies, Inc., Beijing, China) at
37°C for 60 min. The coverslips were subsequently mounted with
DABCO™ mounting medium, and analyzed using an upright microscope
(Carl Zeiss AG, Oberkochen, Germany). Anti-α-actin (catalog no.
sc-130616; 1:100) and anti-MAC-3, a highly glycosylated protein and
a constituent of the lysosomal membrane that may be used as a
macrophage marker, (or LAMP-2, catalog no. sc-81729; 1:100)
antibodies were purchased from Santa Cruz Biotechnology, Inc.
Dallas, TX, USA.
En face analysis of the descending
aorta
The descending aortas were used for en face
lipid staining. The aortas were dissected from the left subclavian
artery to the iliac bifurcation, then opened longitudinally and
stained with Oil Red O to visualize the extent of the lipid
deposition. Aortic images were captured with a Sony DXC-960MD (Sony
Corporation, Tokyo, Japan), and the lesion size was quantitatively
analyzed. Data were analyzed with Image-Pro® Plus-6
software (Media Cybernetics, Inc.).
Cell culture and adhesion assays
CRL-1730 human umbilical vein vascular endothelial
cells (HUVECs) were purchased from the American Type Culture
Collection (Manassas, VA, USA) and were cultured in endothelial
cell basal medium-2 Bullet kit media (Clonetics™; Lonza, Basel,
Switzerland). Cells were grown to confluence at 37°C in 5%
CO2, on 0.2% gelatin-coated culture dishes. Human
monocytoid U937 cells (American Type Culture Collection) were
cultured in RPMI-1640 medium supplemented with 10% fetal bovine
serum (catalog no. 12633-012 and 10082139; Gibco; Thermo Fisher
Scientific Inc., Waltham, MA, USA). To assess monocyte adhesion,
HUVECs were plated onto 6-well gelatin-coated dishes at a density
of 1.2×105 cells/well. The following day, cells were
pretreated with oxidized-LDL (ox-LDL, catalog no. YB-002; Yiyuan
Biotechnology, Guangzhou, China), after which MCP was added at
various concentrations (0.025, 0.05, 0.1 and 0.25%, for 24 h). U937
cells (3×105 cells/well) were subsequently plated onto
each monolayer, and incubated for 10 min under rotating conditions
(63 rpm), at room temperature. Non-adherent cells were removed by
gentle washing with MCDB 131 medium (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA), and monolayers were fixed with
1% paraformaldehyde and observed with an inverted microscope
(CKX41; Olympus Corporation, Tokyo, Japan).
Scanning electron microscopy
(SEM)
Branchiocephalic arteries (BCA) recovered from the
mice were fixed with 2.5% gluteraldehyde in Millonig's phosphate
buffer for 48 h, followed by post-fixation with 1% osmium tetroxide
for 45 min. The specimens were dehydrated using increasing grades
of ethyl alcohol (15 min in 50, 70, 80, and 95% alcohol, followed
by three 10-min periods in 100% alcohol) and subsequently placed in
an LPD-100 critical point dryer (S4800; Hitachi Limited, Inc.,
Tokyo, Japan) for 5 min at 41°C and 1,200 psi CO2. The
tissues were then mounted on aluminum stubs with silver glue,
sputter coated with gold, and examined using an SEM (17,18).
Statistical analysis
All values were presented as the mean ± standard
error of the indicated number of measurements. An unpaired
Student's t-test and analysis of variance were conducted, followed
by post hoc testing with the Tukey procedure to determine
significance. P<0.05 was considered to indicate a statistically
significant difference.
Results
MCP treatment reduces the size of
atherosclerotic lesions in apoE−/− mice
MCP is a naturally occurring inhibitor of galectin-3
carbohydrate binding (1,2). To investigate whether in vivo
galectin-3 inhibition could reduce plaque size, apoE−/−
mice were fed an atherogenic diet, with MCP (1%) supplemented in
their drinking water, for 4 weeks. The size of the aortic lesion
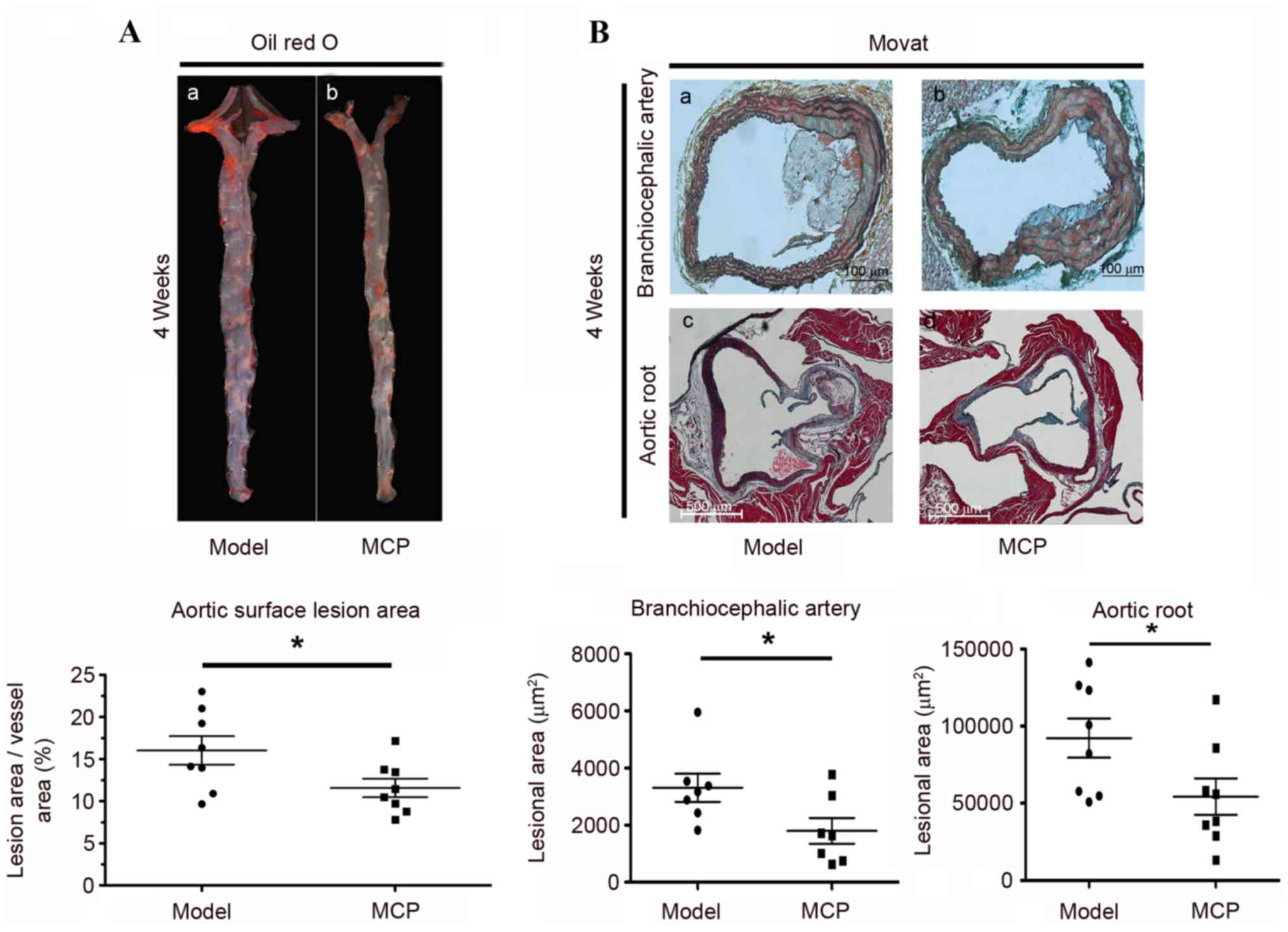
vs. the total arch area was determined, via quantitative
histomorphological analysis of Oil Red O-stained en face
specimens of the descending aorta. The area of lesioned aorta,
compared with the total arch area, was reduced following MCP
administration for 4 weeks (Fig.
1A). Similarly, the lesion size on the BCA and the aortic root
was reduced in the MCP group, compared with the model group, as
evidenced by Movat staining (Fig.
1B). These results indicated that MCP may reduce the size of
the atherosclerotic lesion in apoE−/− mice.
MCP alters plaque structure and
composition in apoE−/− mice
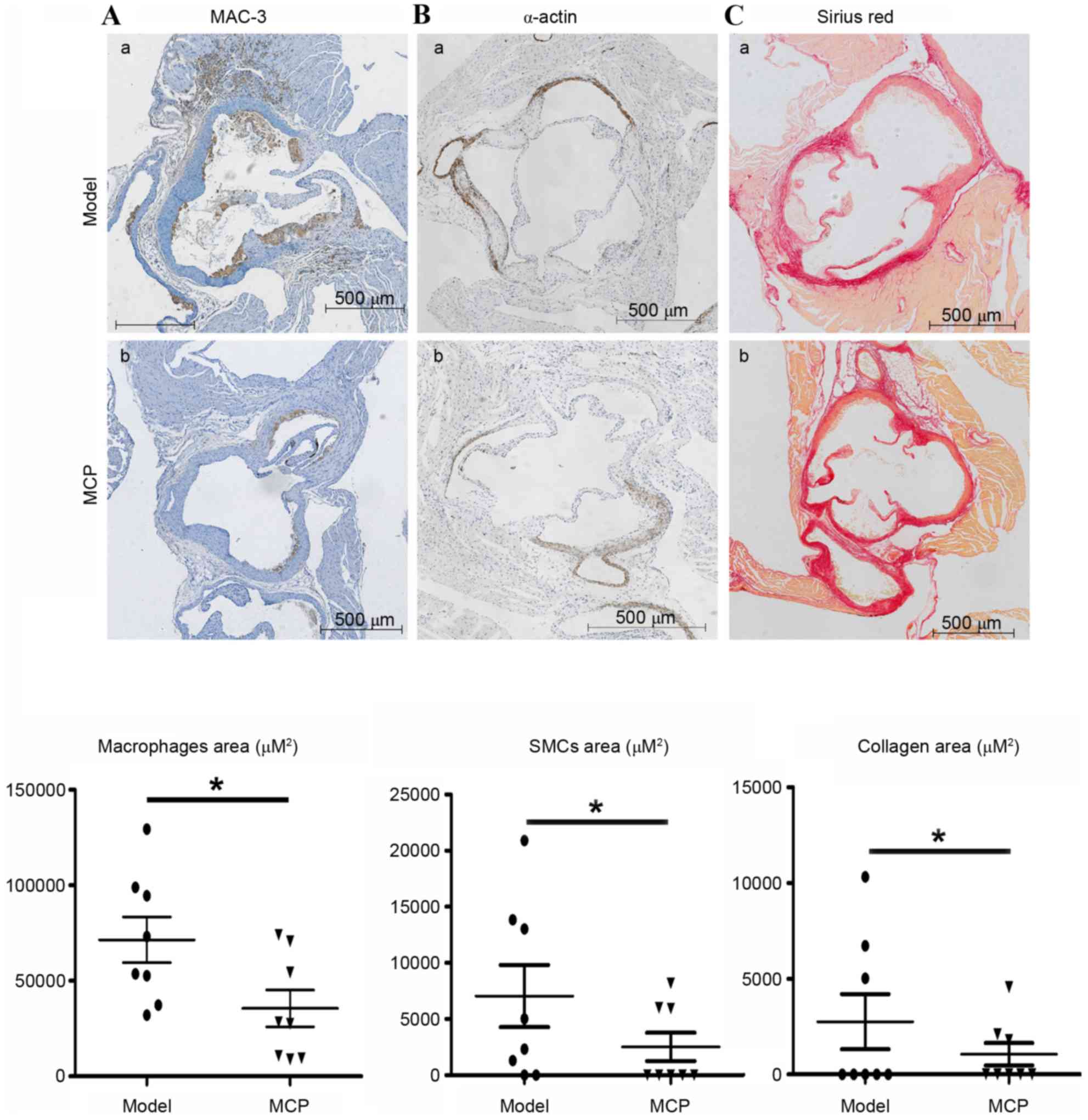
Atherosclerotic lesions are predominantly composed
of macrophages, SMCs and collagen. Immunohistochemical probing for
MAC-3, a marker of macrophages, indicated that MCP treatment
resulted in fewer macrophages in the atherosclerotic lesions
(Fig. 2A). Similarly, the amount
of collagen and the number of SMCs were also reduced in the
MCP-treated aortic root plaques (Fig.
2B and C).
MCP reduces in vitro adhesion of U937
monocytes to HUVECs
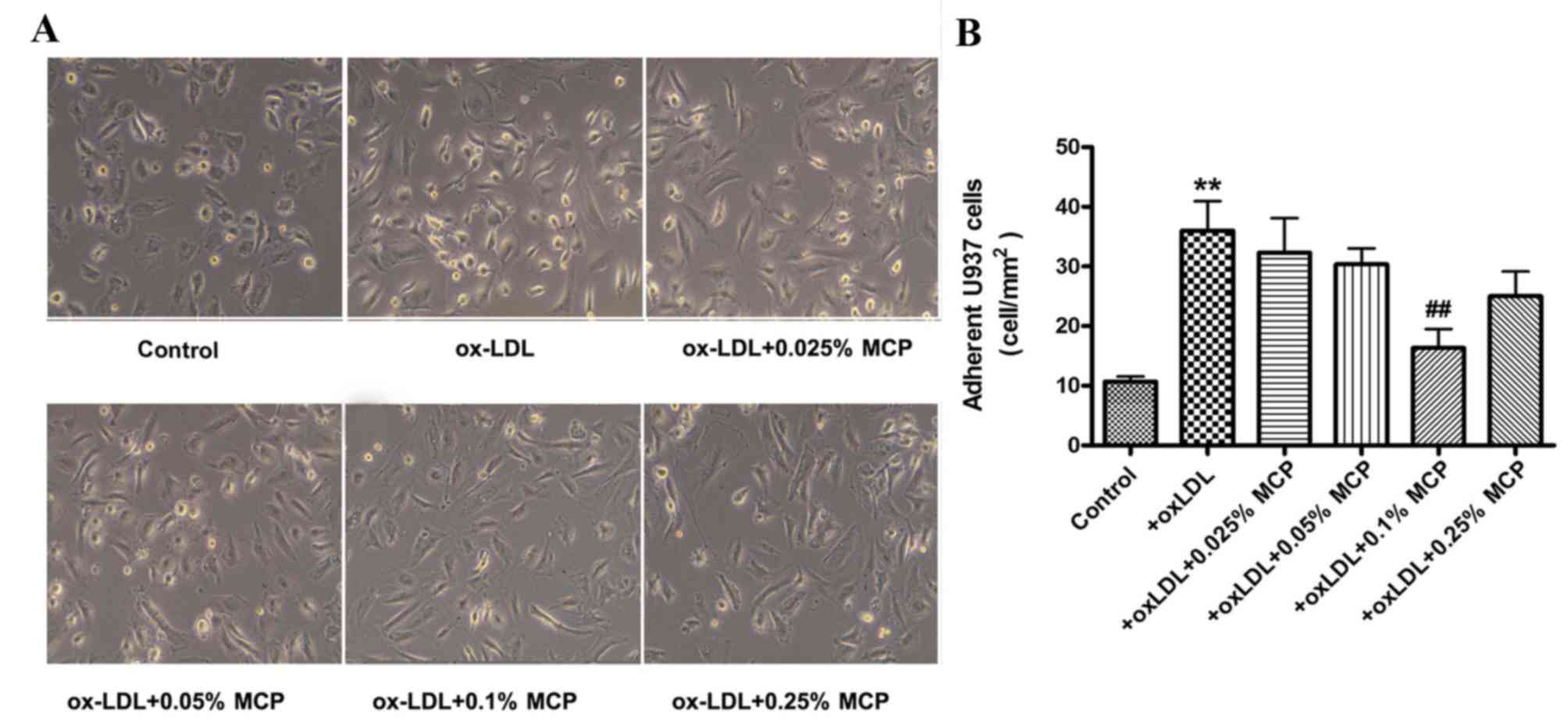
HUVECs were pretreated with ox-LDL and subsequently
co-cultured with U937 cells. Treatment of HUVECs with ox-LDL
increased the number of adherent monocytes (Fig. 3). However, subsequent incubation
with various concentrations of MCP (0.025, 0.05, 0.1 and 0.25%)
reduced the number of adherent U937 monocytes (Fig. 3).
MCP impairs endothelial injury in
apoE−/− mice
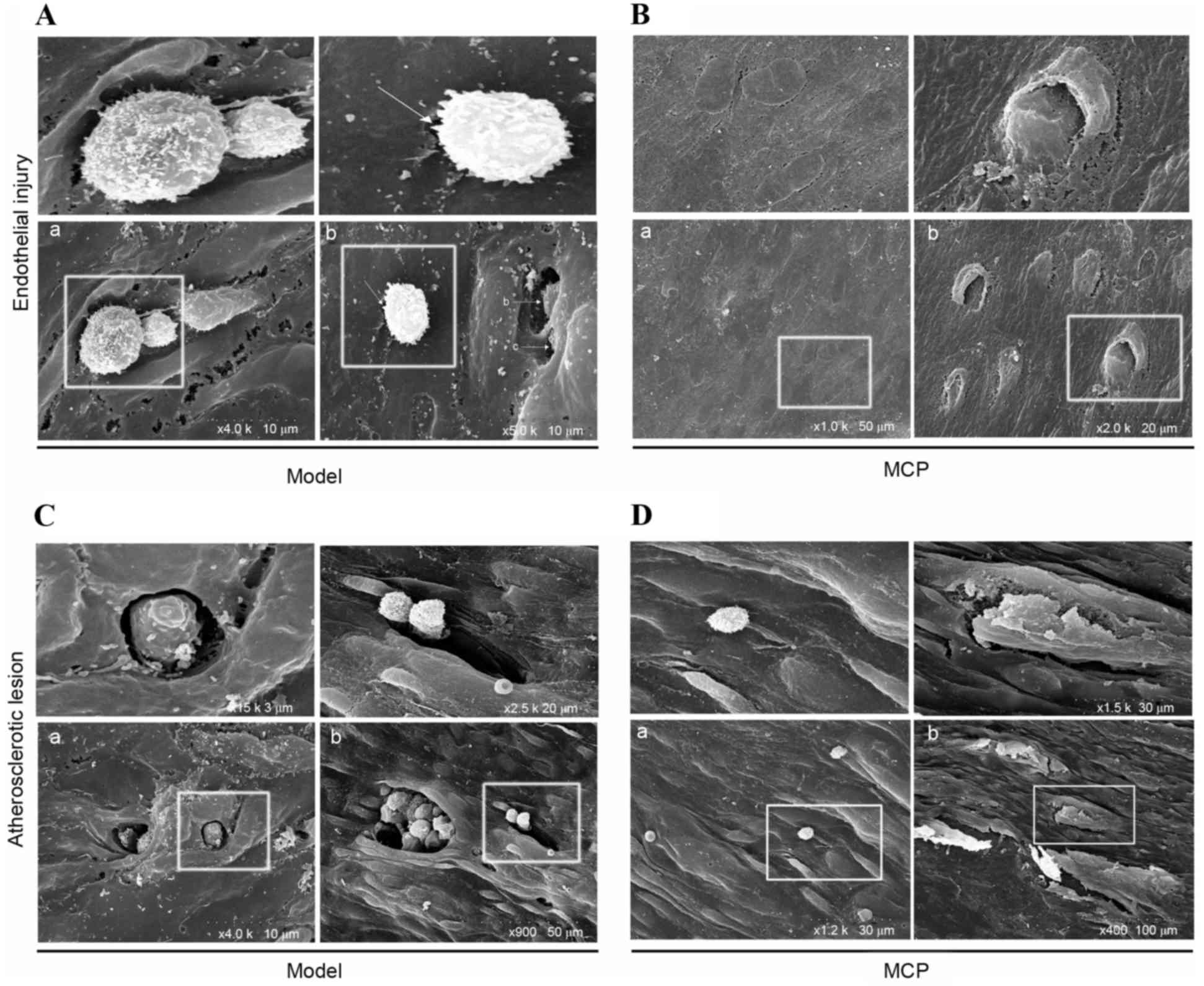
SEM analysis was performed on the surface of the
atheroma-prone vessel wall in apoE−/− mice. Pathological
alterations caused by endothelial injury were observed in the model
group; adherent monocytes were located at breakages in the
endothelial cell junction (Fig.
4A-a). In the MCP group, endothelial junction sites were loose,
however they did not appear to be broken (Fig. 4B-a). Notably, a monocyte was
observed entering the endothelial barrier (Fig. 4B-b). From these results, it was
speculated that the endothelial cell junctions recombined loosely,
following monocyte transmigration, in the MCP-treated group.
Furthermore, the lesion surface in the model group (Fig. 4C-a and -b) contained multiple
disruption holes or ‘craters’, whereas the endothelium of the MCP
group appeared to be of greater integrity (Fig. 4D-a and -b).
Discussion
The present study demonstrated that MCP-treated
apoE−/− mice developed smaller atherosclerotic lesions,
accompanied by fewer macrophages and SMCs and reduced amounts of
collagen. Previous research (13)
indicated that galectin-3-deficient mice had smaller
atherosclerotic plaque volumes, and reduced intralesional
macrophage accumulation and activation. Furthermore, the orally
active inhibitor of galectin-3, MCP, may also reduce the
atherosclerotic area; however, the precise mechanism remains
unclear. The present study was therefore conducted to further
investigate the role of galectin-3 in atherosclerosis.
ApoE−/− mice fed an atherogenic diet (1.25% cholesterol)
for 4 weeks displayed early or initial lesions (14); 1% MCP was therefore supplemented
into the drinking water for 4 weeks, to evaluate the effects of MCP
in early plaque formation. Oil Red O staining of en face
specimens was performed to evaluate the total atherosclerotic area,
and this revealed reduced aortic lesion areas in the MCP group,
compared with the model group. Furthermore, the BCA and aortic root
were examined pathologically, and lesional areas were observed to
be significantly smaller in the MCP group compared with the model
group.
Macrophages are the predominant cell type present in
early atheroma plaques; they release various proinflammatory
cytokines and chemokines, thereby inducing additional circulating
monocytes to gather at the atheroma-prone areas, and to further
differentiate into lesional macrophages, which is followed by
vascular medial SMC migration and collagen synthesis (3). Analysis of the composition of the
MCP-treated group lesions indicated fewer macrophages and SMCs, and
reduced amounts of collagen, compared with the model group, thereby
indicating that MCP may be able to protect against atherosclerotic
development. Macrophages may interact with the ECM, at least
partly, via galectin-3-induced differentiation and maturation
(15). Galectin-3 is also
chemotactic, guiding macrophages to lectin-rich locations in
vivo and in vitro, and, similarly to monocyte
chemoattractant protein-1, it may induce Ca2+ influx in
monocytes; the chemotactic effect and the induction of
Ca2+ influx may involve a PTX-sensitive pathway
(10). Furthermore, galectin-3 is
present on macrophages in atherosclerotic areas, and increased
extracellular galectin-3 expression and secretion is a feature of
alternative macrophage activation; galectin-3 interaction with CD98
results in the activation of phosphoinositide-3 kinase, and thus
the alternative activation of macrophages. In addition, the
interleukin-4-induced alternative activation pathway may be blocked
via a specific inhibitor of extracellular galectin-3 carbohydrate
binding (16). These results
indicated that a galectin-3 feedback loop may induce alternative
macrophage activation. Pharmacological modulation of galectin-3
function may therefore represent a novel therapeutic strategy in
alternatively activated macrophage pathologies (19). The present study detected
significantly reduced macrophage- and SMC-positive areas in
atherosclerotic plaques in MCP-treated apoE−/− mice.
Galectin-3 may promote the expression of monocytic chemokines,
causing monocytes to preferentially migrate to, and accumulate in,
vascular endothelial and arterial plaque lesions, particularly in
lipid cores. This triggers the adhesion of macrophages to vascular
endothelial cells, via interaction with laminin, fibronectin,
elastin and collagen IV fibers (8). The transmigrated macrophages
communicate with SMCs, leading to SMC activation and proliferation,
in turn resulting in the appearance of SMCs in the atherosclerotic
plaque. Since SMCs also synthesize collagen and other ECM
components present in atherosclerotic lesions, a reduction in SMCs
observed in the MCP group was concurrent with decreased amounts of
collagen in the same group. Since lesional macrophages trigger and
expand the accumulation of additional macrophages and medial SMCs
to the lesion, the anti-atheroma effects of MCP may therefore
mainly result from its inhibitory impact on macrophage
adhesion.
The role served by the interaction between
endothelial cells and monocytes/macrophages in atherosclerotic
initiation is well documented (1).
Monocyte and macrophage adhesion and sub-endothelial migration are
the predominant processes associated with atherosclerotic
initiation. Atheroma-prone areas are inflammatory sites; these may
appeal to monocytes leaving the circulation, thus leading to
endothelial cell anchorage via sequential attachments, which are
mediated by several adhesion molecules, including galectin-3. It
has previously been indicated that galectin-3 is expressed by
various cell types including endothelial, epithelial and dendritic
cells, and numerous inflammatory cells such as
monocytes/macrophages, mast cells, neutrophils and eosinophils
(20). Furthermore, galectin-3 is
distributed on the cell surface, cytoplasm, and nucleus, as well as
in the extracellular environment, indicating the
multi-functionality of this lectin (21). Galectin-3 enables cell adhesion via
cell-ECM-protein and cell-cell interactions. ECM proteins include
laminin, fibronectin, proteoglycans, collagen and vitronectin,
which are distributed in endothelial cells, the interstitial space
and the basement membrane, and serve important roles in cell
adherence (22). Galectin-3
participates in the ECM-cell adhesion process, and the association
between galectin-3 and ECM proteins, particularly laminin and
fibronectin, is essential for leucocyte movement. Furthermore, the
interaction of galectin-3 with fibronectin is unique for the
macrophage migratory process (23), indicating that galectin-3 may guide
monocyte-derived macrophages to transmigrate across the endothelium
and reach the arterial media, via its ECM binding ability. Previous
studies have demonstrated that galectin-3 has no impact on the
expression of adhesion molecules, such as selectin, ICAM, VCAM and
very late antigen-4, indicating that galectin-3 is not an
inflammatory modulator. Therefore, galectin-3 may predominantly
guide macrophage movement via its binding affinity with the
redistributed ligands in various vascular tissues. The associations
between leucocytes and endothelial cells are primarily mediated by
the interaction of various adhesion molecules, such as selectins
and VCAM-1 to α4β1 integrin, and ICAM-1 to β2-integrin (24). VCAM-1 may mediate rolling-type
adhesion and firm adhesion, depending on the avidity status of
α4β1-integrin (25). It has been
demonstrated that galectin-3 is capable of associating with β1
integrin, via its carbohydrate recognition domain, in a
lactose-dependent manner (23).
The binding of galectin-3 with α4β1 integrin may further modulate
its receptor state and alter its affinity to VCAM-1; therefore,
galectin-3 may be indirectly involved in α4β1-integrin-VCAM-1
dependent rolling and adhesion. To identify the impact of
galectin-3 on endothelial cell-monocyte adhesion, the present study
treated U937 monocytes with MCP, followed by co-incubation with
ox-LDL stimulated HUVECs. The results revealed significantly
greater numbers of adherent U937 monocytes on ox-LDL stimulated
HUVECs, compared with controls. However, the number of adherent
U937 cells was markedly reduced when they were pretreated with only
0.1% MCP.
The transendothelial migration of monocytes requires
dynamic morphological alterations in endothelial cells. Monocyte
adhesion induces endothelial F-actin contraction, thereby
generating tension and facilitating the necessary changes to
cellular morphology, which results in endothelial deformability
(26). SEM analysis of
transendothelial macrophages revealed large endothelial structure
disruptions in the model group, whereas MCP-treated tissues had
only a loosened endothelial structure. Furthermore, endothelial
cell destruction was frequent in the atherosclerotic surfaces of
the model group, whereas the atherosclerotic plaque surface
remained intact in the MCP-treated group.
In conclusion, the present study demonstrated that
MCP, a galectin-3 inhibitor, reduced atherosclerotic lesions via
the inhibition of leucocyte adhesion to endothelial cells.
Inhibiting galectin-3 function may be a promising therapeutic
strategy for the treatment of atherosclerosis.
Acknowledgements
This study was supported by funding from the
National Basic Research Program of China (grant no. 2012CB517502)
and the National Natural Science Foundation of China (grant nos.
81270887 and 81070634).
References
|
1
|
Chistiakov DA, Revin VV, Sobenin IA,
Orekhov AN and Bobryshev YV: Vascular endothelium: Functioning in
norm, changes in atherosclerosis and current dietary approaches to
improve endothelial function. Mini Rev Med Chem. 15:338–350. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Schäfer A and Bauersachs J: Endothelial
dysfunction, impaired endogenous platelet inhibition and platelet
activation in diabetes and atherosclerosis. Curr Vasc Pharmacol.
6:52–60. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Blankenberg S, Barbaux S and Tiret L:
Adhesion molecules and atherosclerosis. Atherosclerosis.
170:191–203. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Rao SP, Wang Z, Zuberi RI, Sikora L,
Bahaie NS, Zuraw BL, Liu FT and Sriramarao P: Galectin-3 functions
as an adhesion molecule to support eosinophil rolling and adhesion
under conditions of flow. J Immunol. 179:7800–7877. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Soehnlein O: Multiple roles for
neutrophils in atherosclerosis. Circ Res. 110:875–888. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nieminen J, St-Pierre C, Bhaumik P,
Poirier F and Sato S: Role of galectin-3 in leukocyte recruitment
in a murine model of lung infection by Streptococcus pneumoniae. J
Immunol. 180:2466–2473. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Tadokoro T, Ikekita M, Toda T, Ito H, Sato
T, Nakatani R, Hamaguchi Y and Furukawa K: Involvement of
Galectin-3 with vascular cell adhesion molecule-1 in growth
regulation of mouse BALB/3T3 cells. J Biol Chem. 284:35556–35563.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Inohara H, Akahani S, Koths K and Raz A:
Interactions between galectin-3 and Mac-2-binding protein mediate
cell-cell adhesion. Cancer Res. 56:4530–4534. 1996.PubMed/NCBI
|
|
9
|
Sasaki T, Brakebusch C, Engel J and Timpl
R: Mac-2 binding protein is a cell-adhesive protein of the
extracellular matrix which self-assembles into ring-like structures
and binds beta1 integrins, collagens and fibronectin. EMBO J.
17:1606–1613. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara
I, Yamanaka T, Hirashima M and Liu FT: Human galectin-3 is a novel
chemoattractant for monocytes and macrophages. J Immunol.
165:2156–2164. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hopkins PN: Molecular biology of
atherosclerosis. Physiol Rev. 93:1317–1542. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Tekabe Y, Li Q, Rosario R, Sedlar M,
Majewski S, Hudson BI, Einstein AJ, Schmidt AM and Johnson LL:
Development of receptor for advanced glycation end
products-directed imaging of atherosclerotic plaque in a murine
model of spontaneous atherosclerosis. Circ Cardiovasc Imaging.
1:212–219. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Gao X, Zhi Y, Zhang T, Xue H, Wang X,
Foday AD, Tai G and Zhou Y: Analysis of the neutral polysaccharide
fraction of MCP and its inhibitory activity on galectin-3.
Glycoconj J. 29:159–165. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Nachtigal M, Ghaffar A and Mayer EP:
Galectin-3 gene inactivation reduces atherosclerotic lesions and
adventitial inflammation in ApoE-deficient mice. Am J Pathol.
172:247–255. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jacob SS, Shastry P and Sudhakaran PR:
Monocyte-macrophage differentiation in vitro: Modulation by
extracellular matrix protein substratum. Mol Cell Biochem.
233:9–17. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
MacKinnon AC, Farnworth SL, Hodkinson PS,
Henderson NC, Atkinson KM, Leffler H, Nilsson UJ, Haslett C, Forbes
SJ and Sethi T: Regulation of alternative macrophage activation by
galectin-3. J Immunol. 180:2650–2658. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Walski M, Chlopicki S, Celary-Walska R and
Frontczak-Baniewicz M: Ultrastructural alterations of endothelium
covering advanced atherosclerotic plaque in human carotid artery
visualized by scanning electron microscope. J Physiol Pharmacol.
53:713–723. 2002.PubMed/NCBI
|
|
18
|
Nathan L, Pervin S, Singh R, Rosenfeld M
and Chaudhuri G: Estradiol inhibits leukocyte adhesion and
transendothelial migration in rabbits in vivo: Possible mechanisms
for gender differences in atherosclerosis. Circ Res. 85:377–385.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
MacKinnon AC, Liu X, Hadoke PW, Miller MR,
Newby DE and Sethi T: Inhibition of galectin-3 reduces
atherosclerosis in apolipoprotein E-deficient mice. Glycobiology.
23:654–663. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Swarte VV, Mebius RE, Joziasse DH, Van den
Eijnden DH and Kraal G: Lymphocyte triggering via L-selectin leads
to enhanced galectin-3-mediated binding to dendritic cells. Eur J
Immunol. 28:2864–2871. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Funasaka T, Raz A and Nangia-Makker P:
Galectin-3 in angiogenesis and metastasis. Glycobiology.
24:886–891. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hynes RO: The extracellular matrix: Not
just pretty fibrils. Science. 326:1216–1219. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Hashiba K, Sano M, Nio-Kobayashi J, Hojo
T, Skarzynski DJ and Okuda K: Galectin-3 contributes to luteolysis
by binding to Beta 1 integrin in the bovine corpus luteum. Biol
Reprod. 91:22014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Hatley ME, Srinivasan S, Reilly KB, Bolick
DT and Hedrick CC: Increased production of 12/15 lipoxygenase
eicosanoids accelerates monocyte/endothelial interactions in
diabetic db/db mice. J Biol Chem. 278:25369–25375. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cybulsky MI, Iiyama K, Li H, Zhu S, Chen
M, Iiyama M, Davis V, Gutierrez-Ramos JC, Connelly PW and Milstone
DS: A major role for VCAM-1, but not ICAM-1, in early
atherosclerosis. J Clin Invest. 107:1255–1262. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Ingber DE: Cellular tensegrity: Defining
new rules of biological design that govern the cytoskeleton. J Cell
Sci. 104:613–627. 1993.PubMed/NCBI
|