Introduction
Over the last 40 years, the incidence of obesity in
children and adolescents has increased significantly (1), and has become a major chronic disease
endangering the health of children (2). Obesity was recognized as a disease by
the American Medical Association in 2013 (3). Childhood and adolescent obesity can
adversely affect almost every organ system, including the liver and
adipose tissue, and often causes serious consequences, including
dyslipidemia and fatty liver disease. Obesity, as a chronic disease
requiring long-term management, has led to the increasing focus on
the role of adjunctive therapies for obesity, particularly
pharmacotherapy (4).
Metformin is a widely used fist-line antidiabetic
drug, which effectively lowers plasma glucose and improves hepatic
insulin sensitivity, particularly for overweight and obese
individuals. An increasing number of studies have shown that
metformin can reduce the body weight of obese individuals, and
ameliorate lipid accumulation in the liver in hepatic steatosis
(5–7). Although the mechanisms underlying the
action of metformin are complicated, there is accumulating evidence
that demonstrates that the beneficial effects of metformin are
associated with autophagy (8,9).
Autophagy is a cellular self-digestion process,
which removes damaged macromolecules and organelles, and maintains
cellular homeostasis (10). Under
physiological conditions, autophagy is involved in the basal
turnover of lipids by engulfing and degrading lipid droplets.
Autophagy genes are activated by AMP-activated protein kinase
(AMPK), a central energy sensor, as a novel Unc-51-like kinase 1
binding partner, which is a protein kinase activated by an increase
in the AMP/ATP ratio (11).
Several studies have demonstrated that the level of autophagy can
be decreased in hepatocytes in obesity (9). The pharmacological inhibition of
autophagy using 3-methyladenine (3MA) or the knockdown of the
autophagy gene, autophagy related 5 (Atg5), in hepatocytes has been
found to significantly increase hepatocyte triglyceride (TG)
content in the absence or presence of exogenous lipid
supplementation with oleate (12).
In addition, Kovsan et al reported that autophagy is
upregulated in the adipose tissue of obese individuals,
particularly in omental adipose tissue, correlating with the degree
of obesity, visceral fat distribution and adipocyte hypertrophy
(13). The knockdown of Atg7 or
Atg5 in preadipocytes inhibited lipid accumulation, and the
adipocyte-specific mouse knockout of Atg7 generated lean mice with
decreased white adipose mass (14). These experimental data indicate
that autophagy is closely associated with obesity and lipid
metabolism. Based on these findings, the present study investigated
whether metformin affects lipid metabolism in the liver and adipose
tissue of obese individuals, and whether it is associated with
ameliorating autophagy.
Materials and methods
Animals
Male C57BL/6 mice (3-week-old) were obtained from
the Animal Center of Xi'an Jiaotong University (Xi'an, China). The
mice were housed in a specific pathogen-free environment with
controlled room temperature and humidity (22°C; 60% relative
humidity) prior to commencement of the experiments. The mice were
fed adaptively for 1 week, and then randomly divided into three
groups: Normal chow diet group (NCD), n=8; High fat diet (HFD)
group, n=16; HFD+metformin group, n=13. NCD mice were fed a normal
chow diet containing 20.8% fat, 18.3% protein and 60.9%
carbohydrates for 12 weeks. HFD mice were fed a high fat diet
containing 47.5% fat, 18.5% protein and 34.3% carbohydrates for 12
weeks. HFD+metformin mice were treated with metformin (150 mg/kg/d)
by intraperitoneal injection for the final 4 weeks of HFD feeding.
All mice were maintained on a 12:12-h light-dark cycle (lights on
at 06:00 a.m.). Food and water were provided ad libitum.
During the 12-week feeding/treatment period, the body weights of
the mice were recorded weekly. The food intake of the mice was
recorded daily during the 4-week treatment period. Following the
feeding/treatment regimen, the mice were fasted for 8 h prior to
sacrifice for the collection of blood and tissue samples.
Epididymal, perinephric and brown fat depots were dissected and
weighed. The liver was also dissected and weighed. Following
weighing, epididymal adipose and liver tissue samples were either
fixed and embedded for histological evaluation and
immunohistochemistry, or were frozen in liquid nitrogen and stored
at −80°C for further analyses. All the mice were fasted similarly
and used for serum TG and transaminase assessment. All experiments
were performed according to the guidelines of the Committee of
Animal Research at Xi'an Jiaotong University and the
recommendations in the Guide for the Care and Use of Laboratory
Animals of the National Institutes of Health (15).
Drugs
Metformin (1,1-dimethylbiguanide hydrochloride;
Bristol-Myers Squibb Company, Princeton, NJ, USA) was dissolved in
0.9% saline, and administered daily by intraperitoneal injection at
09:00 a.m.
Histological analyses with hematoxylin
and eosin (H&E) staining, and immunohistochemistry
The paraffin-embedded liver and adipose tissue
specimens were cut into sections of 5 µm thickness and H&E
stained to assess the severity of lipid accumulation in the liver
tissues. The surplus sections were used for immunohistochemical
analysis. The sections were dewaxed and rehydrated with
dimethylbenzene and different concentrations of ethanol,
respectively. Subsequently, sections were processed by
microwave-based antigen retrieval for 10 min, and treated with 3%
H2O2 solution to block endogenous peroxidase
activity. After 3 washes in PBS, the sections were treated with
goat serum (ZSGB-BIO, Beijing, China), 37°C for 15 min to block
nonspecific sites and incubated overnight at 4°C with rabbit
polyclonal antibody against LC3 (1:200; bs-8878R) and AMPKα2
(1:100; bs-2771R) (both from BIOSS, Beijing, China) diluted in
double distilled water. Following several rinses in PBS,
peroxidase-labeled goat anti-rabbit secondary antibody working
solution (SP-9001; ZSGB-BIO) was added for 15 min at 37°C. The
peroxidase activity was observed with a Q550CW Image Acquiring
& Analysis System (Leica Microsystems GmbH, Wetzlar, Germany),
using a diaminobenzidine substrate, which yielded a yellow/brown
deposit. Negative controls included sections with the primary
antibodies omitted. The immunohistochemical-stained slides were
selected randomly and were analyzed using Image-Pro plus 6.0
software (Media Cybernetics, Inc., Rockville, MD, USA). For each
selected field, the positive area was calculated automatically
using the software. These results were expressed by volume
fractions, as the percentage of positive area in relation to the
total\area, and expressed as the mean ± standard error of the
mean.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was isolated from ~50 mg samples of
adipose and liver tissue according to the manufacturer's protocol
using Ambion TRIzol (Invitrogen cat. no. 15596-026; Invitrogen;
Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
concentration of total RNA was measured using ultraviolet
spectrophotometry (ND-1000; Thermo Fisher Scientific, Inc.). First
strand-cDNA synthesis was performed with oligo (dT) primer using a
PrimeScript RT regent kit (cat. no. RR820A; Takara Bio, Inc., Otsu,
Japan). The RT-qPCR analysis was performed with a Bio-Rad iQ5q-PCR
system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) using SYBR
Premix Ex Taq II (Takara Bio, Inc.). The primers used are shown in
Table I. Data were normalized to
the housekeeping gene, glyceraldehyde-3-phosphate dehydrogenase
(GAPDH). qPCR reactions contained 25 ng cDNA template, 10 µM
forward and reverse primers, 10 µl 2X SYBR Premix Ex Tag II
(RR820A; Takara Bio, Inc., Otsu, Japan) and 6.4 µl dH2O (Takara
Bio, Inc.) in a total reaction volume of 20 µl. Following a
pre-denaturation step at 95°C for 30 s, qPCR was performed using 40
cycles of denaturation at 95°C for 5 s, annealing at 60°C for 20 s
and elongation at 72°C for 30 s. The results were expressed as the
number of cycles (Cq value) at which the fluorescence signal
exceeded a defined threshold. The difference in Cq values of the
target cDNA and GAPDH were expressed as ΔCq values. The 2-∆∆Cq
method was used for quantification of the results (16).
 | Table I.Primers used for reverse
transcription-quantitative polymerase chain reaction analysis in
the control and treatment mice. |
Table I.
Primers used for reverse
transcription-quantitative polymerase chain reaction analysis in
the control and treatment mice.
| Gene | Primer sequence | Product length
(bp) | Gene bank no. |
|---|
| LC3 | F:
5′-CCCCAGTGGATTAGGCAGAG-3′ | 127 | NM_025735.3 |
|
| R:
5′-CAGCCAGCACCCAAAAGAG-3′ |
|
|
| AMPK | F:
5′-CCACATTCGAAGCCCATGTT-3′ | 199 | NM_178143.2 |
|
| R:
5′-AGGGACAGGTAGGTCACAGAGA-3′ |
|
|
| GAPDH | F:
5′-TGTGTCCGTCGTGGATCTGA-3′ | 150 | NM_008084.2 |
|
| R:
5′-TTGCTGTTGAAGTCGCAGGAG-3′ |
|
|
Statistical analysis
Numeric data are presented as the mean ± standard
error of mean. All statistical analyses were performed using
Student's t test, and analyzed using two-way repeated
measures analysis of variance followed by Bonferroni multiple
comparisons using SPSS 18.0 software (SPSS, Inc., Chicago, IL,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Effects of metformin on body weight
and food intake in HFD-induced obese mice
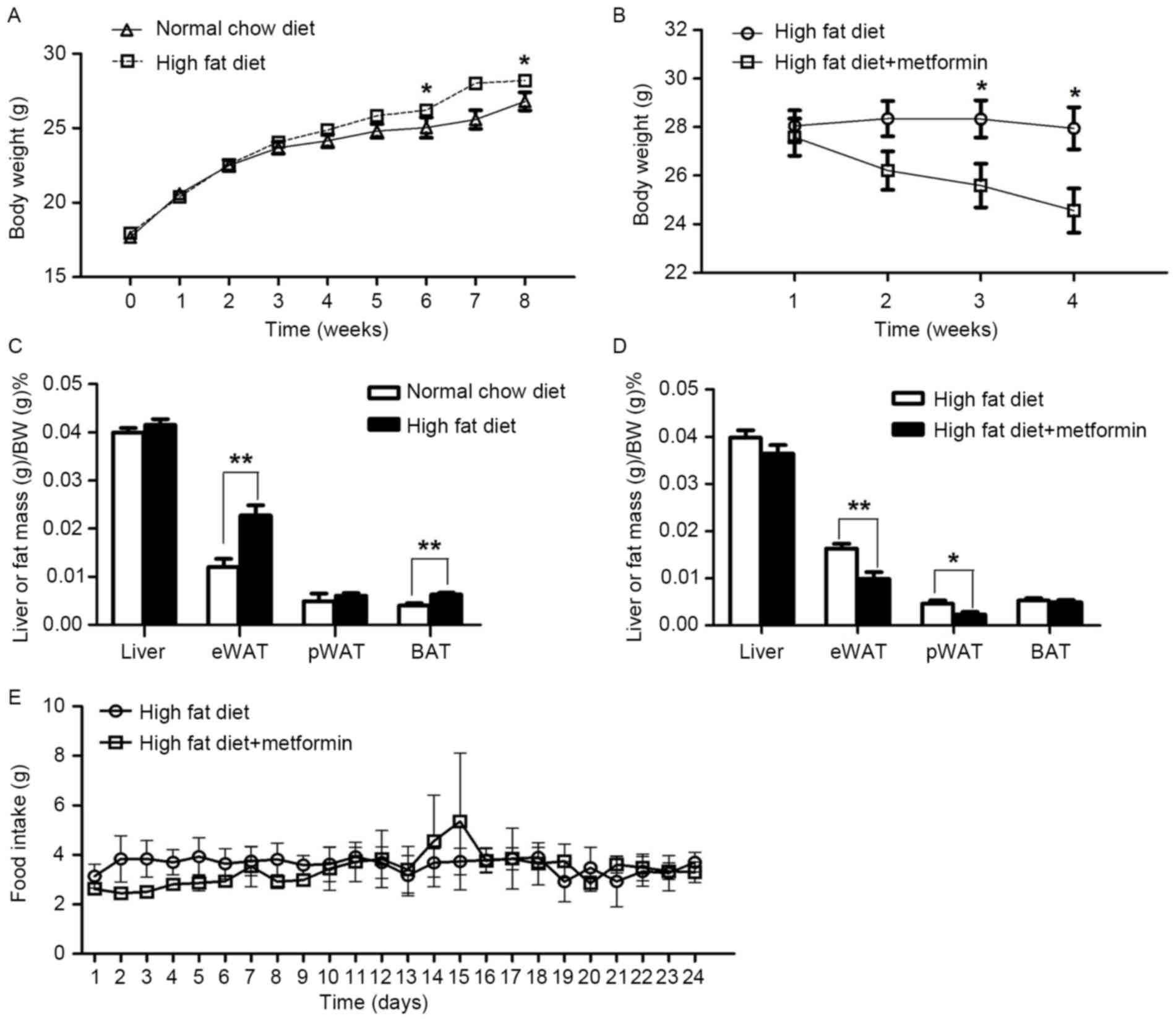
The mice maintained on the HFD for 8 weeks exhibited
significantly increased body weight, compared with the NCD mice
(P<0.05; Fig. 1A). Metformin
treatment at 150 mg/kg daily for 4 weeks significantly decreased
body weight in the obese mice maintained on a HFD (P<0.05;
Fig. 1B). In addition, the
percentage of body fat in the eWAT was significantly reduced in the
HFD+metformin mice following the 4 weeks, compared with that in the
HFD mice (Fig. 1C and D). However,
no significant difference in daily food intake was observed between
the HFD mice and the HFD+metformin mice (P>0.05; Fig. 1E).
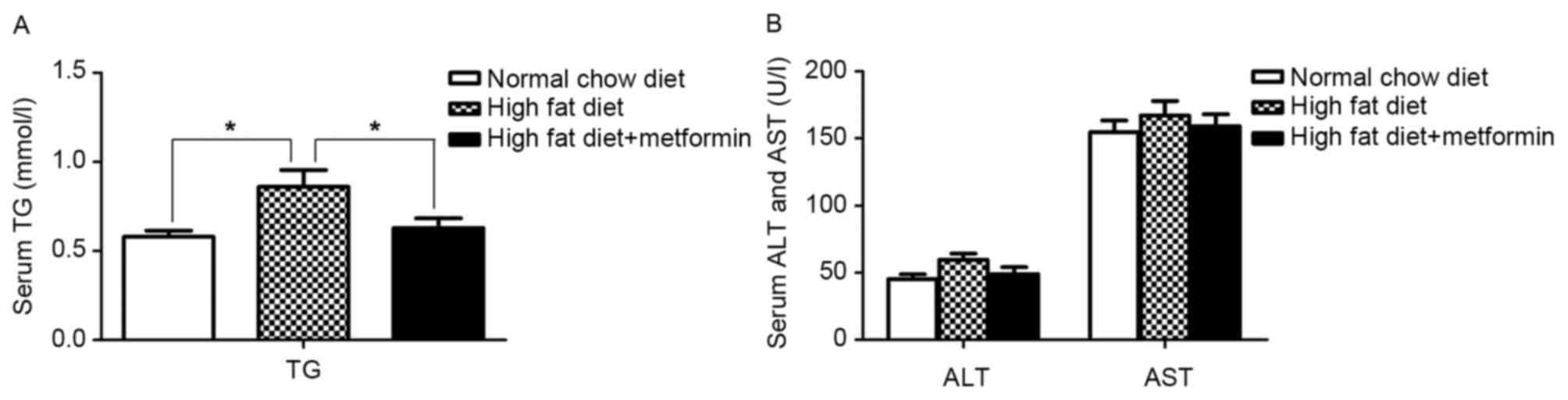
Effect of metformin on serum TG
The metformin-treated mice exhibited a significant
decrease in the severity of the HFD-induced high TG levels
(P<0.05; Fig. 2A). No
differences in the serum levels of aspartate aminotransferase (AST)
or alanine aminotransferase (ALT) were observed between the NCD
mice and HFD mice (P>0.05; Fig.
2B).
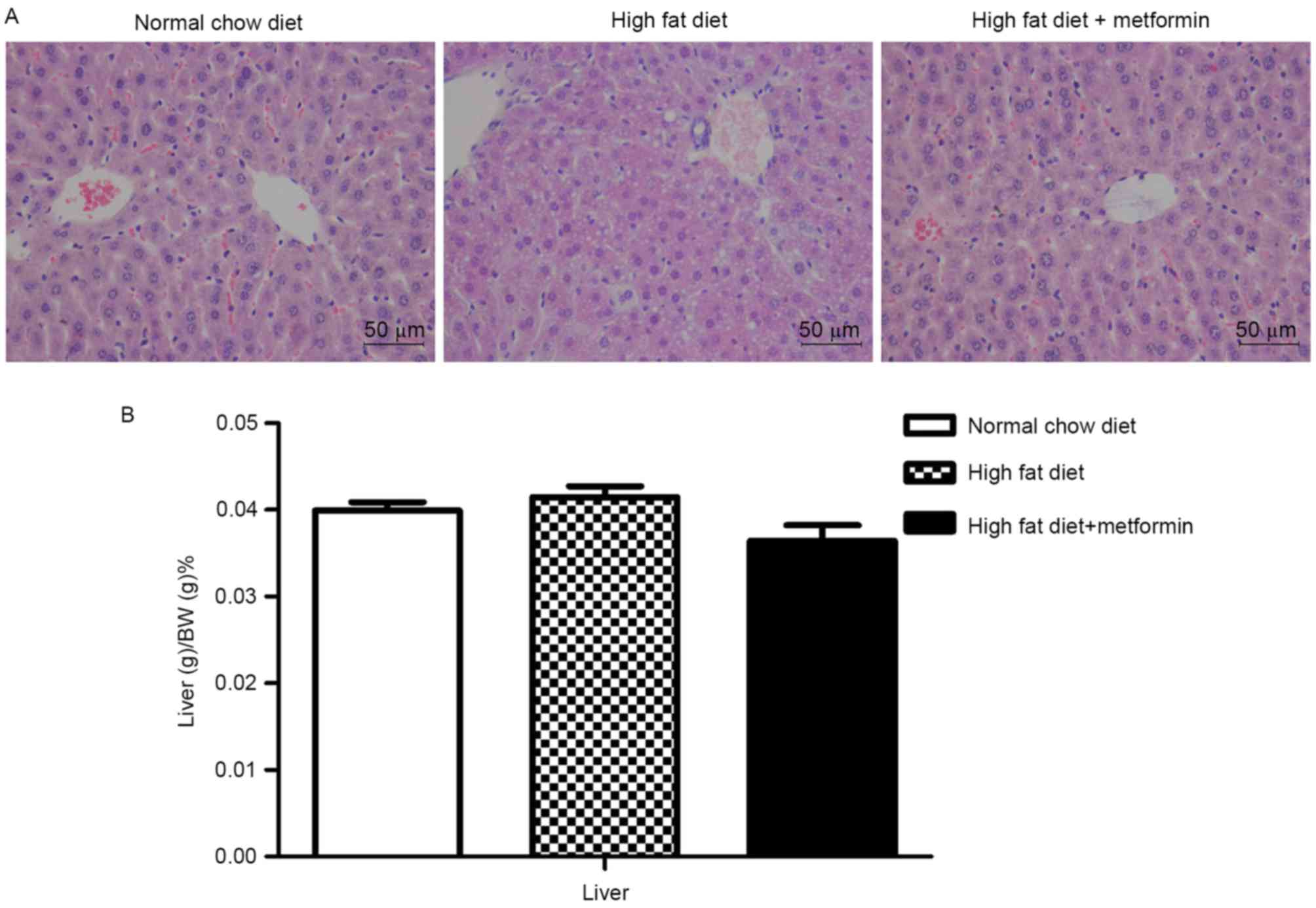
Metformin treatment ameliorates
HFD-induced hepatic lipid accumulation
The HFD mice exhibited lipid accumulation
surrounding the perisinusoidal areas, indicated by changes in the
liver sections stained with H&E (Fig. 3A). In contrast to the increased
body weights, the liver weights of the HFD mice were only
marginally increased, with no significant difference between the
NCD mice and HFD mice (P>0.05; Fig.
3B). Compared with the HFD mice, the HFD+metformin mice
exhibited a marginal decrease in liver weight, which was
accompanied by a marked decrease in lipid accumulation surrounding
the perisinusoidal areas (Fig.
3).
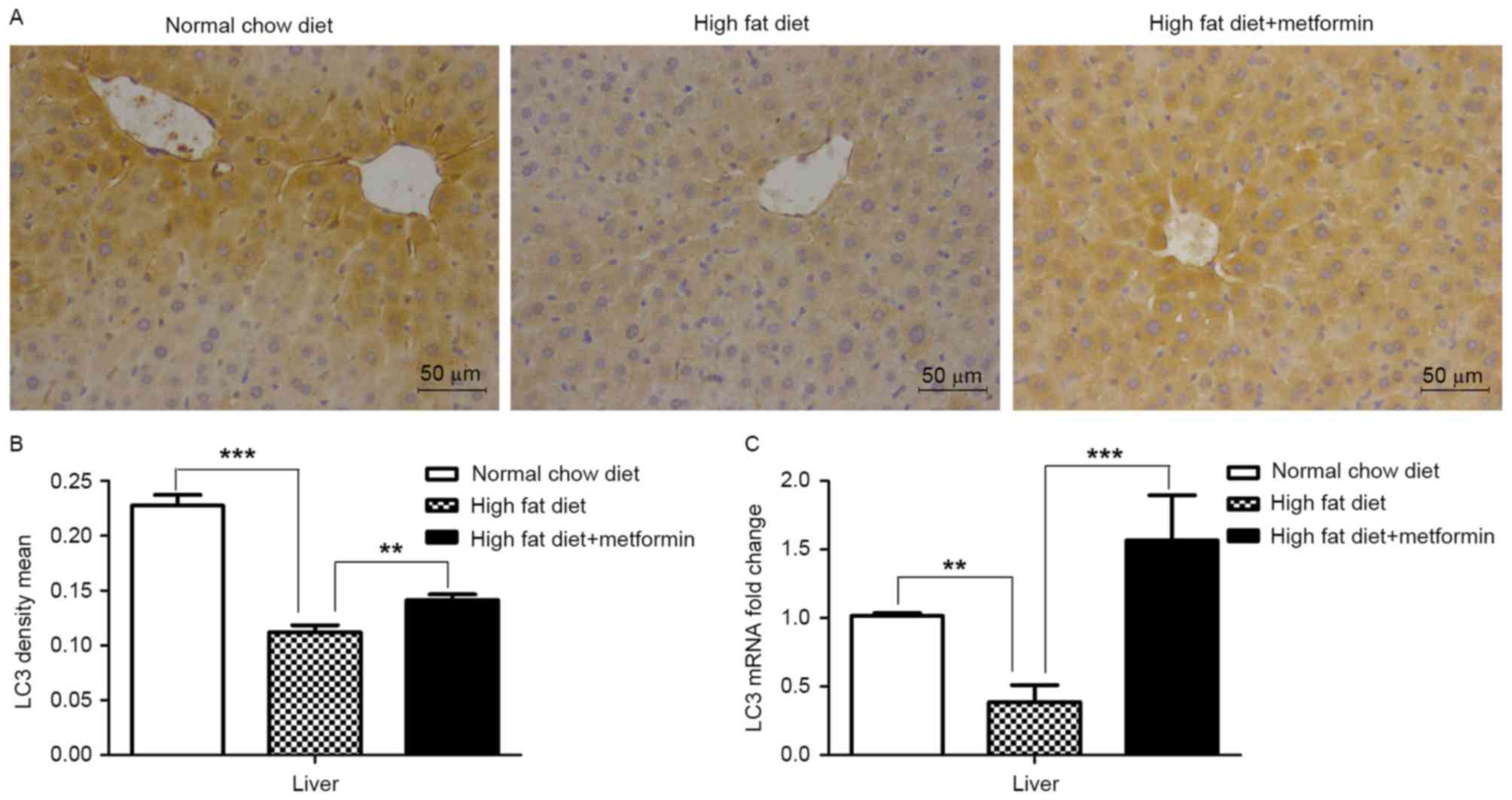
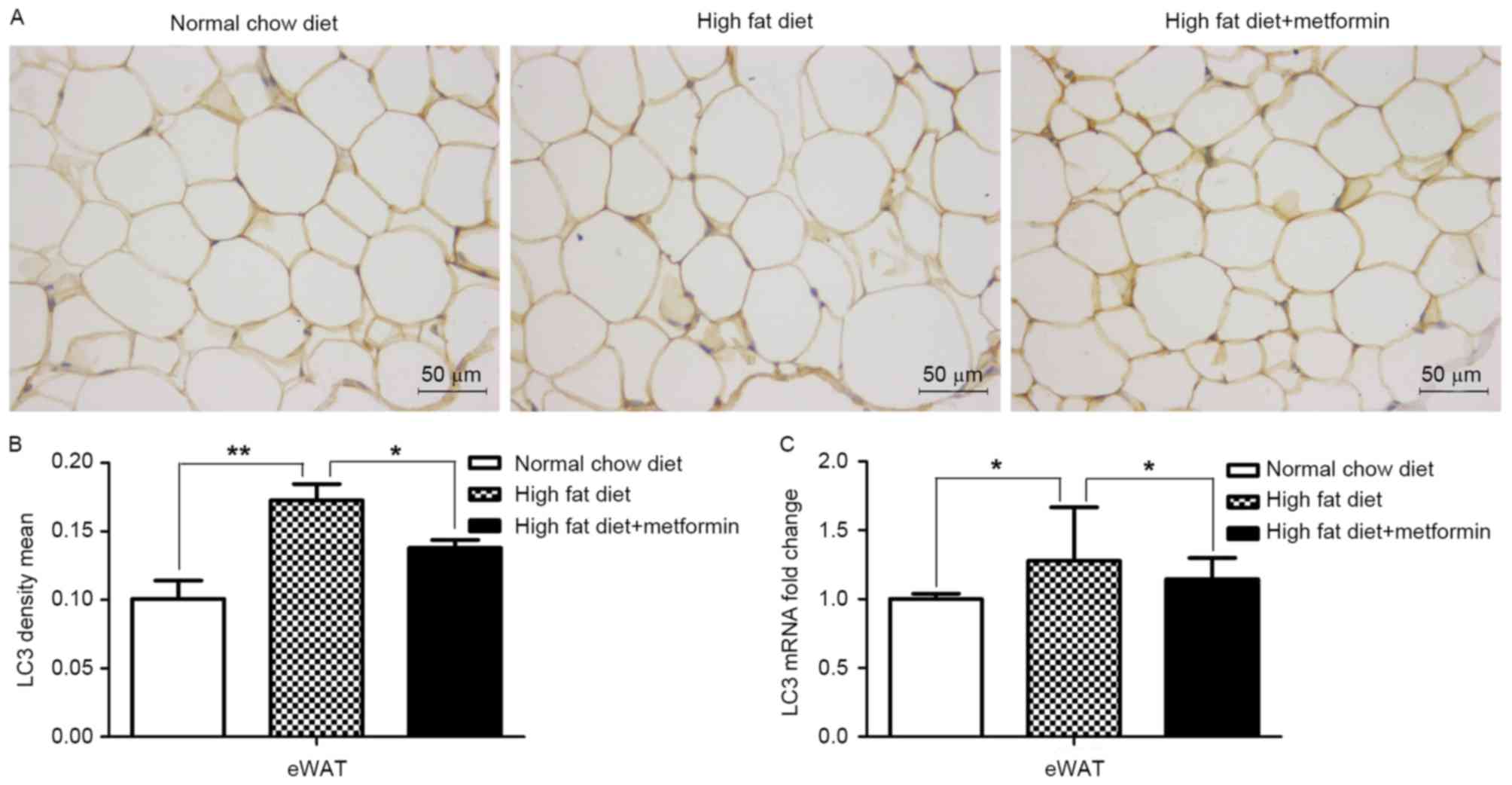
Effects of metformin on the expression
of LC3 in liver and adipose tissues of HFD-induced obese mice
The present study also examined the expression of
LC3 in the liver tissue and eWAT of the HFD mice maintained with
metformin, respectively. Significantly decreased mRNA and protein
levels of LC3 were observed in the liver of the HFD mice, compared
with those of the NCD mice (P<0.001), whereas metformin
treatment upregulated the expression of LC3 in the liver of the HFD
mice (P<0.01; Fig. 4A and B).
However, increased mRNA and protein levels of LC3 were observed in
the adipose tissues of the HFD mice compared with those of the NCD
mice (P<0.01; Fig. 5A-C). Of
note, metformin treatment suppressed the expression of LC3 in the
eWAT, compared with that in the HFD mice (P<0.05; Fig. 5).
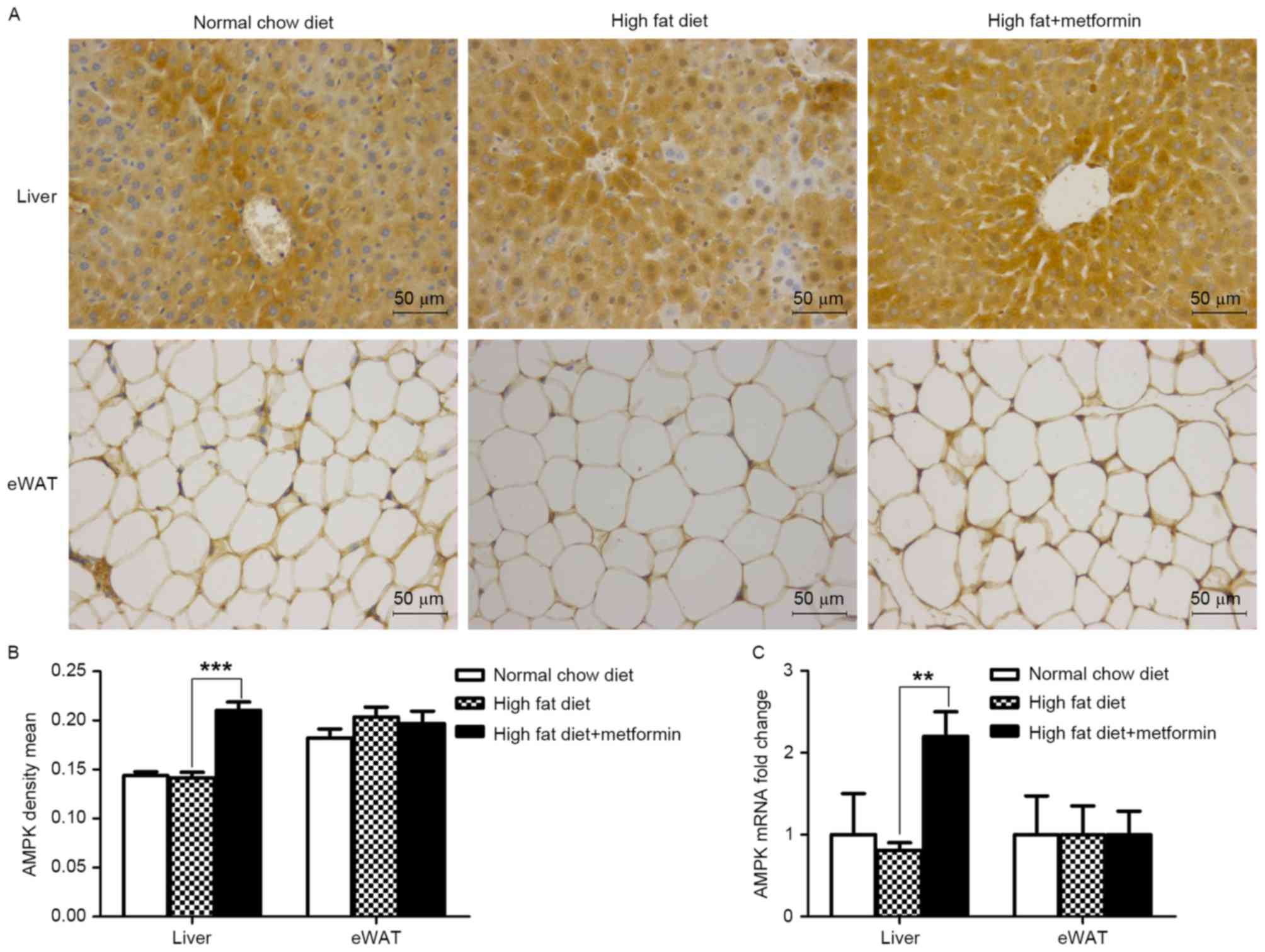
Metformin treatment upregulates the
expression of AMPK in the liver, but not the eWAT of HFD-induced
obese mice
The present study further investigated the effects
of metformin on the protein and mRNA expression levels of AMPK in
the liver and eWAT. As shown in Fig.
6A and B, metformin treatment significantly upregulated the
expression of AMPK in the liver (P<0.001), which was consistent
with the increased expression of LC3 in the liver of the HFD mice.
However, this effect of metformin was not observed in the eWAT of
the HFD+metformin mice.
Discussion
Following feeding of the HFD, C57BL/6 mice developed
obesity and hepatic steatosis. Accordingly, the HFD mice were used
in the present study as a model of obesity and obesity-induced
hepatic steatosis to assess the therapeutic effects of metformin.
Consistent with the results obtained from human subjects and rodent
models (6,7,17,18),
metformin treatment not only decreased the body weight of the
HFD-induced obese mice, particularly adipose tissue weight, it also
caused a marked decrease in the severity of HFD-induced hepatic
steatosis. However, metformin did not alter the food intake of the
obese mice. The results also demonstrated that the protein and mRNA
levels of autophagy markers were significantly decreased in the
liver of the HFD-induced obese mice, but only marginally increased
in adipose tissue. Notably, metformin treatment ameliorated the
decreased protein and mRNA levels of autophagy marker LC3 in the
liver of HFD-induced obese mice, and inhibited the increased
expression of LC3 in the adipose tissue of HFD-induced obese mice.
Therefore, the present study provided evidence to support the
hypothesis that metformin treatment ameliorates obesity and
obesity-associated hepatic steatosis in an adipose tissue
phenotype-dependent manner, without altering food intake.
Metformin is considered to be an activator of AMPK.
The latter, when activated, results in an increase in the induction
of autophagy through inhibition of the mammalian target of
rapamycin (mTOR) pathway (11,19).
Autophagy facilitates the removal of excess lipid, thereby
maintaining cellular lipid homeostasis. In the present study, two
lines of evidence support the hypothesis that metformin acts
through the induction autophagy to reduce hepatic steatosis in
HFD-induced obese mice. Firstly, a decrease in the induction of
autophagy was observed in mice with obesity-induced hepatic
steatosis. Compared with the NCD mice, the protein and mRNA levels
of autophagy marker LC3 were significantly decreased in the liver
of HFD mice. Secondly, metformin treatment significantly
upregulated the expression of LC3 in the HFD mice, and was
consistent with the increase in the expression of AMPK.
The results of the present study also suggested a
potential link between adipose tissue phenotype and autophagy,
which is altered by metformin. As described in widely accepted
concepts, dysfunctional adipose tissue in obesity contributes to
the pathogenesis of hepatic steatosis by increasing the delivery of
fatty acids to the liver to exacerbate hepatic fat deposition
(20). In accordance, the present
study hypothesized that improved adipose tissue phenotype
contributes to the anti-hepatic steatosis effect of metformin. In
the present study, adipose tissue weight was significantly
decreased in response to metformin treatment in the HFD mice.
However, the action of metformin on adipose tissue phenotype
remains controversial. Metformin has been shown to reduce body
weight (adiposity) in human and rodent models, whereas several
studies have demonstrated that metformin does not alter body
weight, including in rodents fed an HFD (21–23).
A number of studies have also shown that the weight-loss effect of
metformin is closely associated with a decrease in food intake
(6). However, in the present
study, it was found that metformin treatment reduced the body
weight of HFD mice without altering the food intake of mice. This
raises the question of whether metformin directly acts on adipose
tissue. The underlying mechanisms remain to be elucidated, however,
they may be attributable to the effect of metformin treatment in
altering the induction of adipose tissue autophagy. Therefore, the
protein and mRNA levels of the autophagy marker, LC3, in adipose
tissues were examined in the present study. Notably, unlike the
liver, HFD-induced autophagy was increased in the eWAT, and
metformin treatment inhibited the induction of autophagy in the
adipose tissue of the HFD mice.
As mentioned above, metformin directly improved the
induction of hepatic autophagy and suppressed the induction of
autophagy in the adipose tissues of the HFD mice. As the
metformin-induced activation of AMPK is associated with inhibition
of mTOR signaling in various cells, which induces autophagy
(8,19), the protein and mRNA levels of AMPK
were detected in the liver and eWAT in the HFD and HFD+metformin
mice. Consistent with the increased expression of LC3 in hepatic
tissue, the expression level of AMPK in the liver of the HFD mice
was upregulated by metformin. When the adipose and liver tissues
were compared, metformin treatment had no effect on the expression
level of AMPK in the adipose tissue of HFD mice. Therefore, it is
possible that a potential anti-autophagy action of metformin acts
on adipose tissue via an AMPK-independent pathway. Although this
requires further validation, the results suggested that metformin
directly targeted the liver to improve features of hepatic
steatosis, and this effect of metformin may be dependent on adipose
tissue phenotype.
In conclusion, the present study provided evidence
to support the beneficial effects of metformin on reducing the body
weight/adiposity and hepatic steatosis of HFD-induced obese mice,
with no significant effect on food intake, and these effects were
dependent on alterations in adipose tissue phenotype.
Mechanistically, the actions of metformin were attributable to its
effects on improving the induction of hepatic autophagy and
inhibiting the induction of adipose tissue autophagy. These results
provide experimental evidence for the inhibition of
obesity-associated hepatic steatosis in mice treated with
metformin. Therefore, metformin supplementation may be an effective
approach for the treatment and/or prevention of HFD-induced obesity
and hepatic steatosis.
Acknowledgements
The authors would like to thank Dr. Yin Chunyan
(Pediatrics department, 2nd Affiliated Hospital of Xi'an Jiaotong
University) for their advice in study design and data analysis; Dr.
Tan Xinrui (Pediatrics department, 2nd Affiliated Hospital of Xi'an
Jiaotong University) for their literature sharing and assistance;
and Dr. Antara Sharma (Health Science Center, Xi'an Jiaotong
University) as a native-English speaker for their editorial
corrections. The authors would also like to thank Professor Zhao
Xiaoge and Dr. Huang Shanlong (Health Science Center, Xi'an
Jiaotong University) for their assistance with pathological
analysis and statistical analysis, respectively. This study was
funded by the National Natural Science Foundation of China (grant
no. 81172689).
References
|
1
|
Ogden CL, Carroll MD, Kit BK and Flegal
KM: Prevalence of childhood and adult obesity in the United States,
2011–2012. JAMA. 311:806–814. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Reed M, Cygan H, Lui K and Mullen M:
Identification, prevention, and management of childhood overweight
and obesity in a pediatric primary care center. Clin Pediatr
(Phila). 55:860–866. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Deiuliis JA: MicroRNAs as regulators of
metabolic disease: Pathophysiologic significance and emerging role
as biomarkers and therapeutics. Int J Obes (Lond). 40:88–101. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sweeting AN, Hocking SL and Markovic TP:
Pharmacotherapy for the treatment of obesity. Mol Cell Endocrinol.
418:173–183. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Lee A and Morley JE: Metformin decreases
food consumption and induces weight loss in subjects with obesity
with type II non-insulin-dependent diabetes. Obes Res. 6:47–53.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kim HJ, Zhang XH, Park EY, Shin KH, Choi
SH, Chun BG and Kim DH: Metformin decreases meal size and number
and increases c-Fos expression in the nucleus tractus solitarius of
obese mice. Physiol Behav. 110–111:213–220. 2013. View Article : Google Scholar
|
|
7
|
Woo SL, Xu H, Li H, Zhao Y, Hu X, Zhao J,
Guo X, Guo T, Botchlett R, Qi T, et al: Metformin ameliorates
hepatic steatosis and inflammation without altering adipose
phenotype in diet-induced obesity. PLoS One. 9:e911112014.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Song YM, Lee YH, Kim JW, Ham DS, Kang ES,
Cha BS, Lee HC and Lee BW: Metformin alleviates hepatosteatosis by
restoring SIRT1-mediated autophagy induction via an AMP-activated
protein kinase-independent pathway. Autophagy. 11:46–59. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lavallard VJ and Gual P: Autophagy and
non-alcoholic fatty liver disease. Biomed Res Int. 2014:1201792014.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ravikumar B, Sarkar S, Davies JE, Futter
M, Garcia-Arencibia M, Green-Thompson ZW, Jimenez-Sanchez M,
Korolchuk VI, Lichtenberg M, Luo S, et al: Regulation of mammalian
autophagy in physiology and pathophysiology. Physiol Rev.
90:1383–1435. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lee JW, Park S, Takahashi Y and Wang HG:
The association of AMPK with ULK1 regulates autophagy. PLoS One.
5:e153942010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Singh R, Kaushik S, Wang Y, Xiang Y, Novak
I, Komatsu M, Tanaka K, Cuervo AM and Czaja MJ: Autophagy regulates
lipid metabolism. Nature. 458:1131–1135. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Kovsan J, Blüher M, Tarnovscki T, Klöting
N, Kirshtein B, Madar L, Shai I, Golan R, Harman-Boehm I, Schön MR,
et al: Altered autophagy in human adipose tissues in obesity. J
Clin Endocrinol Metab. 96:E268–E277. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Singh R, Xiang Y, Wang Y, Baikati K,
Cuervo AM, Luu YK, Tang Y, Pessin JE, Schwartz GJ and Czaja MJ:
Autophagy regulates adipose mass and differentiation in mice. J
Clin Invest. 119:3329–3339. 2009.PubMed/NCBI
|
|
15
|
National Research Council (US) Committee
for the update of the guide for the care and use of laboratory
animals: Guide for the care and use of laboratory animals. 8th.
Washington, DC: National Academies Press; 2011
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Marchesini G, Brizi M, Bianchi G,
Tomassetti S, Zoli M and Melchionda N: Metformin in non-alcoholic
steatohepatitis. Lancet. 358:893–894. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Bugianesi E, Gentilcore E, Manini R,
Natale S, Vanni E, Villanova N, David E, Rizzetto M and Marchesini
G: A randomized controlled trial of metformin versus vitamin E or
prescriptive diet in nonalcoholic fatty liver disease. Am J
Gastroenterol. 100:1082–1090. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Shi WY, Xiao D, Wang L, Dong LH, Yan ZX,
Shen ZX, Chen SJ, Chen Y and Zhao WL: Therapeutic metformin/AMPK
activation blocked lymphoma cell growth via inhibition of mTOR
pathway and induction of autophagy. Cell Death Dis. 3:e2752012.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Singh R and Cuervo AM: Lipophagy:
Connecting autophagy and lipid metabolism. Int J Cell Biol.
2012:2820412012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Song S, Andrikopoulos S, Filippis C,
Thorburn AW, Khan D and Proietto J: Mechanism of fat-induced
hepatic gluconeogenesis: Effect of metformin. Am J Physiol
Endocrinol Metab. 281:E275–E282. 2001.PubMed/NCBI
|
|
22
|
Shin NR, Lee JC, Lee HY, Kim MS, Whon TW,
Lee MS and Bae JW: An increase in the Akkermansia spp. population
induced by metformin treatment improves glucose homeostasis in
diet-induced obese mice. Gut. 63:727–735. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Anthony J, Kelkar A, Wilankar C, Ranjith
V, Bhumra SK, Mutt SJ, Deka N, Sivaramakrishnan H, Sharma S and
Marita AR: Discovery of p1736, a novel antidiabetic compound that
improves peripheral insulin sensitivity in mice models. PLoS One.
8:e779462013. View Article : Google Scholar : PubMed/NCBI
|