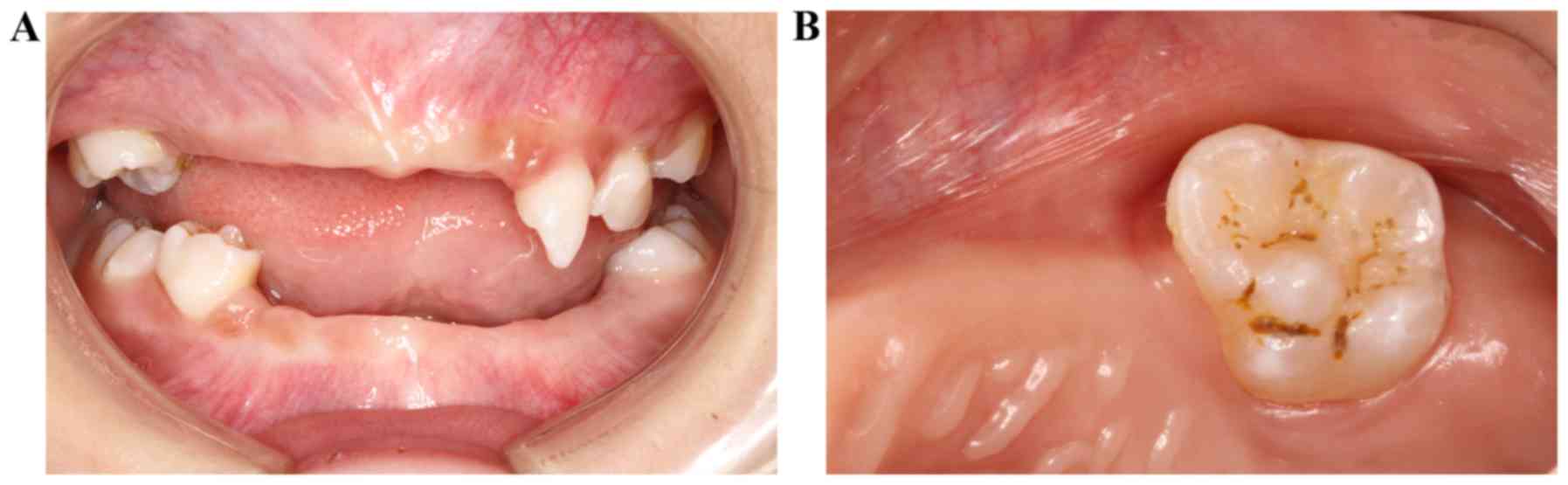
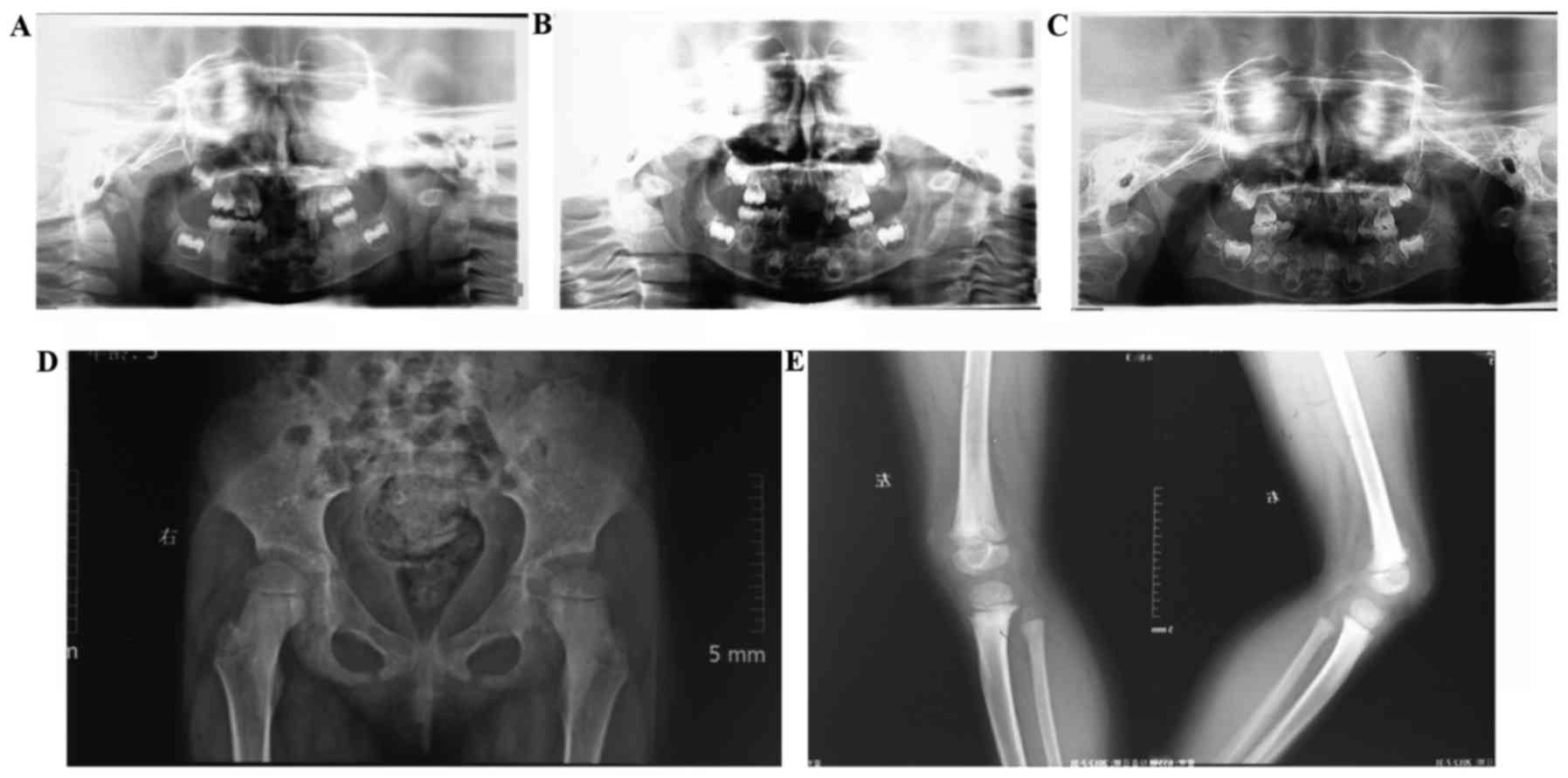
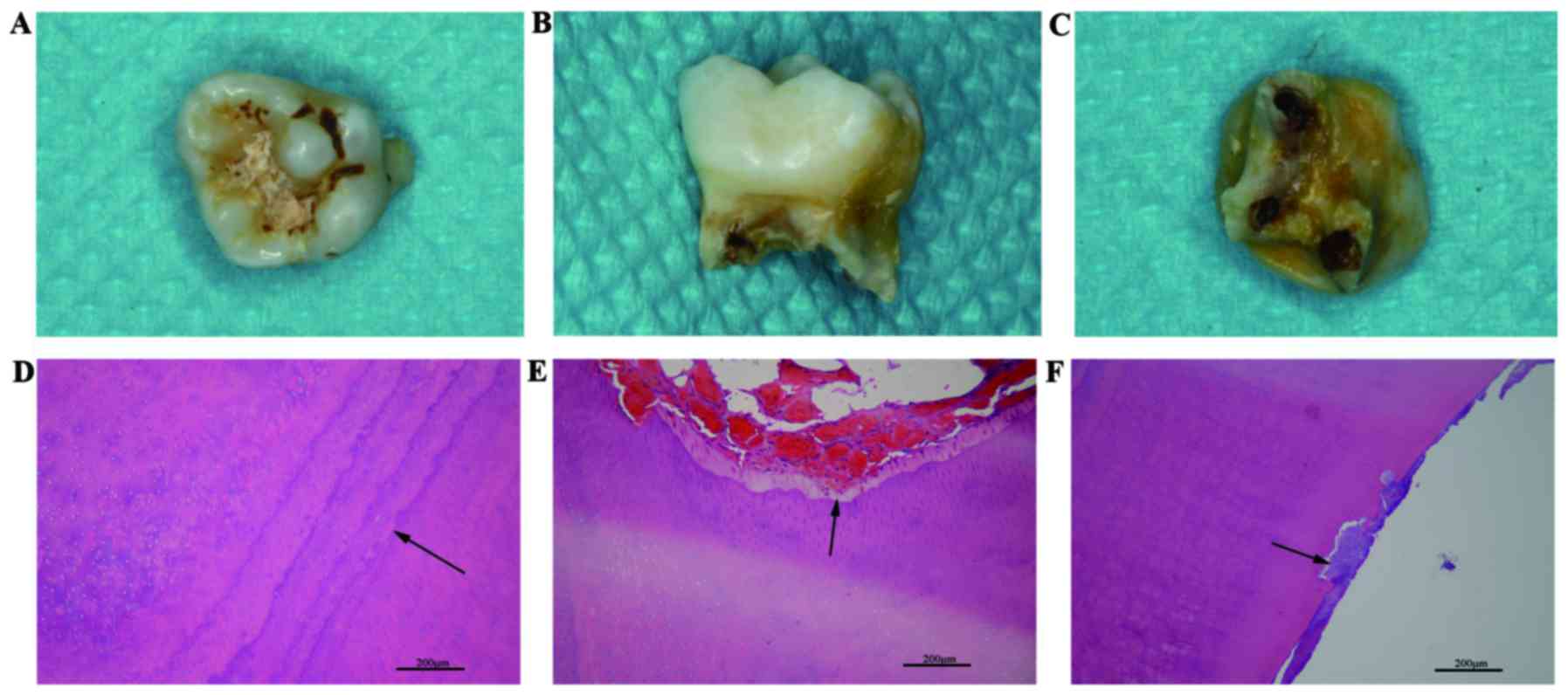
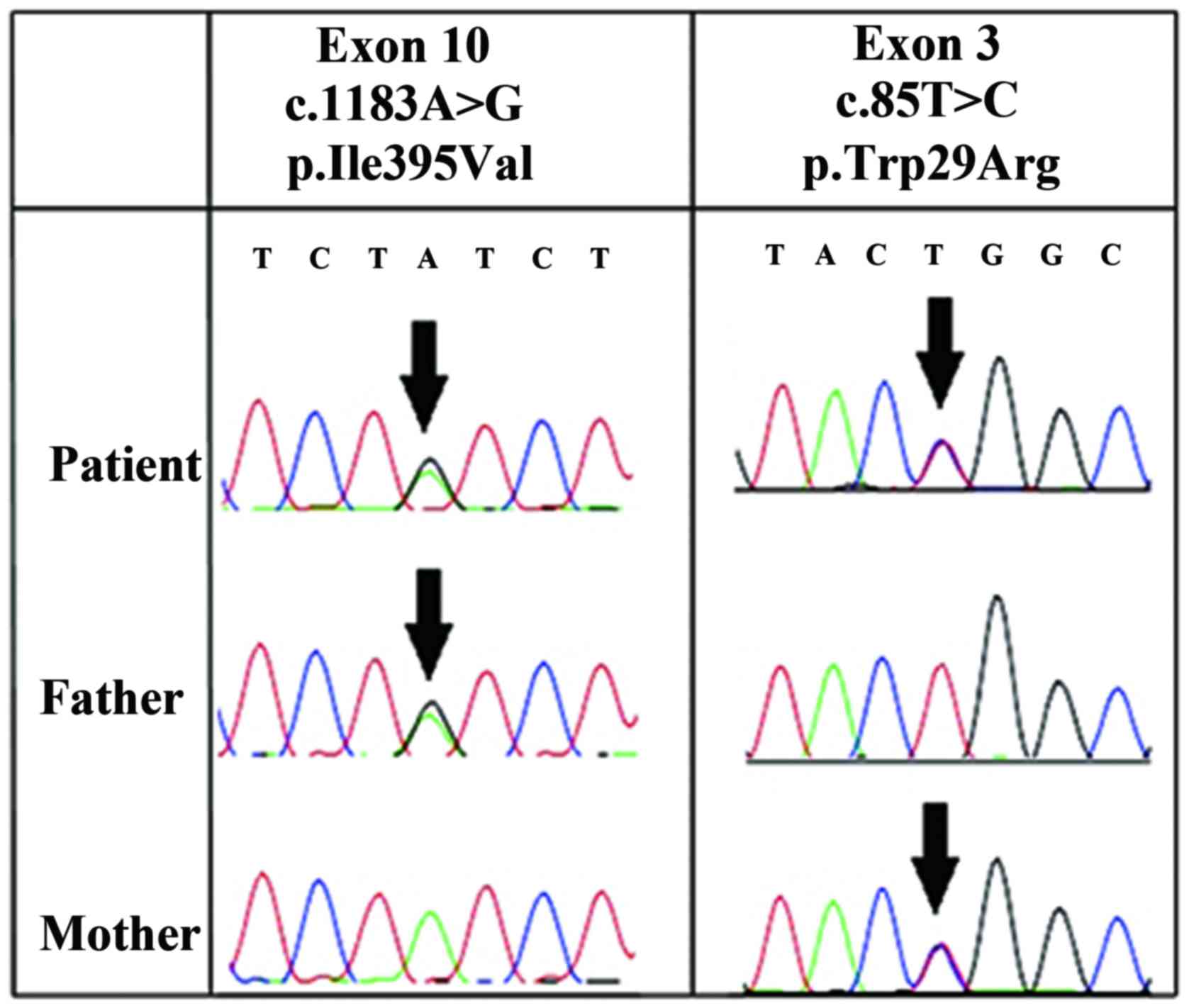
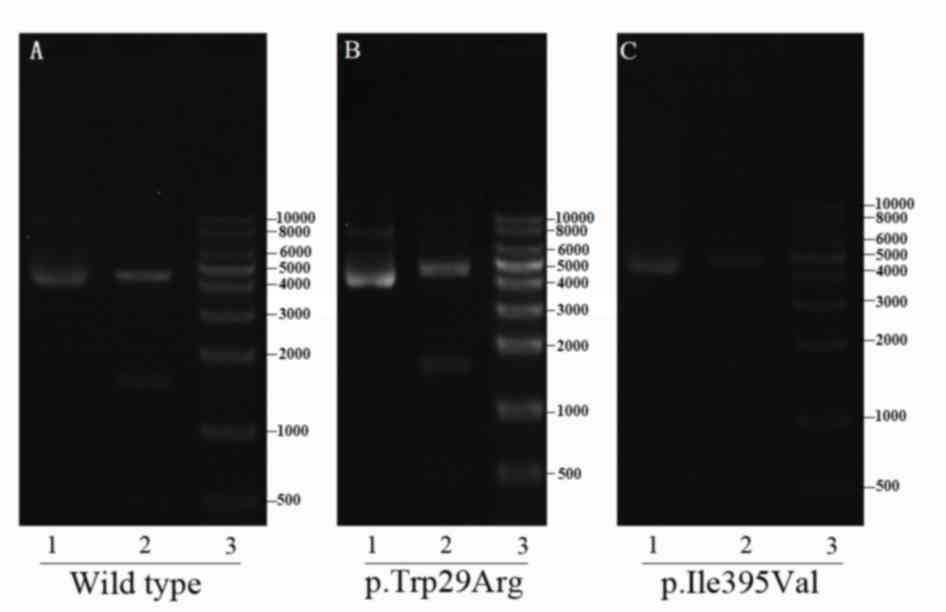
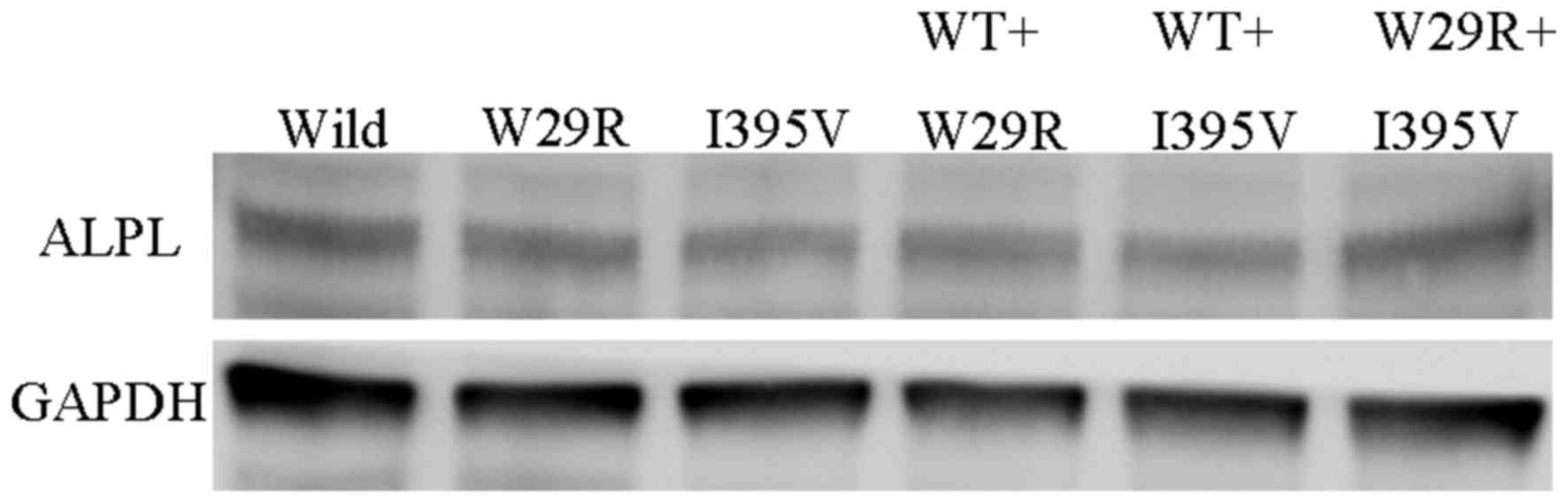
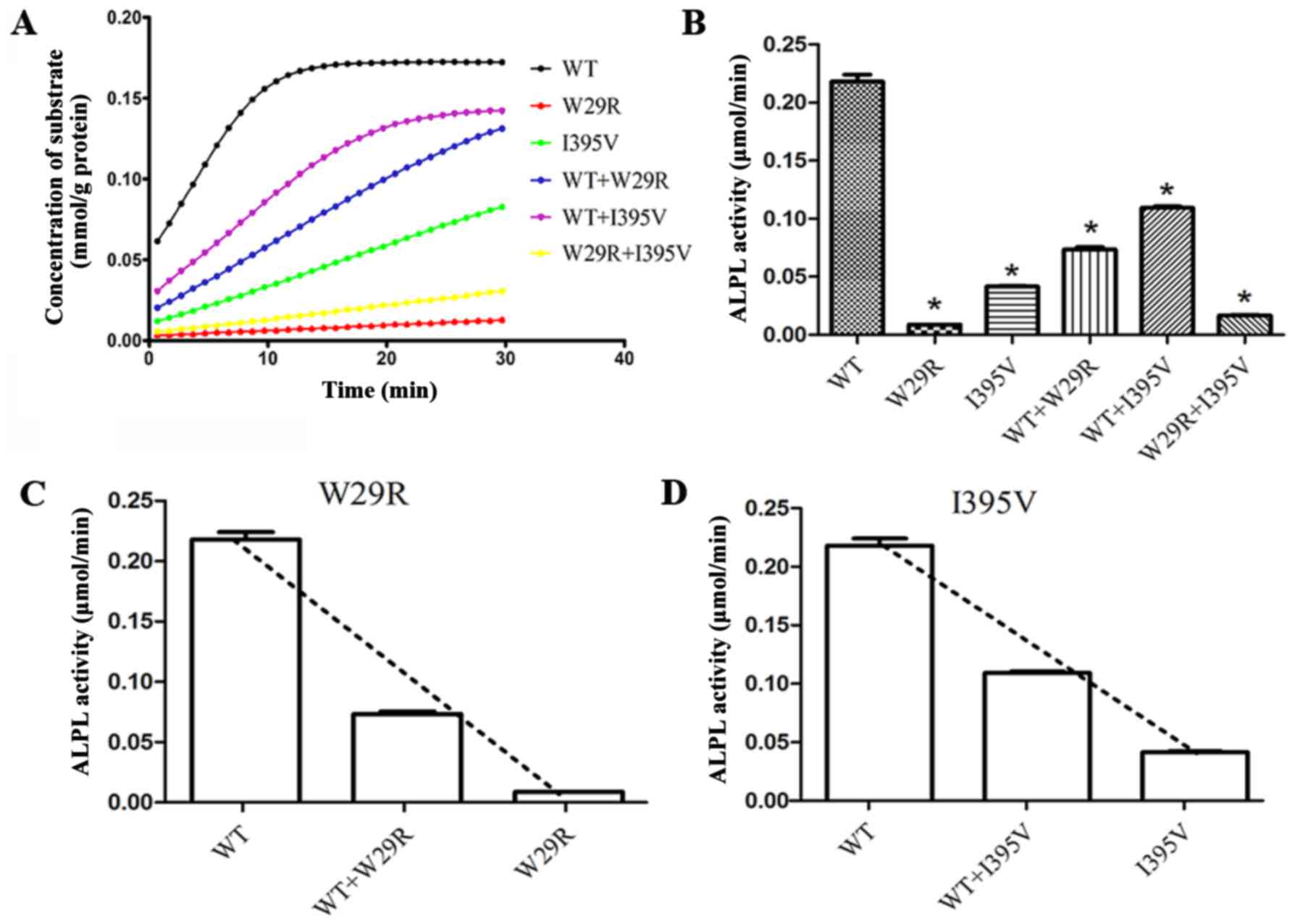
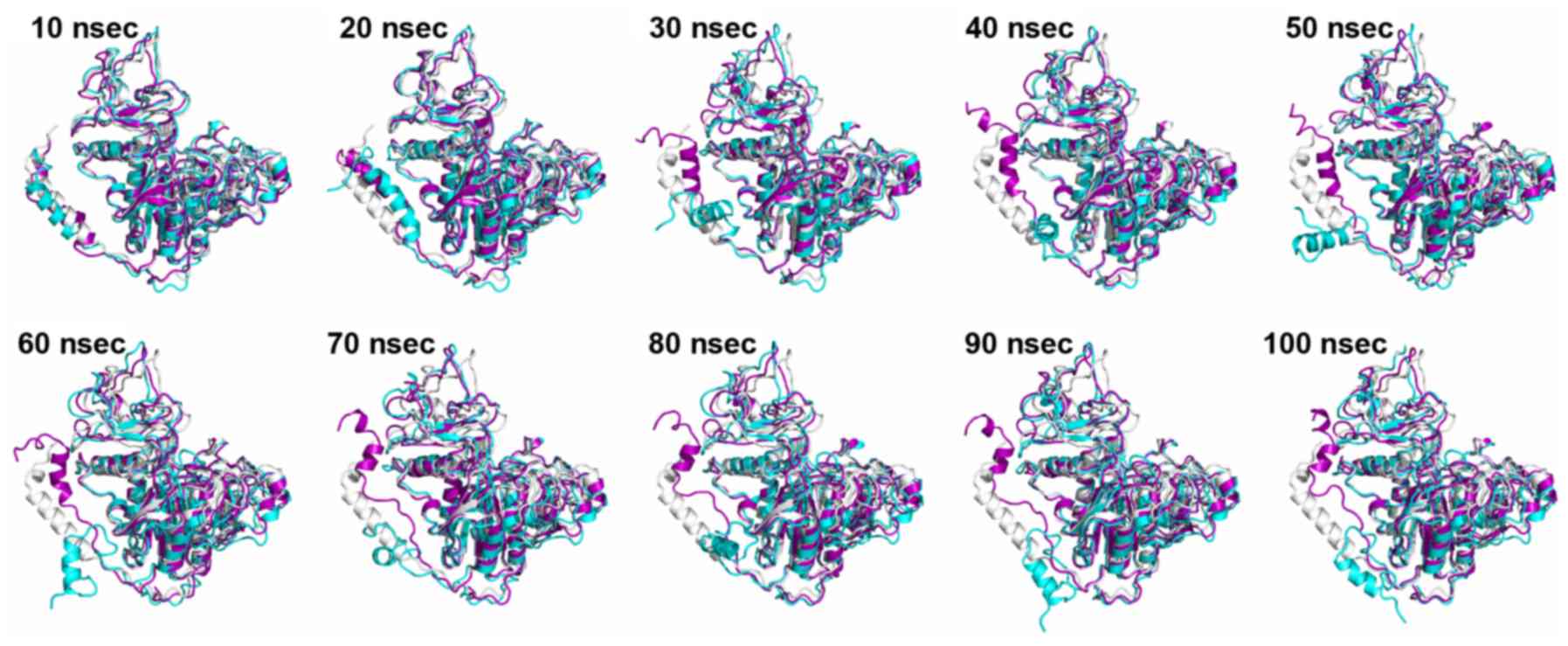
|
1
|
Fraser D: Hypophosphatasia. Am J Med.
22:730–746. 1957. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mornet E, Hofmann C, Bloch-Zupan A,
Girschick H and Le Merrer M: Clinical utility gene card for:
Hypophosphatasia - update 2013. Eur J Hum Genet. 22:2014.doi:
10.1038/ejhg.2013.177. View Article : Google Scholar :
|
|
3
|
Reibel A, Manière MC, Clauss F, Droz D,
Alembik Y, Mornet E and Bloch-Zupan A: Orodental phenotype and
genotype findings in all subtypes of hypophosphatasia. Orphanet J
Rare Dis. 4:62009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Van den Bos T, Handoko G, Niehof A, Ryan
LM, Coburn SP, Whyte MP and Beertsen W: Cementum and dentin in
hypophosphatasia. J Dent Res. 84:1021–1025. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Whyte MP: Hypophosphatasia and the role of
alkaline phosphatase in skeletal mineralization. Endocr Rev.
15:439–461. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zurutuza L, Muller F, Gibrat JF,
Taillandier A, Simon Bouy-B, Serre JL and Mornet E: Correlations of
genotype and phenotype in hypophosphatasia. Hum Mol Genet.
8:1039–1046. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jemmerson R and Low MG:
Phosphatidylinositol anchor of HeLa cell alkaline phosphatase.
Biochemistry. 26:5703–5709. 1987. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kim EE and Wyckoff HW: Structure of
alkaline phosphatases. Clin Chim Acta. 186:175–187. 1990.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Mornet E: Hypophosphatasia. Best Pract Res
Clin Rheumatol. 22:113–127. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhang W, Shen L, Deng Z, Ding Y, Mo X, Xu
Z, Gao Q and Yi L: Novel missense variants of ZFPM2/FOG2 identified
in conotruncal heart defect patients do not impair interaction with
GATA4. PLoS One. 9:e1023792014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ho SN, Hunt HD, Horton RM, Pullen JK and
Pease LR: Site-directed mutagenesis by overlap extension using the
polymerase chain reaction. Gene. 77:51–59. 1989. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Li W, Yang JG, Ren FX, Kang CL and Zhang
SY: PCR site-directed mutagenesis of long QT syndrome KCNQ1 gene in
vitro. Yi Chuan. 26:589–593. 2004.(In Chinese). PubMed/NCBI
|
|
13
|
Witzigmann D, Wu D, Schenk SH,
Balasubramanian V, Meier W and Huwyler J: Biocompatible
polymer-Peptide hybrid-based DNA nanoparticles for gene delivery.
ACS Appl Mater Interfaces. 7:10446–10456. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yang H, Wang L, Geng J, Yu T, Yao RE, Shen
Y, Yin L, Ying D, Huang R, Zhou Y, et al: Characterization of six
missense mutations in the tissue-nonspecific alkaline phosphatase
(TNSALP) gene in Chinese children with hypophosphatasia. Cell
Physiol Biochem. 32:635–644. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Le Du MH, Stigbrand T, Taussig MJ, Menez A
and Stura EA: Crystal structure of alkaline phosphatase from human
placenta at 1.8 A resolution. Implication for a substrate
specificity. J Biol Chem. 276:9158–9165. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sánchez R and Sali A: Comparative protein
structure modeling. Introduction and practical examples with
modeller. Methods Mol Biol. 143:97–129. 2000.PubMed/NCBI
|
|
17
|
Hess B, Kutzner C, van der Spoel D and
Lindahl E: GROMACS 4: Algorithms for highly efficient,
load-balanced, and scalable molecular simulation. J Chem Theory
Comput. 4:435–447. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lindorff-Larsen K, Piana S, Palmo K,
Maragakis P, Klepeis JL, Dror RO and Shaw DE: Improved side-chain
torsion potentials for the Amber ff99SB protein force field.
Proteins. 78:1950–1958. 2010.PubMed/NCBI
|
|
19
|
Jorgensen WL, Chandrasekhar J and Madura
JD: Comparison of simple potential functions for simulating liquid
water. J Chem Phys. 79:9261983. View
Article : Google Scholar
|
|
20
|
Hess B, Bekker H, Berendsen HJC and
Fraaije JG: LINCS: A linear constraint solver for molecular
simulations. J Computation Chem. 18:1463–1472. 1998. View Article : Google Scholar
|
|
21
|
Bussi G, Donadio D and Parrinello M:
Canonical sampling through velocity rescaling. J Chem Phys.
126:0141012007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Parrinello M and Rahman A: Crystal
Structure and Pair Potentials: A Molecular-Dynamics Study. Phys Rev
Lett. 45:1196–1199. 1980. View Article : Google Scholar
|
|
23
|
Wenkert D, McAlister WH, Coburn SP, Zerega
JA, Ryan LM, Ericson KL, Hersh JH, Mumm S and Whyte MP:
Hypophosphatasia: Nonlethal disease despite skeletal presentation
in utero (17 new cases and literature review). J Bone Miner Res.
26:2389–2398. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
24
|
Silvent J, Gasse B, Mornet E and Sire JY:
Molecular evolution of the tissue-nonspecific alkaline phosphatase
allows prediction and validation of missense mutations responsible
for hypophosphatasia. J Biol Chem. 289:24168–24179. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hoylaerts MF, Ding L, Narisawa S, Van
Kerckhoven S and Millan JL: Mammalian alkaline phosphatase
catalysis requires active site structure stabilization via the
N-terminal amino acid microenvironment. Biochemistry. 45:9756–9766.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Silva I, Castelao W, Mateus M and Branco
JC: Childhood hypophosphatasia with myopathy: Clinical report with
recent update. Acta Reumatol Port. 37:92–96. 2012.PubMed/NCBI
|