Introduction
The role of inflammation in tumorigenesis and
development is now well established in the biomedical literature.
There is mounting evidence that an inflammatory microenvironment is
an essential component of most tumor types, including some in which
the causal relationship with inflammation remains to be elucidated
(1). In some cancer types,
inflammatory conditions precede development of malignancy; in
others, oncogenic change induces a tumor-promoting inflammatory
milieu (1,2). Globally, there are large variations
in bladder cancer mortality (3,4),
suggesting an important role of environmental factors in the
etiology of bladder cancer. The common known risk factors of
bladder cancer include a number of occupations with exposures to
aromatic amines (for example, industrial dye manufacturing), the
drug cyclophosphamide, cigarette smoking, and chronic infection
(for example, urinary tract infections and schistosomiasis)
(5). The unifying principle that
underlies these risk factors is chronic inflammation, which is an
aberrantly prolonged form of a protective response to the loss of
tissue homeostasis (6). A previous
study from our group reported great numbers of immune cells
infiltrated in bladder cancer tissue from patients (7), suggesting a strong relationship
between bladder cancer and chronic inflammation. Immune cells that
infiltrate tumors engage in a dynamic crosstalk with cancer cells,
resulting in the inflammatory tumor microenvironment. Understanding
the underlying mechanisms of how immune cells function in different
tumor types, and which molecular inflammatory mediators might drive
or potentially prevent carcinogenesis, is a subject of intense
research.
Lymphotoxin β receptor (LTβR) was initially
discovered in the context of lymph node development, and it has
since been demonstrated that LTβR signaling participates in the
initiation and/or development of inflammation-induced
carcinogenesis (8). LTβR is
expressed in a wide range of tumor types, including breast,
colorectal, lung, stomach, melanoma and bladder cancer (9,10),
while its ligands, lymphotoxin (LT) a1b2 and TNF superfamily member
14 (TNFSF14; also known as LIGHT), are mainly expressed on the
surface of immune cells (11).
Thus, LTβR signaling might enable the communication between
infiltrating immune cells and tumor cells (8). Triggering LTβR induces the canonical
and noncanonical nuclear factor (NF)-κB signaling pathways, which
are linked to inflammation-induced carcinogenesis (12). Sustained LTβR signaling leads to
NF-κB-mediated chronic inflammation and hepatocellular carcinoma
(HCC) development (13). Long-term
suppression of LTβR with a LTβR agonistic antibody significantly
reduces chronic hepatitis incidence and prevents the transition
from chronic hepatitis to HCC in mouse models (13). By contrast, LTβR functions as a
death receptor that mediates tumor cell apoptosis in colon
carcinoma, mammary carcinoma and sarcoma (14); Yang et al (15) and Winter et al (16) reported that LTβR induces cytotoxic
T lymphocyte-mediated antitumor cytotoxicity. Because of these
contrasting observations, the function of LTβR signaling might be
tumor type and cellular context-dependent. In addition, the
function of LTβR remains unclear in bladder cancer. Therefore, the
present study aimed to investigate the effect of LTβR activation on
the mRNA expression levels of the NF-κB main members, RELA
proto-oncogene (RelA; also known as p65) and RELB proto-oncogene
(RelB) and to analyze the function of LTβR in the proliferation and
the pro-inflammatory response in bladder cancer cells.
Materials and methods
Cell culture
Human bladder cancer 5,637 cells were purchased from
the Cell Bank of Type Culture Collection of the Chinese Academy of
Sciences (Shanghai, China) and maintained in RMPI-1640 (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% fetal bovine serum (FBS; Biowest, Nuaillé, France) at 37°C
humidified atmosphere with 5% CO2. For the present
study, 5,637 cells were seeded onto 96-well plates, 24-well plates
and 6-well plates at densities of 4–5×104 cells/ml,
2–5×105 cells/ml and 1–2×106 cells/ml
respectively. Cells were then divided into two experimental groups,
LTβR-activated and LTβR-silenced. Activation of LTβR was induced by
addition of the functional ligand, lymphotoxin (LT) α1β2 (R&D
Systems, Inc., Minneapolis, MN, USA), at a final concentration of
100 ng/ml. Silencing of LTβR was performed by specific LTβR small
hairpin RNA (shRNA). Prior to each experiment, cells were grown in
RPMI-1640 without FBS for 6 h.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from 5,637 cells using
TRIzol reagent (Thermo Fisher Scientific, Inc.), following the
manufacturer's instructions. Total RNA of each sample (2 µg) was
subjected to oligo-dT-primed RT using the RevertAid First Strand
cDNA Synthesis kit (Thermo Fisher Scientific, Inc.), according to
the manufacturer's instructions. The resulting cDNA was diluted
1:20 used for qPCR, using a SYBR-Green Real-Time PCR Master mix on
a 7500 Real-Time PCR System (both from Applied Biosystems; Thermo
Fisher Scientific, Inc.). Primer sequences were as follows: human
LTβR, sense 5′-GCACAAGCAAACGGAAGACC-3′ and antisense
5′-GACCTTGGTTCTCACACCTGGT-3′; LTα, sense 5′-CTGCTCACCTCATTGGAGAC-3′
and antisense 5′-CCTGGGAGTAGACGAAGTAGAT-3′; LTβ, sense
5′-CAGCTGCCCACCTCATAGG-3′ and antisense 5-GCGTCCGAGAACTGCGTC-3′;
LIGHT, sense 5′-ATCTCACAGGGGCCAACT-3′ and antisense
5′-ACGGACGACCACCTTCTC-3′; tumor necrosis factor (TNF)α, sense
5′-GCCCATGTTGTAGCAAACC-3′ and antisense 5′-GGTAGGAGACGGCGATG-3′;
IL-6, sense 5′-AATTCGGTACATCCTCGACGGC-3′ and antisense
5′-GCCAGTGCCTCTTTGCTGCTTT-3′; interleukin (IL)-1β, sense
5′-GAAATGATGGCTTATTACAGTGGCA-3′ and antisense
5′-GTAGTGGTGGTCGGAGATTCGTAG-3′; CyclinD1, sense
5′-TGCATCTACACCGACAACTCC-3′ and antisense
5′-GGGCGGATTGGAAATGAACT-3′; and Survivin, sense
5′-GACCACCGCATCTCTACATTCA-3′ and antisense
5′-AATTCACCAAGGGTTAATTCTTCAA-3′. qPCR was performed under the
following conditions: initial denaturation at 95°C for 5 min, then
45 cycles of 95°C for 30 sec and 60°C for 30 sec. Melting curves
were recorded to verify the singularity of the PCR product. In each
sample, the level of cDNA was normalized to the level of GAPDH
(sense 5′-GTCAACGGATTTGGTCGTATTG-3′ and antisense
5′-CTGGAAGATGGTGATGGGATT-3′). Relative fold changes in mRNA
expression were calculated using the formula 2−ΔΔCq
(17).
LTβR gene silencing by shRNA. The shRNA
sequences were designed to exhibit sequence homology to the two
LTβR transcripts (NM_0,01270987.1, NM_002342.2) at http://rnaidesigner.invitrogen.com/rnaiexpress/rnaiDesign.jsp
(BLOCK-iT™ RNAi Designer; Thermo Fisher Scientific, Inc.). Bladder
cancer 5,637 cells were transfected with LTβR-specific shRNA
(5′-GCACCTATGTCTCAGCTAAAT-3′, loop sequence is CGAA) plasmid as the
tested group (shRNA-T), in parallel with scramble shRNA
(5′-CTACACAAATCAGCGATTT-3′, loop sequence is CGAA) plasmid as the
control group (shRNA-C). The day prior to transfection, 5,637 cells
were seeded into 24-well (7–9×105 cells/ml) or 6-well
(1–2×106 cells/ml) culture plates in complete media. The
next day, shRNA (at final concentration 0.8 and 4 µM for 24-well
and 6-well culture plates, respectively) were introduced into cells
using Lipofectamine 2000 (Thermo Fisher Scientific, Inc.) following
the manufacturer's instructions. At 4 h post-transfection, media
were replaced with regular complete culture media. The cells were
cultured for 72 h prior to analysis of the gene-silencing effects.
The expression of LTβR in the shRNA-transfected cells was
characterized by RT-qPCR and western blotting.
Western blotting
For determining the silencing effect of the shRNA
plasmid, 5,637 cells were cultured in 6-well plates
(1–2×106 cells/ml) and transfected with the shRNA for 72
h. Total cells were lysed on ice with radioimmunoprecipitation
assay lysis buffer (cat. no. P0013B; Beyotime Institute of
Biotechnology, Nantong, China) supplemented with a protease
inhibitor cocktail (cat. no. I3786; Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) for 10–15 min. The concentration of protein in
the lysates was measured by an Enhanced Bicinchoninic Assay kit
(cat. no. P0010S; Beyotime Institute of Biotechnology). A total of
30 µg lysate was loaded onto each lane of 5% stacking gel and 8%
separating gel and separated by SDS-polyacrylamide gel
electrophoresis. Following SDS-PAGE, proteins were transferred onto
polyvinylidene fluoride membranes (Solarbio Science and Technology
Co., Ltd., Beijing, China). Membranes were subsequently blocked
with 5% bovine serum album (BSA) (Generay Biotech Co., Ltd.,
Shanghai, China) diluted in TBST (10 mM Tris-HCL, 100 mM NaCl, 0.2%
Tween-20) at 37°C for 2 h. After blocking, membranes were incubated
with primary antibodies against LTβR (cat. no. 20331-1-AP;
ProteinTech Group, Inc., Chicago, IL, USA) diluted 1:500 in TBST
containing 5% BSA and β-actin (cat. no. GTX124213; GeneTex, Inc.,
Irvine, CA, USA) diluted 1:5,000 in TBST containing 5% BSA
overnight at 4°C. Membranes were subsequently probed with a horse
radish peroxidase-conjugated goat anti-rabbit immunoglobulin G
secondary antibody (cat. no. A0208; Beyotime Institute of
Biotechnology) diluted 1:1,000 in TBST containing 5% BSA at room
temperature for 2 h. Immunoreactivity was visualized using an
Enhanced Chemiluminescence reagent (cat. no. K-12045-C20; Advansta,
Inc., CA, USA). Densitometric analysis was performed using Quantity
One software version 4.6.2 (Bio-Rad Laboratories, Inc., CA,
USA).
Cell viability assay
Cell viability was measured using the Cell Counting
Kit-8 (CCK-8) (Dojindo Molecular Technologies, Inc., Kumamoto,
Japan), according to the manufacturer's protocol. Briefly, 5,637
cells were seeded in 96-well plates at a density of
4–5×104 cells/ml and stimulated with LTα1β2 for 24 and
48 h. Control cells were treated with equal volumes of sterile PBS.
At the indicated time point, the culture media was aspirated and
fresh media supplemented with 10% (v/v) CCK-8 was added to the
cells. Cells were cultured at 37°C for an additional 3 h and the
absorbance was then measured at a wavelength of 450 nm. Cell
viability was expressed as % of the control unstimulated culture
measurement.
Statistical analysis
All statistical analyses were performed using SPSS
version 15.0 software (SPSS, Inc., Chicago, IL, USA). Independent t
tests between two groups were performed to determine statistical
significance. P<0.05 was considered to indicate a statistically
significant difference. Experiments were repeated three times.
Results
Activation of LTβR enhances the mRNA
expression of RelA, but not of RelB
Activation of NF-κB signaling occurs by two
pathways, the canonical pathway and the noncanonical signaling
pathway. In order to examine the effect of LTβR activation on the
expression of the main members of the canonical and noncanonical
NF-κB signaling pathways, the RelA and RelB genes were selected as
target genes, respectively. As demonstrated in Fig. 1A, RelA mRNA expression levels were
upregulated by 2.5-fold following activation of LTβR in bladder
cancer 5,637 cells, compared with control unstimulated cells
(P=0.003). No significant difference was observed in the expression
of RelB mRNA following LTβR activation compared with control
unstimulated cells (P=0.254; Fig.
1A).
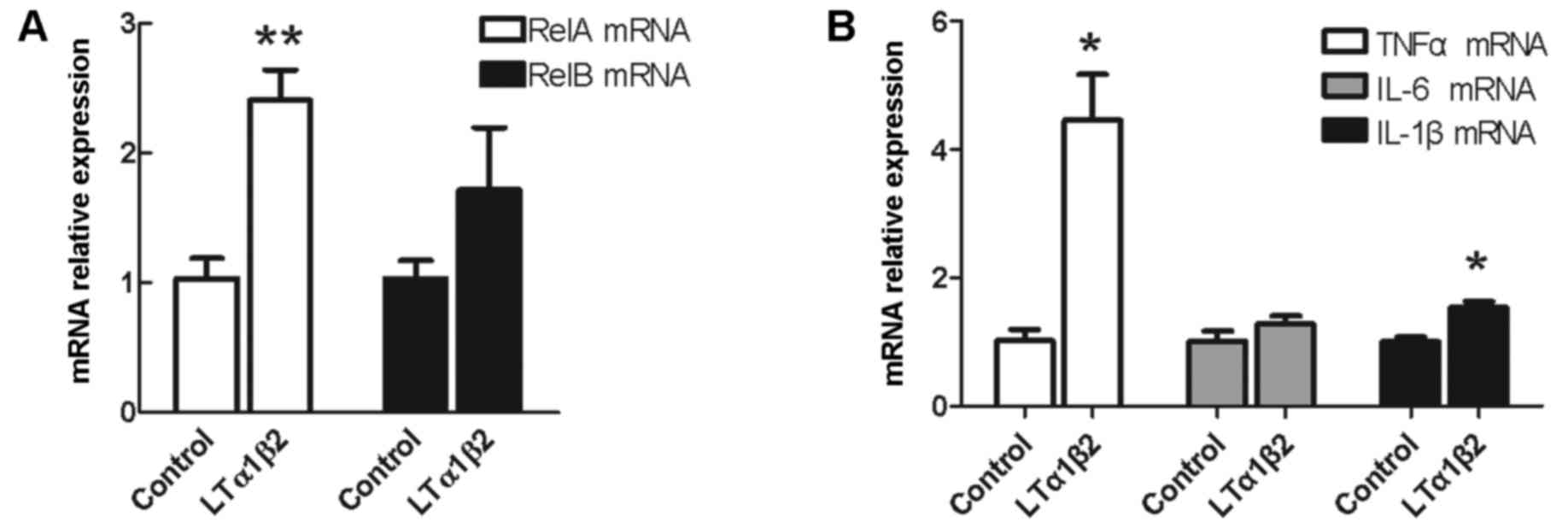 | Figure 1.Effect of LTβR activation on mRNA
expression of RelA/p65, RelA, and pro-inflammatory cytokines TNFα,
IL-6 and IL-1β. Bladder cancer 5,637 cells were stimulated with 100
ng/ml LTα1β2 and mRNA expression levels were examined for (A) RelA
and RelB, (B) TNFα, IL-6 and IL-1β mRNA. *P<0.05 and
**P<0.01, compared with control unstimulated cells. Error bars
represented the standard deviation. LTβR, lymphotoxin β receptor;
RelA, RELA proto-oncogene NF-κB subunit; RelB, RELB proto-oncogene
NF-κB subunit; TNFα, tumor necrosis factor αl; IL, interleukin; LT,
lymphotoxin. |
Activation of LTβR promotes the mRNA
expression of pro-inflammatory cytokines TNFα and IL-1β
It is known that cytokines are major mediators of
communication between cancer cells and immune cells in the
inflammatory tumor microenvironment (2). The hypothesis that major
pro-inflammatory cytokines, TNFα, IL-6 and IL-1β, which are also
target genes of NF-κB signaling, might be regulated by LTβR was
examined. As demonstrated in Fig.
1B, mRNA expression levels of TNFα were increased by ~5-fold
(P=0.034) and IL-1β by 1.5-fold (P=0.013) following LTβR activation
compared with control unstimulated cells. No effect on the
expression of IL-6 mRNA was observed (P=0.334; Fig. 1B).
LTβR-induced upregulation of RelA,
TNFα and IL-1β is reversed by LTβR silencing
To determine whether activation of LTβR has a
causative role in the upregulation of NF-κB signaling members and
pro-inflammatory cytokines, LTβR was silenced in 5,637 cells by
shRNA. LTβR-specific shRNA (shRNA-T group) and non-silencing shRNA
plasmids (shRNA-C group) were transfected into the 5,637 cells. The
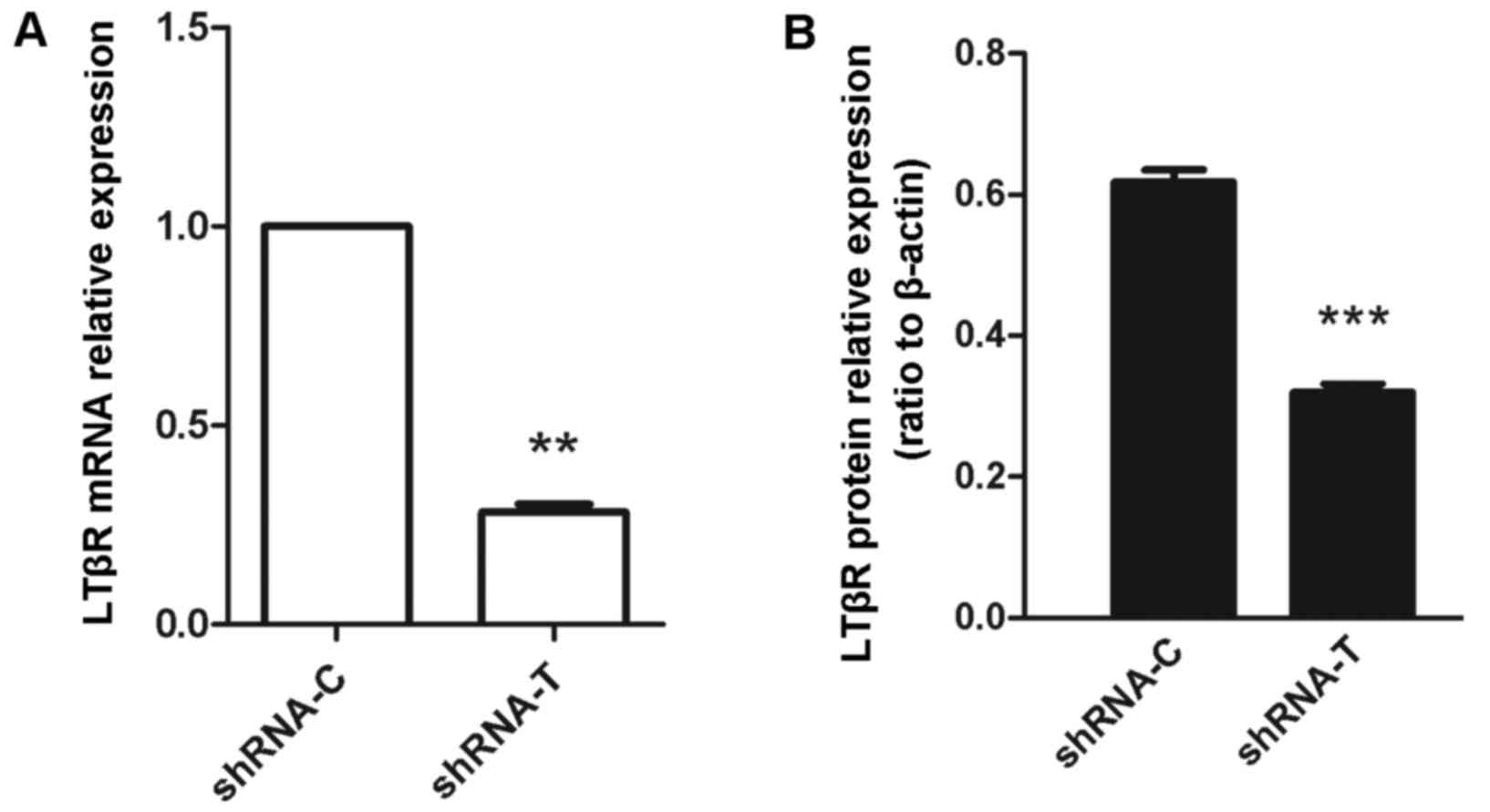
efficiency of LTβR silencing was confirmed at the mRNA and protein
level by RT-qPCR and western blot, respectively (Fig. 2). Compared with the control group,
expression of LTβR in cells transfected with LTβR-specific shRNA
was reduced by 65–75% at the mRNA level (P=0.001; Fig. 2A) and ~50% at the protein level
(P<0.001; Fig. 2B). Compared
with the shRNA-C group, RelA mRNA expression was downregulated by
~33% (P=0.012), TNFα by 27% (P=0.011) and IL-1β by 26% (P=0.011) in
the shRNA-T group following activation of LTβR (Fig. 3).
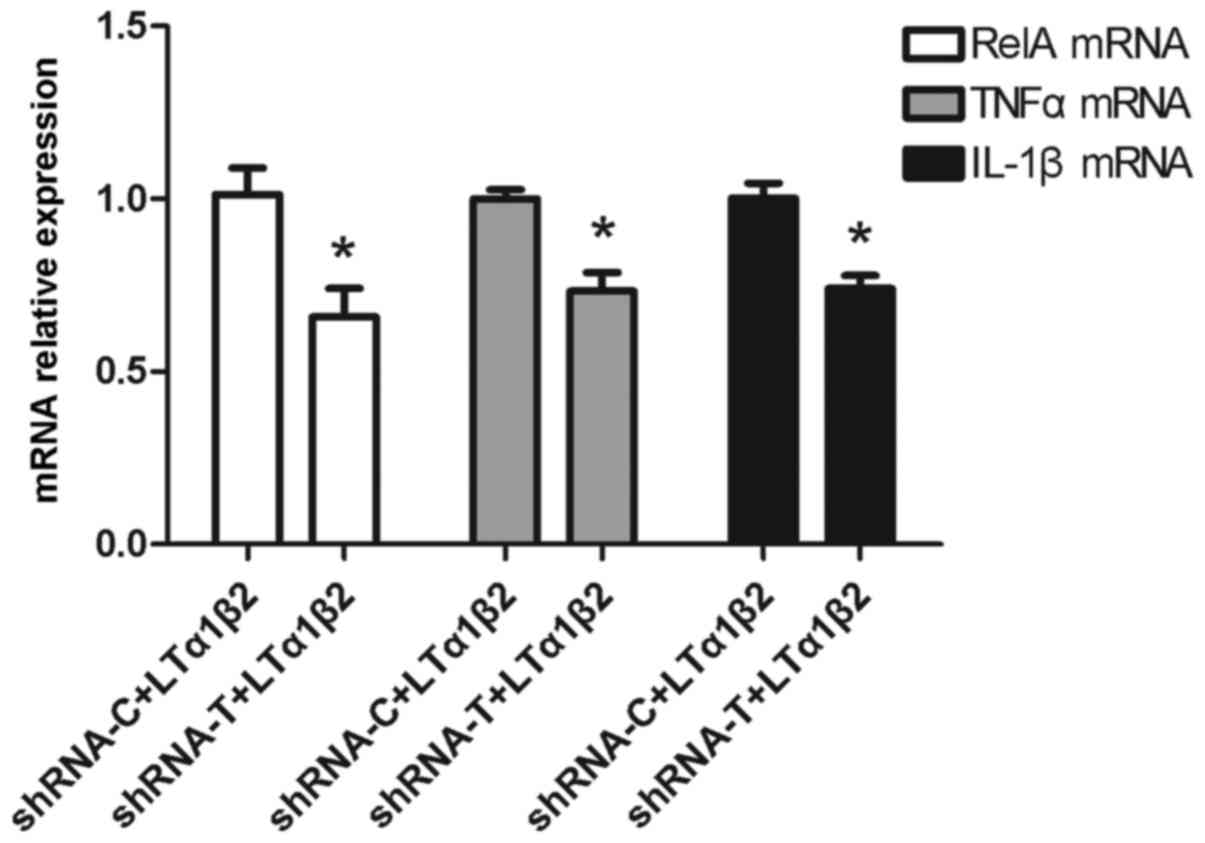 | Figure 3.Effect of LTβR silencing on the
LTα1β2-mediated overexpression of RelA, TNFα and IL-1β. Following
LTβR silencing by shRNA transfection for 72 h, the bladder cancer
5,637 cells were treated with 100 ng/ml LTα1β2 for activation of
LTβR. *P<0.05, compared with shRNA-C for each gene. Error bars
represented the standard deviation. LTβR, lymphotoxin β receptor;
LT, lymphotoxin; RelA, RELA proto-oncogene NF-κB subunit; TNFα,
tumor necrosis factor αl; IL, interleukin; shRNA-C, control
scramble short hairpin RNA; shRNA-T, LTβR-specific shRNA. |
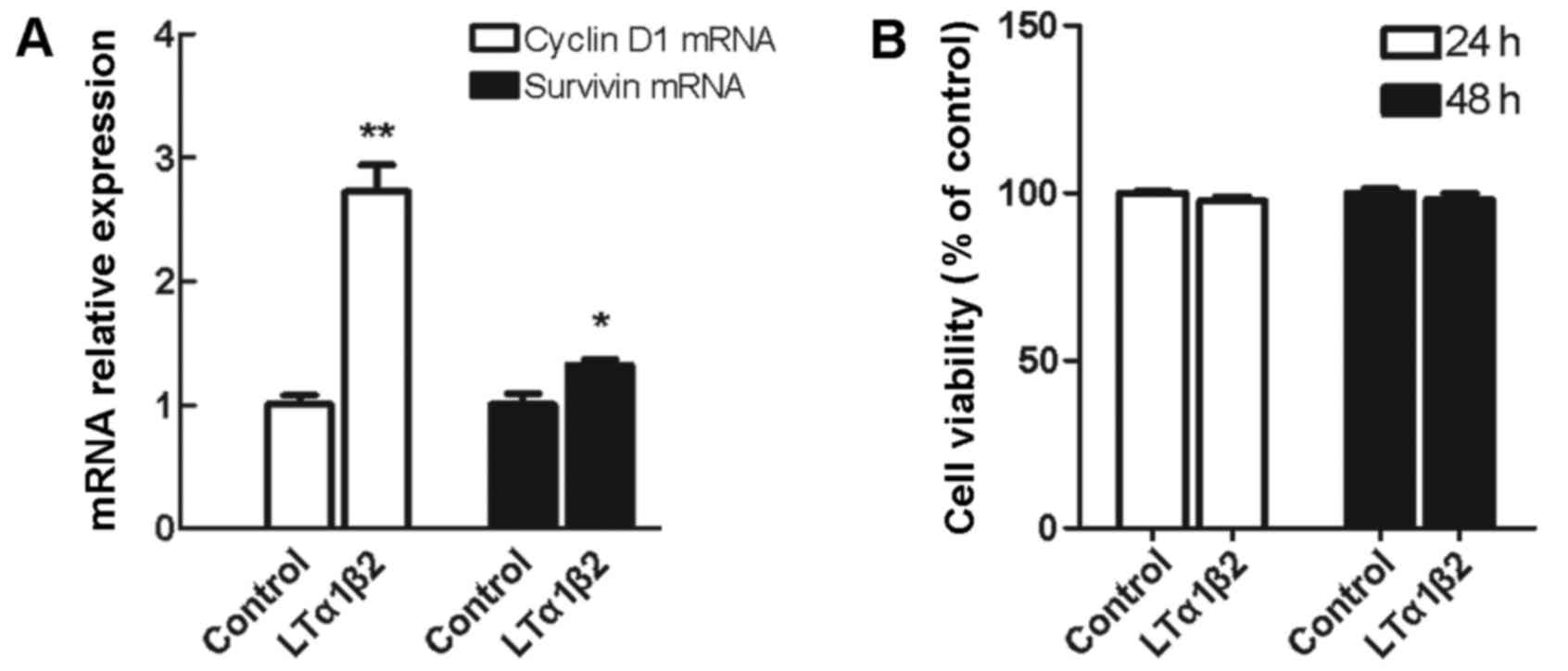
Activation of LTβR has no effect on
5,637 cell growth despite increased mRNA expression of
proliferation-related genes CyclinD1 and Survivin
There are contrasting observations regarding the
role of LTβR signaling pathway in tumor cell apoptosis and growth
promotion. In the present study, LTβR activation in 5,637 cells
resulted in a 2.7-fold upregulation of CyclinD1 mRNA expression
levels (P=0.002) and a 1.3-fold upregulation of Survivin mRNA
expression levels (P=0.035), compared with control unstimulated
cells (Fig. 4A). However, when
cell viability was measured by CCK-8 assay, no significant change
was observed in the % of cell viability in cells stimulated with
LTα1β2 for 24 or 48 h, compared with control unstimulated cells
(Fig. 4B).
Discussion
It is well documented that NF-κB signaling
represents a critical link between inflammation and cancer. NF-κB
signaling can be activated by a great variety of stimuli, including
inflammatory mediators and stress response. The diversity of
stimuli endows NF-κB signaling with a great complexity and
multiplicity as to the biology processes it regulates. Activation
of NF-κB signaling (usually assessed by the presence of nuclear
RelA) has been observed in many types of cancer, including colon
cancer, hepatocellular carcinoma, prostate cancer, pancreatic
cancer, various types of leukemia and melanoma (18). In addition, it has been reported
that LTβR is critical in NF-κB-dependent promotion of HCC (13) and prostate cancer (19). Higher expression levels of RelA and
RelB have been observed in bladder cancer and bladder inflammation
tissues, compared with normal tissues by RT-PCR, suggesting a
potential link between NF-κB signaling and the development of
bladder cancer (8). Thus, it was
hypothesized that LTβR activation may enhance the expression levels
of these main members of NF-κB signaling in bladder cancer cells.
LTβR activation participates in both the canonical and noncanonical
NF-κB signaling pathways (20).
Notably, bladder cancer 5,637 cells exhibited a significant
increase in RelA transcripts following LTα1β2 treatment compared
with unstimulated cells, but only a non-significant trend toward
increased RelB mRNA levels was observed. These findings suggested
that the NF-κB canonical signaling member RelA may be the main
target gene of LTβR activation.
It is estimated that underlying infections and
inflammatory reactions are linked to 25% of all cancer cases
(21). Infections with hepatitis B
(HBV) or C (HCV) virus increase the risk of HCC (13), infections with Schistosoma
or Bacteroides species are linked to bladder cancer and
colon cancer, respectively (22,23),
and inflammatory bowel disease (IBD) greatly increases the risk of
colorectal cancer (24). It is now
well established that the inflammatory microenvironment is
important in the development of cancer, which is why it was added
as the seventh hallmark of cancer (25). The inflammation present on the
tumor microenvironment is characterized by infiltration of
leukocytes (such as lymphocytes and neutrophils), inflammatory
mediators (such as cytokines and chemokines) and persistent
activation of molecular signaling pathways (such as NF-κB and
protein kinase B signaling) (26).
These elements in the microenvironment are often subject to a
feed-forward loop; for example, activation of NF-κB in immune cells
induces production of cytokines which then activate NF-κB in cancer
cells, resulting in the release of chemokines that attract more
inflammatory cells into the tumor tissue (27).
In the present study, activation of LTβR was
demonstrated to promote the mRNA expression of cytokines TNFα and
IL-1β. TNFα, IL-6 and IL-1β are target genes of NF-κB signaling
(27), and overproduction of TNFα
and IL-1β are stimuli in the persistent activation of NF-κB
signaling (28). Popivanova et
al (29) have reported that
increased TNFα expression and increased numbers of infiltrating
leukocytes expressing its major receptor, p55 (TNF-Rp55), are
followed by the initiation and progression of colitis-associated
colon carcinogenesis. Tu et al (30) have demonstrated that
stomach-specific expression of human IL-1β in transgenic mice leads
to spontaneous gastric inflammation and cancer, and this effect is
dependent on early recruitment of myeloid-derived suppressor cells
(MDSCs) to the stomach, which are activated by IL-1β in a
IL-1R/NF-κB-dependent manner. Based on these previous observations,
it is possible that the LTβR overexpression of TNFα and IL-1β
observed in 5,637 bladder cancer cells may be involved in the
persistent activation of NF-κB signaling, resulting in the
feed-forward loop of chronic inflammation in bladder cancer.
Further studies will be required to explore the underlying
molecular links and mechanisms.
Activation of LTβR has been demonstrated to induce
both tumor growth inhibition and promotion. By contrast,
LTα1β2-induced activation of LTβR activates NF-κB to induce chronic
inflammation. In HCC, sustained activation of LTβR signaling
results in chronic inflammation response, which promotes HCC
development in a NF-κB-dependent manner (13). It has also been reported that the
expression of pro-angiogenic chemokine C-X-C motif ligand 2 is
increased in mouse fibrosarcoma cells, paralleled by enhanced solid
tumor growth, when stimulated with an agonistic anti-LTβR antibody
(31). By contrast, LTβR
activation effectively inhibits human colorectal tumor growth in a
xenograft mouse model (9). LTβR
directly mediates cytotoxic lymphocyte-directed tumor rejection
(15,16). Hu et al (14) determined that LTβR mediates
caspase-dependent tumor cell apoptosis in colon carcinoma, mammary
carcinoma and sarcoma, and that LTβR-activated NF-κB potentially
functions as a tumor suppressor. In the present study, activation
of LTβR had no effect on bladder cancer cell growth, despite
increasing the mRNA expression levels of proliferation-related
genes CyclinD1 and Survivin. Since tumor development involves a
variety of molecular signaling pathways and complex processes, a
limitation in the present study was that only two
proliferation-related genes were examined, and therefore the exact
regulation mechanism and function of LTβR signaling in
proliferation was not fully assessed. Further studies will be
needed to investigate the molecular mechanism of LTβR in the
regulation of cell proliferation.
In summary, the present study indicated a potential
role of LTβR signaling in inducing expression of NF-κB canonical
pathway members and pro-inflammatory mediators. Further studies are
needed to investigate in greater detail the link between LTβR
signaling and the biological processes leading to the development
and progression of bladder cancer, potentially guiding in the
future the development of novel targeted drugs therapies.
Acknowledgements
The present study was supported by Zhejiang
Provincial Natural Science Foundation of China (grant no.
Y2110555), Zhejiang Medical Technology and Education Foundation of
China (grant no. 2016KYB200) and Wenzhou Science and Technology
Plan Project (grant no. Y20140609).
References
|
1
|
Mantovani A, Allavena P, Sica A and
Balkwill F: Cancer-related inflammation. Nature. 454:436–444. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Candido J and Hagemann T: Cancer-related
inflammation. J Clin Immunol. 33:(Suppl 1). 79–84. 2013. View Article : Google Scholar
|
|
3
|
Silverman DT, Hartge P, Morrison AS and
Devesa SS: Epidemiology of bladder cancer. Hematol Oncol Clin North
Am. 6:1–30. 1992.PubMed/NCBI
|
|
4
|
Devesa SS, Grauman DJ, Blot WJ and
Fraumeni JF Jr: Cancer surveillance series: Changing geographic
patterns of lung cancer mortality in the United States, 1950
through 1994. J Natl Cancer Inst. 91:1040–1050. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Michaud DS: Chronic inflammation and
bladder cancer. Urol Oncol. 25:260–268. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Medzhitov R: Origin and physiological
roles of inflammation. Nature. 454:428–435. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shen M, Zhou LL, Zhou P and Lin XY:
Expression and clinical pathologic significance of CD4~+,CD8~+ and
CD20~+ lymphocytes in tissue of bladder cancer. Chinese J Health
Lab Technol. 25:1112–1114. 2015.
|
|
8
|
Wolf MJ, Seleznik GM, Zeller N and
Heikenwalder M: The unexpected role of lymphotoxin beta receptor
signaling in carcinogenesis: From lymphoid tissue formation to
liver and prostate cancer development. Oncogene. 29:5006–5018.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lukashev M, LePage D, Wilson C, Bailly V,
Garber E, Lukashin A, Ngam-ek A, Zeng W, Allaire N, Perrin S, et
al: Targeting the lymphotoxin-beta receptor with agonist antibodies
as a potential cancer therapy. Cancer Res. 66:9617–9624. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shen M, Duan X, Zhou P, Zhou W, Wu X, Xu
S, Chen Y and Tao Z: Lymphotoxin β receptor activation promotes
bladder cancer in a nuclear factor-κB-dependent manner. Mol Med
Rep. 11:783–790. 2015.PubMed/NCBI
|
|
11
|
Norris PS and Ware CF: The LT beta R
signaling pathway. Adv Exp Med Biol. 597:160–172. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Greten FR and Karin M: The IKK/NF-kappaB
activation pathway-a target for prevention and treatment of cancer.
Cancer Lett. 206:193–199. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Haybaeck J, Zeller N, Wolf MJ, Weber A,
Wagner U, Kurrer MO, Bremer J, Iezzi G, Graf R, Clavien PA, et al:
A lymphotoxin-driven pathway to hepatocellular carcinoma. Cancer
Cell. 16:295–308. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Hu X, Zimmerman MA, Bardhan K, Yang D,
Waller JL, Liles GB, Lee JR, Pollock R, Lev D, Ware CF, et al:
Lymphotoxin β receptor mediates caspase-dependent tumor cell
apoptosis in vitro and tumor suppression in vivo despite induction
of NF-κB activation. Carcinogenesis. 34:1105–1114. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yang D, Din UDN, Browning DD, Abrams SI
and Liu K: Targeting lymphotoxin beta receptor with tumor-specific
T lymphocytes for tumor regression. Clin Cancer Res. 13:5202–5210.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Winter H, Van Den Engel NK, Poehlein CH,
Hatz RA, Fox BA and Hu HM: Tumor-specific T cells signal tumor
destruction via the lymphotoxin beta receptor. J Transl Med.
5:142007. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Naugler WE and Karin M: NF-kappaB and
cancer-identifying targets and mechanisms. Curr Opin Genet Dev.
18:19–26. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ammirante M, Luo JL, Grivennikov S,
Nedospasov S and Karin M: B-cell-derived lymphotoxin promotes
castration-resistant prostate cancer. Nature. 464:302–305. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dejardin E, Droin NM, Delhase M, Haas E,
Cao Y, Makris C, Li ZW, Karin M, Ware CF and Green DR: The
lymphotoxin-beta receptor induces different patterns of gene
expression via two NF-kappaB pathways. Immunity. 17:525–535. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Eiró N and Vizoso FJ: Inflammation and
cancer. World J Gastrointest Surg. 4:62–72. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Karin M: Nuclear factor-kappaB in cancer
development and progression. Nature. 441:431–436. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Wu S, Rhee KJ, Albesiano E, Rabizadeh S,
Wu X, Yen HR, Huso DL, Brancati FL, Wick E, McAllister F, et al: A
human colonic commensal promotes colon tumorigenesis via activation
of T helper type 17 T cell responses. Nat Med. 15:1016–1022. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Waldner MJ and Neurath MF:
Colitis-associated cancer: The role of T cells in tumor
development. Semin Immunopathol. 31:249–256. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Colotta F, Allavena P, Sica A, Garlanda C
and Mantovani A: Cancer-related inflammation, the seventh hallmark
of cancer: Links to genetic instability. Carcinogenesis.
30:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Vendramini-Costa DB and Carvalho JE:
Molecular link mechanisms between inflammation and cancer. Curr
Pharm Des. 18:3831–3852. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Maeda S and Omata M: Inflammation and
cancer: Role of nuclear factor-kappaB activation. Cancer Sci.
99:836–842. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Blonska M, You Y, Geleziunas R and Lin X:
Restoration of NF-kappaB activation by tumor necrosis factor alpha
receptor complex-targeted MEKK3 in receptor-interacting
protein-deficient cells. Mol Cell Biol. 24:10757–10765. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Popivanova BK, Kitamura K, Wu Y, Kondo T,
Kagaya T, Kaneko S, Oshima M, Fujii C and Mukaida N: Blocking
TNF-alpha in mice reduces colorectal carcinogenesis associated with
chronic colitis. J Clin Invest. 118:560–570. 2008.PubMed/NCBI
|
|
30
|
Tu S, Bhagat G, Cui G, Takaishi S,
Kurt-Jones EA, Rickman B, Betz KS, Penz-Oesterreicher M, Bjorkdahl
O, Fox JG and Wang TC: Overexpression of interleukin-1beta induces
gastric inflammation and cancer and mobilizes myeloid-derived
suppressor cells in mice. Cancer Cell. 14:408–419. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Hehlgans T, Stoelcker B, Stopfer P, Müller
P, Cernaianu G, Guba M, Steinbauer M, Nedospasov SA, Pfeffer K and
Männel DN: Lymphotoxin-beta receptor immune interaction promotes
tumor growth by inducing angiogenesis. Cancer Res. 62:4034–4040.
2002.PubMed/NCBI
|