Introduction
Microglia are resident immune cells of the central
nervous system and respond to microenvironmental changes (1). Activation of microglia contributes to
inflammatory responses in certain neuronal disorders, such as
ischemic stroke (2). In response
to injury, microglia become activated and secrete inflammatory
factors, including interleukin (IL)-1β, tumor necrosis factor
(TNF)-α, IL-6 and nitric oxide (NO) (3). Previous studies demonstrated that
inhibition of microglia activation reduces the severity and
improves the neurological outcomes of neurodegenerative diseases
(4).
Lipopolysaccharides (LPS), a primary constituent of
Gram-negative bacteria, are key in the initiation of inflammation
mediated by Toll-like receptor 4 (5). LPS activates BV2 microglial cells by
releasing IL-1β, IL-6, TNF-α and inducible NO synthase (iNOs)
(6). Thus, the LPS-stimulated BV2
microglial model is useful for investigating the underlying
mechanisms of neurodegenerative diseases that are mediated by
pro-inflammatory cytokines (7).
The thromboxane A2 receptor (TXA2R) is a
seven-transmembrane G-protein-coupled receptor localized on the
cell membrane and in intracellular compartments (8). In the central nervous system,
numerous types of cells, such as microglia (9), oligodendrocytes (10) and astrocytes (11) express TXA2R. Stimulation of TXA2R
in astrocytes results in the secretion of the inflammatory
cytokine, IL-6 (12). Furthermore,
previous studies demonstrated that the TXA2R agonist U46619 may
activate BV2 microglia to release inflammatory cytokines, whereas
the release of inflammatory cytokines may be inhibited by SQ29548
(13). SQ29548 is a highly
selective TXA2R receptor antagonist, which abolishes the majority
of the biological actions of TXA2R. In a previous study, human
platelet reactions to collagen and epinephrine, involving the
generation of endogenous TXA2R, were effectively antagonized by
SQ29548, and platelet aggregation was induced by adding arachidonic
acid (14).
The function of TXA2R antagonists in LPS-stimulated
BV2 microglial cells remains unknown. Thus, the current study
focuses on the anti-inflammatory effects of SQ29548 on
LPS-stimulated BV2 microglial cells and its molecular mechanisms.
The aim of the study was to determine the toxicity of SQ29548 on
BV2 microglial cells and elucidate the effects of the TXA2R
antagonist on the LPS-induced inflammatory response in BV2
microglial cells.
Materials and methods
Reagents
The TXA2R receptor antagonist (SQ29548), LPS and
dimethyl sulfoxide (DMSO) were purchased from Sigma-Aldrich (Merck
KGaA, Darmstadt, Germany). SQ29548 was dissolved in DMSO.
BV2 microglial cell culture
The BV2 microglial cell line was provided by the
Institute of Neurology, Rui Jin Hospital (Shanghai, China). The
cells were cultured in Dulbecco's modified Eagle's medium (Gibco;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented with
10% heat-inactivated fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.) at 55°C for 30 min, and 1%
penicillin/streptomycin at 37°C for 48 h in a humidified incubator
with an atmosphere of 5% CO2.
Cell viability assay
The BV2 cells were seeded at 4.0×103
cells/ml in 96-well plates and treated with various doses of
SQ29548 (0.1, 0.5, or 1.0 µM) for 30 min, followed by treatment
with LPS (100 ng/ml) for 24 h at 37°C. Cell viability was examined
using a Cell Counting kit (CCK)-8 assay (Nanjing KeyGen Biotech
Co., Ltd., Nanjing, China) according to the manufacturer's
instructions.
NO measurement
The BV2 cells were seeded at 1.0×105
cells/ml in 24-well plates and treated with various SQ29548
concentrations (0.1, 0.5 or 1.0 µM) for 30 min, followed by LPS
(100 ng/ml) for 24 h. Following incubation at 37°C for 24 h,
culture supernatants were collected, and the NO concentration was
measured using an NO Griess reaction assay kit (Beyotime Institute
of Biotechnology, Haimen, China) according to the manufacturer's
instructions.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
BV2 cells, seeded at 2.0×105 cells/ml in
12-well plates, were pre-treated with SQ29548 (0.1 µM) for 30 min,
followed by LPS (100 ng/ml) treatment for 6 or 18 h at 37°C. Total
RNA from BV2 cells was isolated using TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA) and
was reverse transcribed to cDNA using the PrimeScript RT Reagent
kit (Takara Bio, Inc., Otsu, Japan). qPCR was performed on cDNA
using an SYBR Green kit (Takara Bio, Inc.) on an Applied Biosystems
7500 Real-Time PCR system (Applied Biosystems; Thermo Fisher
Scientific, Inc.), according to the manufacturer's instructions
under the following conditions: Denaturation at 95°C for 10 sec,
followed by 40 cycles at 95°C for 5 sec and at 60°C for 30 sec.
Relative gene expression was quantified according to the
comparative Cq method (15), and
the results were expressed as fold-difference normalized to the
ribosomal phosphoprotein P0 (Rplp0). The following primer sequences
were used: IL-1β, forward GCA ACT GTT CCT GAA CTC AACT, reverse ATC
TTT TGG GGC GTC AACT; IL-6, forward TAG TCC TTC CTA CCC CAA TTT CC,
reverse TTG GTC CTT AGC CAC TCC TTC; TNF-α, forward CCC TCA CAC TCA
GAT CAT CTT CT reverse GCT ACG ACG TGG GCT ACAG; iNOS, forward ATG
TCC GAA GCA AAC ATCAC, reverse TAA TGT CCA GGA AGT AGG TG; and
Rplp0, forward AGA TTC GGG ATA TGC TGT TGGC and reverse TCG GGT CCT
AGA CCA GTG TTC.
Western blot analysis
BV2 cells, seeded at 4×105 cells/ml in
6-well plates, were pre-treated with SQ29548 (0.1 µM) for 30 min,
followed by treatment with LPS (100 ng/ml) for 30 min. Proteins
were extracted from BV2 cells following lysis in
radioimmunoprecipitation assay lysis buffer (Cell Signaling
Technology, Inc., Danvers, MA, USA) containing protease and
phosphatase inhibitor cocktail at 4°C for 30 min. The protein
concentration was measured using a bicinchoninic acid protein assay
kit (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. Equal amounts of extracted protein
samples (40 µg) were separated by 10% SDS-PAGE and transferred to
nitrocellulose membranes (300 mA for 70 min). The membranes were
blocked with 5% bovine serum albumin for 1 h at room temperature
and incubated with primary antibodies overnight at 4°C. The primary
antibodies used were as follows: phosphorylated (p)-p38 (cat. no.
4511), P-38 (cat. no. 8690), p-c-Jun N-terminal kinases (JNK; cat.
no. 4668), JNK (cat. no. 9252), p-extracellular signal-regulated
kinase (ERK; cat. no. 4370), ERK (cat. no. 4695), p-inhibitor (I)
κB (cat. no. 2859), p-p65 nuclear factor (NF) κB (cat. no. 3033),
p65 NFκB (cat. no. 8242; 1:1,000; Cell Signaling Technology, Inc.)
and β-tubulin (cat. no. T8328; 1:2,000; Sigma-Aldrich; Merck KGaA).
Membranes were washed three times with TBS containing 0.1% Tween-20
(Beyotime Institute of Biotechnology) and incubated with
anti-rabbit (cat. no. 7074) or anti-mouse (cat. no. 7076)
horseradish peroxidase-conjugated secondary antibodies (1:1,000;
Cell Signaling Technology, Inc.) for 1 h at room temperature. The
specific bands were visualized by enhanced chemiluminescence
(Thermo Fisher Scientific, Inc.) using a ChemiDoc™XRS+ imaging
system (Bio-Rad Laboratories, Inc., Hercules, CA, USA). Blots were
semi-quantified by densitometry using ImageJ software version 1.41
(National Institutes of Health, Bethesda, MA, USA).
Statistical analysis
All statistical analyses were performed using
GraphPad Prism software version 5.0 (GraphPad Software, Inc. La
Jolla, CA, USA). The statistical significance of the differences
between groups was assessed using Student's t test for pair-wise
comparisons or a one-way analysis of variance followed by a post
hoc Student-Newman-Keuls test for multiple comparisons. Data are
expressed as the mean ± standard error of the mean of 3 independent
experiments. P<0.05 was considered to indicate a statistically
significant difference.
Results
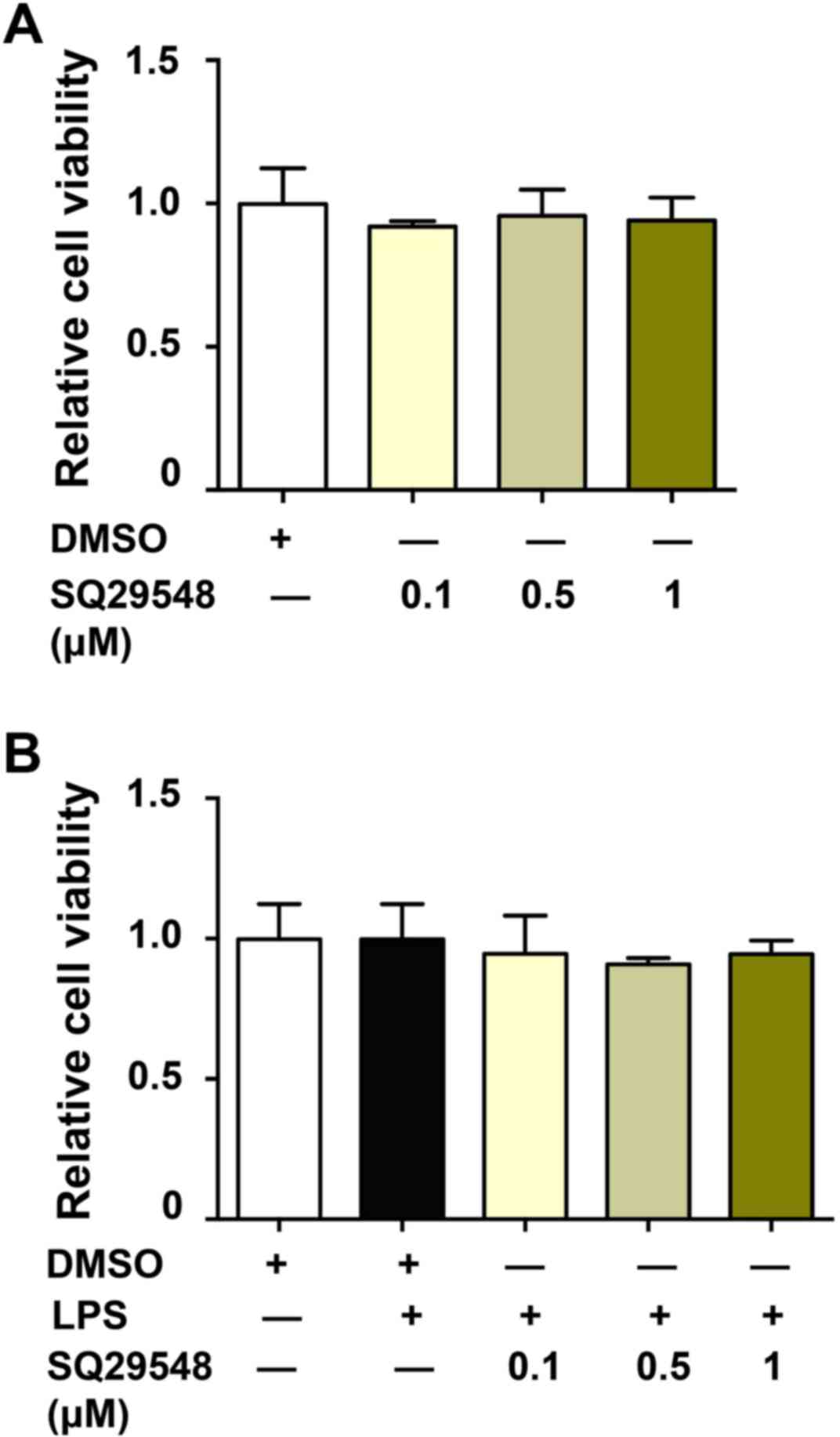
SQ29548 is not toxic to BV2 microglial
cells
To investigate the potential cytotoxicity of
SQ29548, the viability of BV2 microglial cells was investigated.
Cells were incubated with SQ29548 (0.1, 0.5 or 1.0 µM) for 24 h,
and cell viability was evaluated using a CCK-8 assay kit. The data
indicated that cell viability was unaffected by the SQ29548
treatments (Fig. 1A). In addition,
LPS treatment (100 ng/ml) alone or with SQ29548 (0.1, 0.5 or 1.0
µM) did not affect cell viability for 24 h (Fig. 1B).
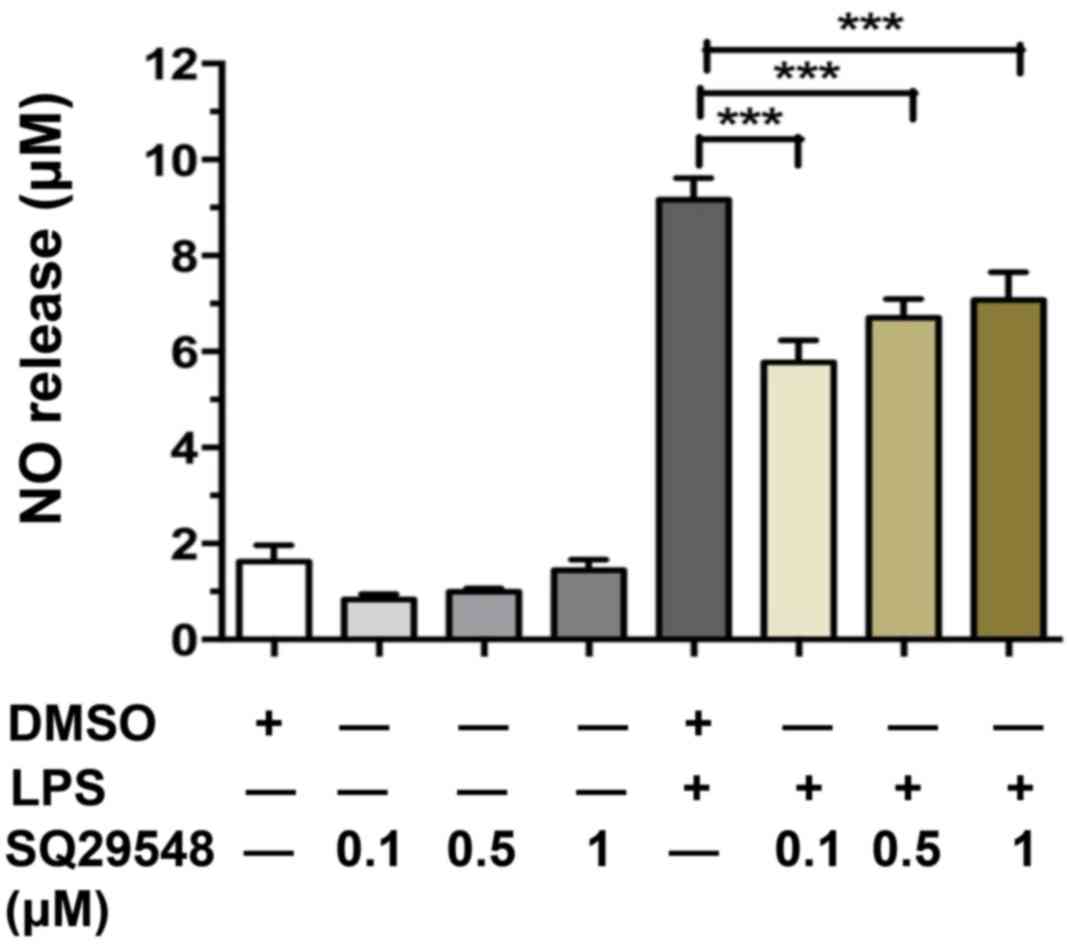
SQ29548 reduced LPS-induced NO
release
NO is generated from L-arginine by iNOS and high
levels of NO are associated with the progression of
neurodegenerative diseases (16).
To investigate the effect of SQ29548 on LPS-stimulated BV2
microglial cell activation, the effects of SQ29548 on LPS-induced
NO release were observed. In the current study, BV2 cells were
pre-treated with SQ29548 (0.1, 0.5 or 1.0 µM) for 30 min and
stimulated with LPS (100 ng/ml) for another 24 h. NO concentrations
in the culture supernatants were measured using an NO assay kit.
SQ29548 significantly decreased the LPS-stimulated production of NO
in the BV2 microglial cells (Fig.
2). SQ29548 at 0.1 µM was the most effective at inhibiting NO
release; thus, this concentration was selected for subsequent
experiments.
SQ29548 reduced LPS-induced expression
of inflammatory cytokines
Inflammatory cytokines released from activated
microglia exacerbate cell damage and LPS may activate BV2
microglial cells, which are major sources of various inflammatory
cytokines, such as IL-1β, IL-6, TNF-α and iNOS (6). In the present study, LPS stimulation
caused a significant increase in the expression levels of IL-1β,
IL-6, TNF-α and iNOS mRNA in BV2 microglial cells. Furthermore,
pretreatment with SQ29548 markedly suppressed their expression
levels (Fig. 3).
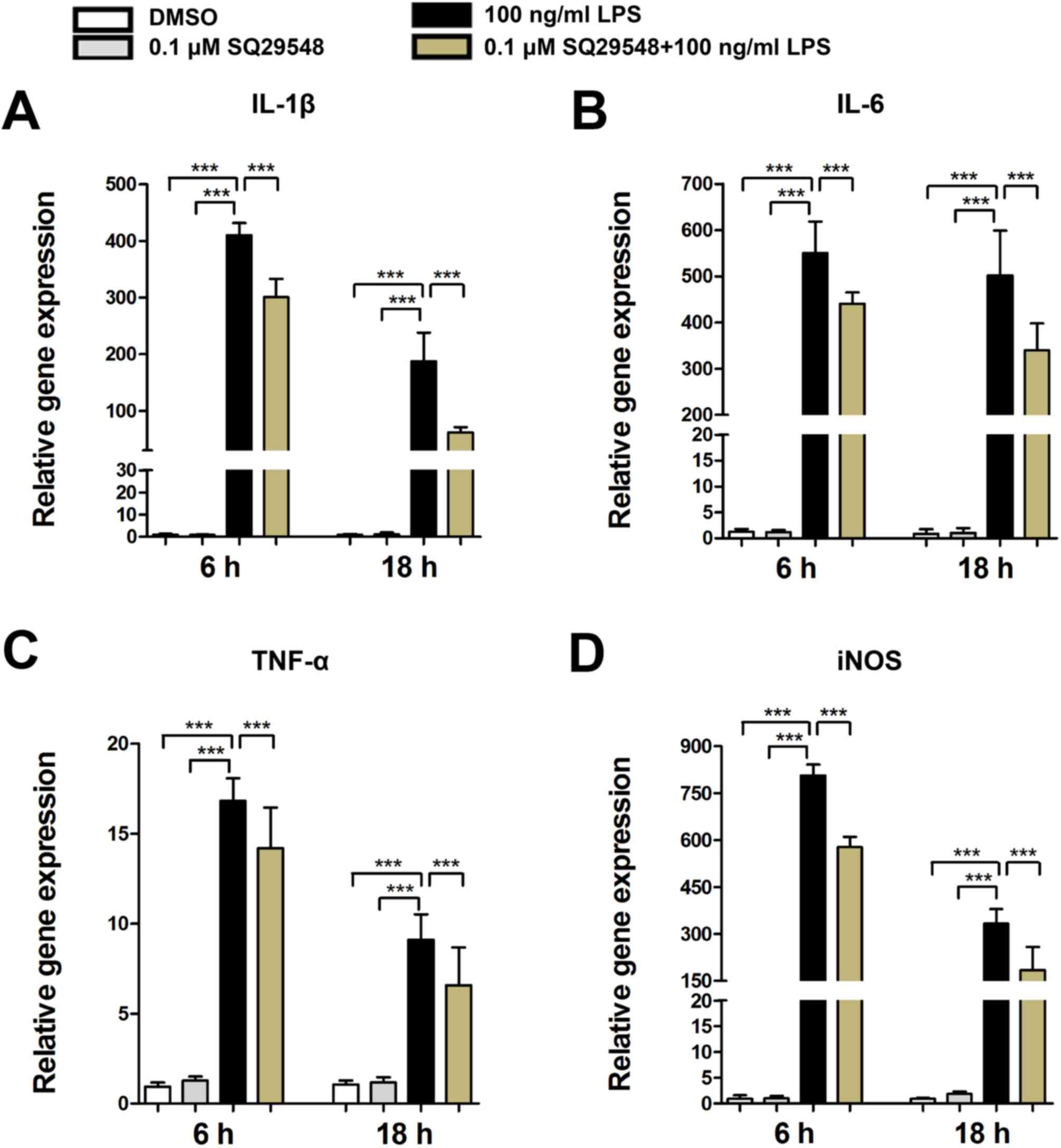 | Figure 3.SQ29548 suppresses the expression of
IL-1β, IL-6, TNF-α and iNOS in BV2 microglial cells. BV2 cells were
incubated at 37°C in the absence or presence of SQ29548 (0.1 µM)
for 30 min and stimulated with LPS (100 ng/ml). After 6 or 18 h,
the mRNA expression levels of (A) IL-1β, (B) IL-6, (C) TNF-α and
(D) iNOS were measured by reverse transcription-quantitative
polymerase chain reaction. Values are presented as the mean ±
standard error of the mean from at least three independent
experiments. ***P<0.001, as indicated. IL, interleukin; TNF-α,
tumor necrosis factor-α; iNOS, inducible nitric oxide synthase;
LPS, lipopolysaccharide; DMSO, dimethyl sulfoxide. |
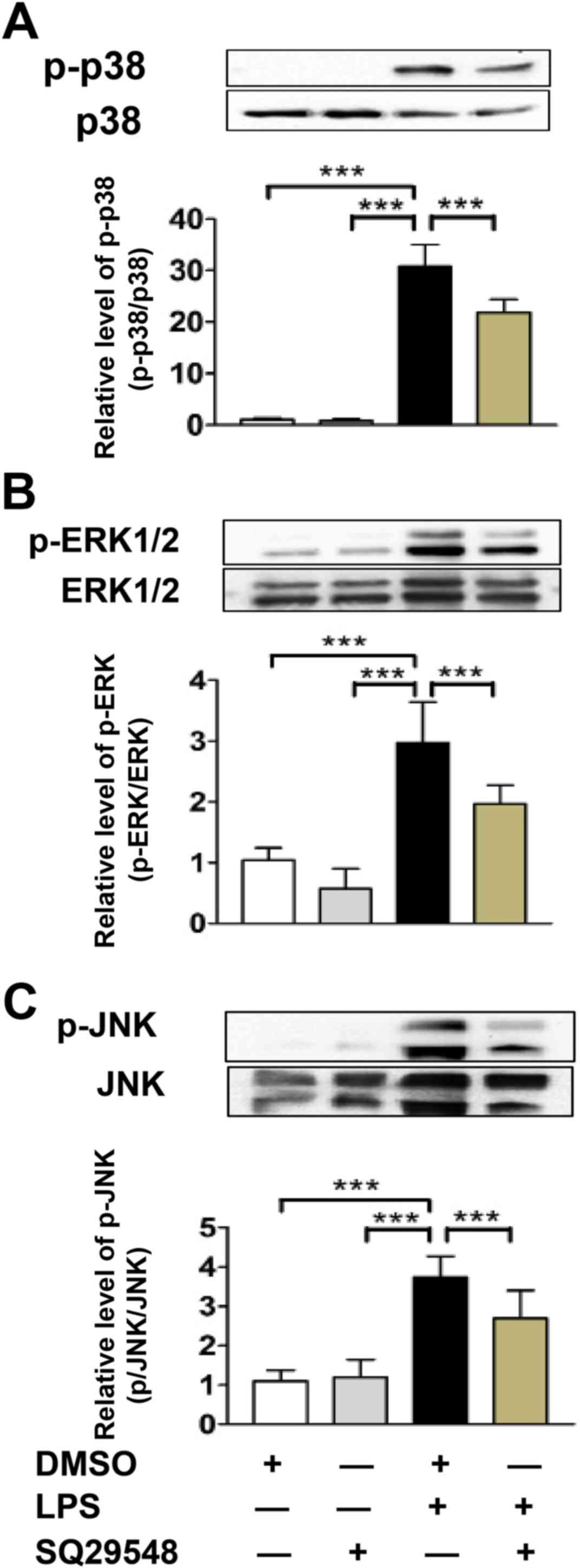
SQ29548 inhibited LPS-induced
activation of mitogen activated protein kinases (MAPKs)
MAPKs are key regulators in the early stages of
LPS-stimulated inflammatory responses in BV2 microglial cells
(17). To determine the underlying
mechanisms of the SQ29548-mediated suppression of inflammatory
cytokine production, BV2 microglial cells were treated with or
without SQ29548 for 30 min and subsequently stimulated with LPS
(100 ng/ml) for 30 min. LPS caused a significant increase in the
phosphorylation of p38, ERK and JNK, which was markedly inhibited
by SQ29548 pretreatment (Fig. 4).
These results indicated that SQ29548 inhibited the LPS-induced
expression of IL-1β, IL-6, TNF-α and iNOS by inhibiting the
activation of MAPK signaling pathways.
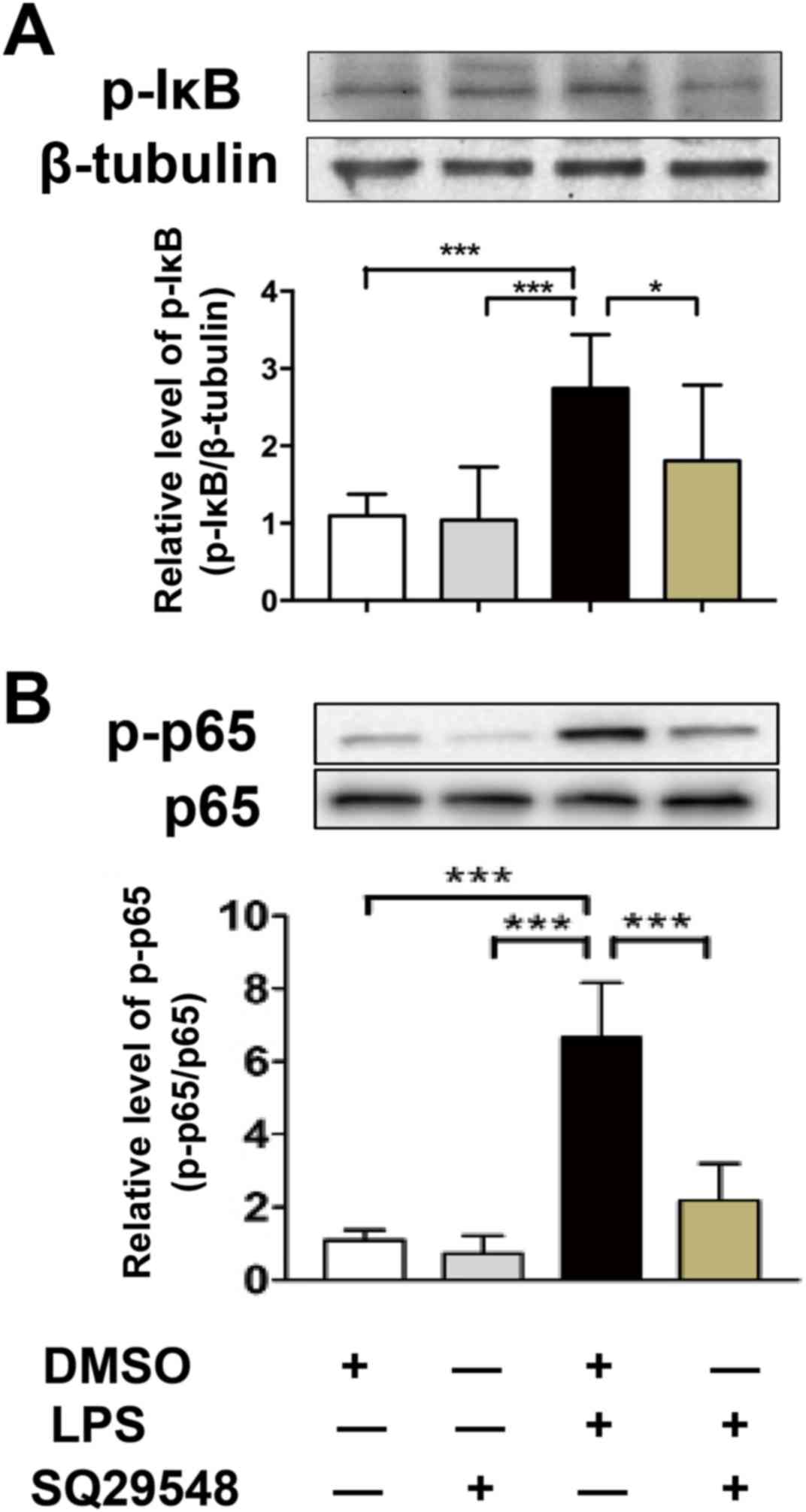
SQ29548 inhibited LPS-induced NF-κB
activation
The NF-κB signaling pathway is another crucial
mediator of the LPS-stimulated inflammatory response in BV2
microglial cells (18). Thus, the
NF-κB signaling pathway was evaluated as a possible mechanistic
target for the inhibitory action of SQ29548 during inflammation. In
the current study, the effects of SQ29548 on LPS-induced NF-κB
activation were observed. The results demonstrated that treatment
with SQ29548 did not exert an effect on the phosphorylation of IκB
and NF-κB-p65. Following LPS treatment, the expression levels of
p-IκB and p-p65 were increased. Pretreatment with SQ29548 inhibited
the phosphorylation of IκB and NF-κB-p65 induced by LPS (Fig. 5).
Discussion
In the present study, the data demonstrated that
SQ29548, a TXA2R antagonist, significantly inhibits LPS-stimulated
microglial activation and the mRNA expression levels of IL-1β,
IL-6, TNF-α and iNOS. Furthermore, SQ29548 significantly inhibited
LPS-induced phosphorylation of p38, ERK, JNK, IκB, and NF-κB-p65 in
BV2 microglial cells.
Microglia are resident immune cells that are
important in host defense and immune surveillance under normal
conditions (19). Activated
microglia release inflammatory cytokines, such as IL-1β, TNF-α,
IL-6 and iNOS. These inflammatory factors are known to contribute
to activated microglia-mediated neurodegeneration (20). Therefore, the inhibition of
microglial activation has been proven as a useful therapeutic
method for slowing the progression of neurodegenerative diseases.
In the present study, the results indicated that SQ29548
significantly reduced LPS-induced microglial activation and the
release of inflammatory cytokines.
LPS is one of the most potent stimuli for activation
of the mouse BV2 microglial cell line (7). Activation of BV2 microglial cells
produces increased expression levels of the inflammatory mediators
IL-1β, IL-6, TNF-α and iNOS (6).
The present study demonstrated that LPS causes a significant
increase in the expression levels of IL-1β, IL-6, TNF-α, and iNOS,
which was reduced by SQ29548, indicating that SQ29548 suppresses
the production of IL-1β, IL-6, TNF-α, and iNOS via downregulation
of their gene expression in LPS-stimulated microglial cells. To
further investigate the underlying mechanisms for the reduction of
inflammatory cytokine release by SQ29548, various different signal
transduction pathways that are activated by TLR4 signaling via LPS
were evaluated. MAPKs and NF-κB signaling pathways are crucial
regulators of LPS-induced IL-1β, IL-6, TNF-α and iNOS expression in
microglia (16). The MAPK family
consists of p38, ERK and JNK. Inhibition of these signaling
pathways reduces the release of inflammatory mediators in
LPS-stimulated BV2 cells (21). In
the current study, the phosphorylation of p38, ERK, JNK, IκB and
NF-κB was observed to occur within 30 min of LPS stimulation and
may be markedly reduced by SQ29548, indicating that inhibition of
neuroinflammation signaling cascades occurs via TXA2R inhibition.
These results indicate that SQ29548 inhibits LPS-induced
inflammatory responses in BV2 microglial cells by inhibiting MAPK
and NF-κB activation.
In conclusion, the current study demonstrated that
SQ29548 markedly reduced the LPS-induced microglial activation and
release of the inflammatory cytokines IL-1β, IL-6, TNF-α and iNOS
in BV2 microglial cells. These effects may be mediated by the
inhibition of MAPKs and NF-κB signaling pathways. These findings
suggested that SQ29548 may have potential as a novel
anti-inflammatory agent to suppress the inflammatory response
during microglia-mediated neurodegeneration. However, further
studies are required to fully elucidate the effects of SQ29548 on
microglia and to investigate the exact molecular mechanisms that
are involved in these actions.
Acknowledgements
The present study was supported by the Science and
Technology Commission of Shanghai Municipality (grant no.
12ZR1418600), the Chinese Ministry of Science and Technology (grant
no. 2013CB945604) and the SJTU Interdisciplinary Research Programme
(grant no. YG2012ZD05).
References
|
1
|
Saijo K and Glass CK: Microglial cell
origin and phenotypes in health and disease. Nat Rev Immunol.
11:775–787. 2011. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Iadecola C and Anrather J: The immunology
of stroke: From mechanisms to translation. Nat Med. 17:796–808.
2011. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Lawrence T and Natoli G: Transcriptional
regulation of macrophage polarization: Enabling diversity with
identity. Nat Rev Immunol. 11:750–761. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sehgal N, Agarwal V, Valli RK, Joshi SD,
Antonovic L, Strobel HW and Ravindranath V: Cytochrome P4504f, a
potential therapeutic target limiting neuroinflammation. Biochem
Pharmacol. 82:53–64. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Firdous AP, Kuttan G and Kuttan R:
Anti-inflammatory potential of carotenoid meso-zeaxanthin and its
mode of action. Pharm Biol. 53:961–967. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Abate W, Alghaithy AA, Parton J, Jones KP
and Jackson SK: Surfactant lipids regulate LPS-induced
interleukin-8 production in A549 lung epithelial cells by
inhibiting translocation of TLR4 into lipid raft domains. J Lipid
Res. 51:334–344. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Dai XJ, Li N, Yu L, Chen ZY, Hua R, Qin X
and Zhang YM: Activation of BV2 microglia by lipopolysaccharide
triggers an inflammatory reaction in PC12 cell apoptosis through a
toll-like receptor 4-dependent pathway. Cell Stress Chaperones.
20:321–331. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Nakahata N: Thromboxane A2:
Physiology/pathophysiology, cellular signal transduction and
pharmacology. Pharmacol Ther. 118:18–35. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kitanaka J, Hashimoto H, Sugimoto Y,
Sawada M, Negishi M, Suzumura A, Marunouchi T, Ichikawa A and Baba
A: cDNA cloning of a thromboxane A2 receptor from rat astrocytes.
Biochim Biophys Acta. 1265:220–223. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Blackman SC, Dawson G, Antonakis K and Le
Breton GC: The identification and characterization of
oligodendrocyte thromboxane A2 receptors. J Biol Chem. 273:475–483.
1998. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Nakahata N, Miyamoto A, Ohkubo S, Ishimoto
H, Sakai K, Nakanishi H, Ohshika H and Ohizumi Y: Gq/11
communicates with thromboxane A2 receptors in human astrocytoma
cells, rabbit astrocytes and human platelets. Res Commun Mol Pathol
Pharmacol. 87:243–251. 1995.PubMed/NCBI
|
|
12
|
Obara Y, Kurose H and Nakahata N:
Thromboxane A2 promotes interleukin-6 biosynthesis mediated by an
activation of cyclic AMP-response element-binding protein in 1321N1
human astrocytoma cells. Mol Pharmacol. 68:670–679. 2005.PubMed/NCBI
|
|
13
|
Yang W, Yan A, Zhang T, Shao J, Liu T,
Yang X, Xia W and Fu Y: Thromboxane A2 receptor stimulation
enhances microglial interleukin-1b and NO biosynthesis mediated by
the activation of ERK pathway. Front Aging Neurosci. 8:82016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Ogletree ML, Harris DN, Greenberg R,
Haslanger MF and Nakane M: Pharmacological actions of SQ 29,548, a
novel selective thromboxane antagonist. J Pharmacol Exp Ther.
234:435–441. 1985.PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Boje KM: Nitric oxide neurotoxicity in
neurodegenerative diseases. Front Biosci. 9:763–776. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kacimi R, Giffard RG and Yenari MA:
Endotoxin-activated microglia injure brain derived endothelial
cells via NF-kB, JAK-STAT and JNK stress kinase pathways. J Inflamm
(Lond). 8:72011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Min S, More SV, Park JY, Jeon SB, Park SY,
Park EJ, Yoon SH and Choi DK: EOP, a newly synthesized ethyl
pyruvate derivative, attenuates the production of inflammatory
mediators via p38, ERK and NF-kappaB pathways in
lipopolysaccharide-activated BV-2 microglial cells. Molecules.
19:19361–19375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kreutzberg GW: Microglia: A sensor for
pathological events in the CNS. Trends Neurosci. 19:312–318. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Le W, Rowe D, Xie W, Ortiz I, He Y and
Appel SH: Microglial activation and dopaminergic cell injury: An in
vitro model relevant to Parkinson's disease. J Neurosci.
21:8447–8455. 2001.PubMed/NCBI
|
|
21
|
Yu Z, Tang L, Chen L, Li J, Wu W and Hu C:
Capillarisin suppresses lipopolysaccharide-induced inflammatory
mediators in BV2 microglial cells by suppressing TLR4-mediated
NF-kappa B and MAPKs signaling pathway. Neurochem Res.
40:1095–1101. 2015. View Article : Google Scholar : PubMed/NCBI
|