Introduction
Congenital heart disease (CHD), which commonly
refers to a developmental abnormality in the structure of the heart
or intrathoracic great blood vessel, is the most common type of
birth defect in humans, accounting for one-third of all major
congenital malformations worldwide (1). There are 1.35 million neonates born
with CHD worldwide every year, with a global incidence of ~1% in
live births and as high as 10% in early miscarriages (1–3). At
present, CHD remains the leading noninfectious cause of infant
morbidity and mortality, and is estimated to account for ~27% of
all birth defect-associated cases of mortality (2). Despite the high prevalence and
important clinical significance, the causes of CHD remain largely
unclear.
The heart is the first functional organ to develop
during vertebrate embryogenesis (4). The development of the cardiovascular
system is a complex biological process that involves cell
proliferation, differentiation, polarization and migration, and is
extremely sensitive to environmental and genetic risk factors
(4,5). Errors during the process of
cardiovascular morphogenesis may result in a broad spectrum of
congenital cardiovascular anomalies, including ventricular septal
defect (VSD), atrial septal defect, tetralogy of Fallot, double
outlet right ventricle (DORV), transposition of the great arteries
and endocardial cushion defect (6–8).
Although there are nongenetic risk factors associated with CHD,
including parental characteristics and conditions, maternal drug
use in the first trimester of pregnancy, and long-term exposure to
toxicants and ionizing radiation (9), an increasing body of evidence in the
field of human genetics has demonstrated that genetic defects serve
a key role in the pathogenesis of CHD, and to date, a large number
of mutations in >60 genes have been causally linked to CHD
(6–8,10–35).
Among these established CHD-associated genes, the majority encode
cardiac transcription factors (6–8,36).
However, CHD is a heterogeneous disorder, and the genetic
components underlying CHD in the majority of patients remain
unclear.
Mesoderm posterior 1 (MESP1), also known as class C
basic helix-loop-helix (bHLH) protein 5, is expressed in the
posterior part of the embryonic mesoderm and is an important
transcription factor expressed in cardiac progenitors during
embryogenesis. It is also the earliest marker identified once a
cell has been committed towards cardiac development (37,38).
In mice, inactivation of the MESP1 gene leads to mortality
due to abnormalities in heart tube formation and heart looping,
resulting in various degrees of cardiac bifida (39,40).
By contrast, overexpression of MESP1 in mouse embryonic stem
cells results in abnormal expression of specific gene sets and,
subsequently, accelerated cardiovascular specification and
premature appearance of beating cells (41). Among these upregulated genes are
crucial cardiovascular transcription factors including NKX2
homeobox 5 (NKX2-5), GATA binding protein 4 (GATA4),
T-box 20 (TBX20), heart and neural crest derivatives
expressed 2 (HAND2) and myocardin (MYOCD) (41), of which certain genes
(NKX2-5, GATA4, TBX20 and HAND2) are
involved in the pathogenesis of CHD in humans (13,19,24,27,31–36,42–44).
In humans, mutations in MESP1 have been demonstrated to
contribute to various types of CHD, including VSD, atrial septal
defect, tetralogy of Fallot, coarctation of the aorta and aortic
atresia (41,45). However, the prevalence and variety
of the MESP1 mutations in Chinese patients with CHD have yet
to be investigated.
Materials and methods
Study patients and control
individuals
In the present study, a cohort of 178 Chinese Han
unrelated patients diagnosed with non-syndromic CHD were recruited
from the following hospitals: Shanghai Tenth People's Hospital,
Tongji Hospital, Renji Hospital and Shanghai Chest Hospital
(Shanghai, China), between February 2013 and March 2015. The
available family members of the index patient carrying an
identified MESP1 mutation were also included. A total of 95
males and 83 females, of which 9 had a positive family history of
CHD were included. A total of 200 ethnically-matched healthy
individuals, who had no CHD diagnosis, family history of CHD or any
other heart disease, were recruited as controls from the following
hospitals: Shanghai Tenth People's Hospital, Tongji Hospital, Renji
Hospital and Shanghai Chest Hospital (Shanghai, China), between
February 2013 and March 2015. A total of 107 males and 93 females,
with no family history of CHD were recruited. All study
participants underwent detailed clinical evaluation, including
medical histories, physical examination (including assessment for
shortness of breath, cyanosis, heart murmur, under-development of
limbs or poor growth), 12-lead electrocardiogram and
two-dimensional transthoracic echocardiography with color flow
Doppler. Transesophageal echocardiography or cardiac
catheterization procedure was performed only when clinically
indicated. Diagnosis of CHD was confirmed by imaging and/or direct
view during cardiac surgery. Subjects with a recognizable syndromic
CHD at the time of enrollment, including Down syndrome, Holt-Oram
syndrome, Di George syndrome and Turner syndrome, were excluded
from the study. The present study was conducted in accordance with
the ethical principles outlined in the Declaration of Helsinki. The
study protocol was approved by the Institutional Ethical Committee
of the Tongji Hospital, Tongji University School of Medicine, China
[project no. LL (H)-09-07]. Informed written consent was obtained
from the guardians of the patients with CHD and the control
individuals prior to the collection of blood samples.
DNA isolation and mutation
analysis
Peripheral venous blood samples were drawn from all
study subjects. Genomic DNA was isolated from whole blood cells
using the Wizard Genomic DNA Purification kit (Promega Corporation,
Madison, WI, USA), according to the manufacturer's recommendation.
Based on the referential genomic DNA sequence of the MESP1
gene (GenBank accession no.: NC_000015.10), which was derived from
the National Center for Biotechnology Information (NCBI; http://www.ncbi.nlm.nih.gov/), the primer pairs used
to amplify the coding exons and splicing junctions of MESP1
by polymerase chain reaction (PCR) were designed as presented in
Table I. DNA samples were
amplified using HotStar Taq DNA Polymerase (Qiagen GmbH, Hilden,
Germany) on a Verti Thermal Cycler (Applied Biosystems; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) with standard
thermocycling conditions. The total volume of the PCR mixture was
25 µl, comprising 11.25 µl deionized water, 2.5 µl 10X buffer, 5 µl
5X Q solution, 2 µl dNTPs (2.5 mM each), 1 µl of each primer (20
µM), 0.25 µl (5 U/µl) HotStar Taq DNA Polymerase (Qiagen, Hilden,
Germany) and 2 µl of genomic DNA (200 ng/µl). Thermocycling
conditions were as follows: Initial denaturation at 95°C for 15
min, followed by 35 cycles of denaturation at 94°C for 30 sec,
annealing at 62°C for 30 sec and extension at 72°C for 1 min, with
a final extension at 72°C for 10 min. PCR products were purified
with a QIAquick PCR Purification kit (Qiagen GmbH), according to
the manufacturer's protocol, and subjected to PCR-sequencing with a
BigDye® Terminator v3.1 Cycle Sequencing kit (Applied
Biosystems; Thermo Fisher Scientific, Inc.) under an ABI PRISM 3130
XL DNA Analyzer (Applied Biosystems; Thermo Fisher Scientific,
Inc.). The total volume of the PCR sequencing mixture was 20 µl,
including 8 µl deionized water, 8 µl Premix, 1 µl forward or
reverse primer (2 µM) and 3 µl of the purified DNA products (20
ng/µl). The sequencing PCR program was set as follows: A total of
35 cycles of denaturation at 95°C for 20 sec, annealing at 50°C for
15 sec and extension at 60°C for 1 min. The obtained sequences were
compared with the genomic reference sequence (GenBank accession
no.: NC_000015.10) using the Basic Local Alignment Search Tool
(BLAST; https://blast.ncbi.nlm.nih.gov/Blast.cgi?PAGE_TYPE=BlastSearch&PROG_DEF=blastn&BLAST_PROG_DEF=blastn&BLAST_SPEC=
GlobalAln&LINK_LOC=BlastHomeLink; National Center for
Biotechnology Information, Bethesda, MD, USA). For an identified
sequence variation, a second independent PCR analysis was
performed, as aforementioned, to confirm it. The position of an
exonic variation is described according to the guidelines of the
human genome variation society using the NCBI reference sequence of
the MESP1 mRNA (NM_018670.3). For a newly discovered genetic
variation, the public databases for human sequence variations,
including single nucleotide polymorphism (SNP; http://www.ncbi.nlm.nih.gov/SNP), the 1000
Genomes Project (1000 GP; http://www.1000genomes.org/) and human gene mutation
(HGM; http://www.hgmd.org) databases, were consulted to
confirm its novelty.
 | Table I.Primers for amplification of the
coding exons and flanking introns of the Mesoderm Posterior 1
gene. |
Table I.
Primers for amplification of the
coding exons and flanking introns of the Mesoderm Posterior 1
gene.
| Exon | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Amplicon (base
pairs) |
|---|
| 1-a |
ACCTCGGGCTCGGCATAAAG |
GCAGTTTCTCCCGCTCACTG | 340 |
| 1-b |
TCGTCTCGTCCCCAGACTCA |
GGTGCCTGGTCCTCACCTT | 699 |
| 2 |
GAAGGGCAGGCGATGGAGC |
GAGGCCAAAAAGCCTCGGTG | 450 |
Multiple alignments of the MESP1
protein across species
The human MESP1 protein was autonomously aligned
with those of a chimpanzee, monkey, dog, mouse and rat, using
Multiple Sequence Comparison by Log-Expectation (MUSCLE; http://www.ebi.ac.uk/Tools/msa/muscle/;
European Bioinformatics Institute, Hinxton, UK).
Expression plasmids and site-directed
mutagenesis
Human heart cDNAs were prepared as described
previously (46). The full-length
wild-type cDNAs of the human MESP1 gene were amplified by
PCR using the pfuUltra high-fidelity DNA polymerase (Stratagene;
Agilent Technologies, Inc., Santa Clara, CA, USA) and the following
primers: Forward, 5′-ATGGCTAGCTGGAAGGGGCCACTTCACAC-3′ and reverse,
5′-CATTCTAGAGACGGCGTCAGTTGTCC-3′. The total volume of the PCR
mixture was 50 µl, including 36 µl deionized water, 5 µl 10X
buffer, 4 µl dNTPs (2.5 mM each), 1 µl of each primer (20 µM), 1 µl
(2.5 U/µl) pfuUltra high-fidelity DNA polymerase and 2 µl cDNA (100
ng/µl). Thermocycling conditions were as follows: Initial
denaturation at 95°C for 2 min, followed by 35 cycles of
denaturation at 95°C for 1 min, annealing at 62°C for 30 sec and
extension at 72°C for 1 min, with a final elongation at 72°C for 10
min. The PCR fragment was doubly digested by endonucleases
NheI and XbaI (Takara Biotechnology Co., Ltd.,
Dalian, China). The digested product (40 µl/lane), with a length of
912 base pairs, was fractionated by 1.5% agarose gel
electrophoresis, purified with the QIAquick Gel Extraction kit
(Qiagen GmbH), according to the manufacturer's protocol, and then
inserted into pcDNA3.1 (Promega Corporation) to construct the
eukaryotic expression vector, pcDNA3.1-MESP1. Similarly, the
full-length wild-type cDNAs of the human E47 gene [also
termed transcription factor 3 (TCF3) isoform 2] were
amplified by PCR using the following primers: Forward,
5′-GGTGCTAGCGGTTTCCAGGCCTGAGGTGC-3′ and reverse,
5′-CCATCTAGACAAAGTGTATGTTTTGTTGC-3′, and doubly digested by
NheI and XbaI (Takara Biotechnology Co., Ltd.).
Thermocycling conditions were as follows: Initial denaturation at
95°C for 2 min, followed by 35 cycles of denaturation at 95°C for 1
min, annealing at 62°C for 30 sec and extension at 72°C for 2 min,
with a final elongation at 72°C for 10 min. The digested product,
with a length of 2,044 base pairs, was subcloned into pcDNA3.1
(Promega Corporation) to construct the recombinant plasmid
pcDNA3.1-E47. The Ebox-luc [pGL4.23-Dickkopf WNT signaling pathway
inhibitor 1 (DKK1)-11] plasmid, which contains a triplicate repeat
of the E-box region acCATATGgt located ~11.6 kb upstream of
DKK1 and expresses Firefly luciferase (47), was constructed as described
previously (41). The identified
mutation was introduced into wild-type MESP1 by
site-directed mutagenesis using a QuickChange II XL Site-Directed
Mutagenesis kit (Stratagene; Agilent Technologies, Inc.) with the
following mutagenic primers: Forward,
5′-GTGGCGCCCGCGGGCTAGAGCCTGACCAAGA-3′ and reverse,
5′-TCTTGGTCAGGCTCTAGCCCGCGGGCGCCAC-3′. The mutant was selected by
DpnI (Takara Biotechnology Co., Ltd.) digestion and then
sequenced for verification. Sequencing was performed as
aforementioned using the following primers at either side of the
multiple cloning sites of the pcDNA3.1 plasmid: Forward,
5′-TAATACGACTCACTATAGGG-3′ and reverse,
5′-TAGAAGGCACAGTCGAGG-3′.
Dual-luciferase reporter assays
Human embryonic kidney (HEK) 293 cells were
purchased from the Shanghai Institute of Biochemistry and Cell
Biology, Shanghai Institutes for Biological Sciences, Chinese
Academy of Sciences (Shanghai, China) were cultured in Dulbecco's
modified Eagle's medium supplemented with 10% fetal calf serum
(Invitrogen; Thermo Fisher Scientific, Inc.) at 37°C, and plated at
a density of 1×105 cells/well on 24-well plates 24 h
prior to transfection. Cells were cotransfected with either 100 ng
empty pcDNA3.1 vector (−), 100 ng wild-type MESP1-pcDNA3.1 vector
(MESP1), 100 ng Q118X-mutant MESP1-pcDNA3.1 vector (Q118X) or 50 ng
wild-type MESP1-pcDNA3.1 vector plus 50 ng Q118X-mutant
MESP1-pcDNA3.1 vector (MESP1 + Q118X), together with 250 ng
pcDNA3.1-E47, 100 ng Ebox-luc vector and 0.4 ng pGL4.75 (a
Renilla luciferase reporter vector, which was used as an
internal control to normalize transfection efficiency) using the
PolyFect Transfection Reagent (Qiagen GmbH), according to the
manufacturer's protocol. Transfected cells were incubated for 48 h
at 37°C with 5% CO2, then lysed with Passive Lysis
Buffer (Promega Corporation) and assayed with the Dual-Glo
luciferase assay kit (Promega Corporation) on a GloMax®
96 Luminometer (Promega Corporation) according to the
manufacturer's protocol. Firefly luciferase and Renilla
luciferase activities were analyzed using the GloMax® 96
Microplate Luminometer software version 1.9.3 (Promega
Corporation), and the activity of the Ebox promoter was presented
as fold activation of Firefly luciferase relative to Renilla
luciferase. A minimum of three independent cotransfection
experiments were performed in triplicate to calculate the average
values and standard deviations.
Statistical analysis
Statistical analyses were performed using the SPSS
software version 17.0 (SPSS, Inc., Chicago, IL, USA). Continuous
variables are expressed as the mean ± standard deviation, and
categorical variables are expressed as numbers and percentages. The
statistical significance of the differences between groups was
assessed using a one-way analysis of variance followed by a post
hoc Bonferroni test for multiple comparisons. P<0.05 was
considered to indicate a statistically significant difference.
Results
Clinical characteristics of the study
subjects
A cohort of 178 unrelated patients with was
clinically evaluated in comparison with a total of 200 unrelated
non-CHD control individuals. All of the patients had confirmed CHD
diagnoses; while the control individuals had no congenital
anomalies in cardiovascular structures. There was no statistical
difference in age, gender or ethnicity between the patient and
control groups. The baseline clinical features of the 178 patients
with CHD are summarized in Table
II.
| Table II.Clinical characteristics of the
patients with congenital heart disease. |
Table II.
Clinical characteristics of the
patients with congenital heart disease.
| Variable | Number | Percentage (%) or
range |
|---|
| Gender |
|
Male | 95 | 53 |
|
Female | 83 | 47 |
| Age (years;
mean) |
4 | 0–21 |
| Positive family
history of CHD |
9 | 5 |
| Distribution of
various CHDs |
|
Isolated CHD | 92 | 52 |
|
VSD | 30 | 17 |
|
ASD | 14 | 8 |
|
DORV | 10 | 6 |
|
PS |
8 | 4 |
|
TGA |
6 | 3 |
|
TA |
6 | 3 |
|
HLV |
5 | 3 |
|
PA |
4 | 2 |
|
TAPVC |
3 | 2 |
|
AS |
3 | 2 |
|
IAA |
2 | 1 |
|
AVSD |
1 | 1 |
| Complex CHD | 86 | 48 |
|
TOF | 28 | 16 |
|
VSD + ASD | 17 | 10 |
|
DORV + VSD | 15 | 8 |
|
VSD + PDA | 12 | 7 |
|
TGA + VSD |
8 | 4 |
|
TA + VSD |
6 | 3 |
| Incidence of
arrhythmias |
|
Atrioventricular block |
6 | 3 |
| Atrial
fibrillation |
4 | 2 |
| Treatment |
|
Surgical repair | 109 | 61 |
|
Percutaneous closure | 41 | 23 |
|
Follow-up | 28 | 16 |
Identification of a novel MESP1
mutation in CHD
Direct PCR-sequencing of the MESP1 gene in
178 unrelated patients with CHD identified a heterozygous nonsense
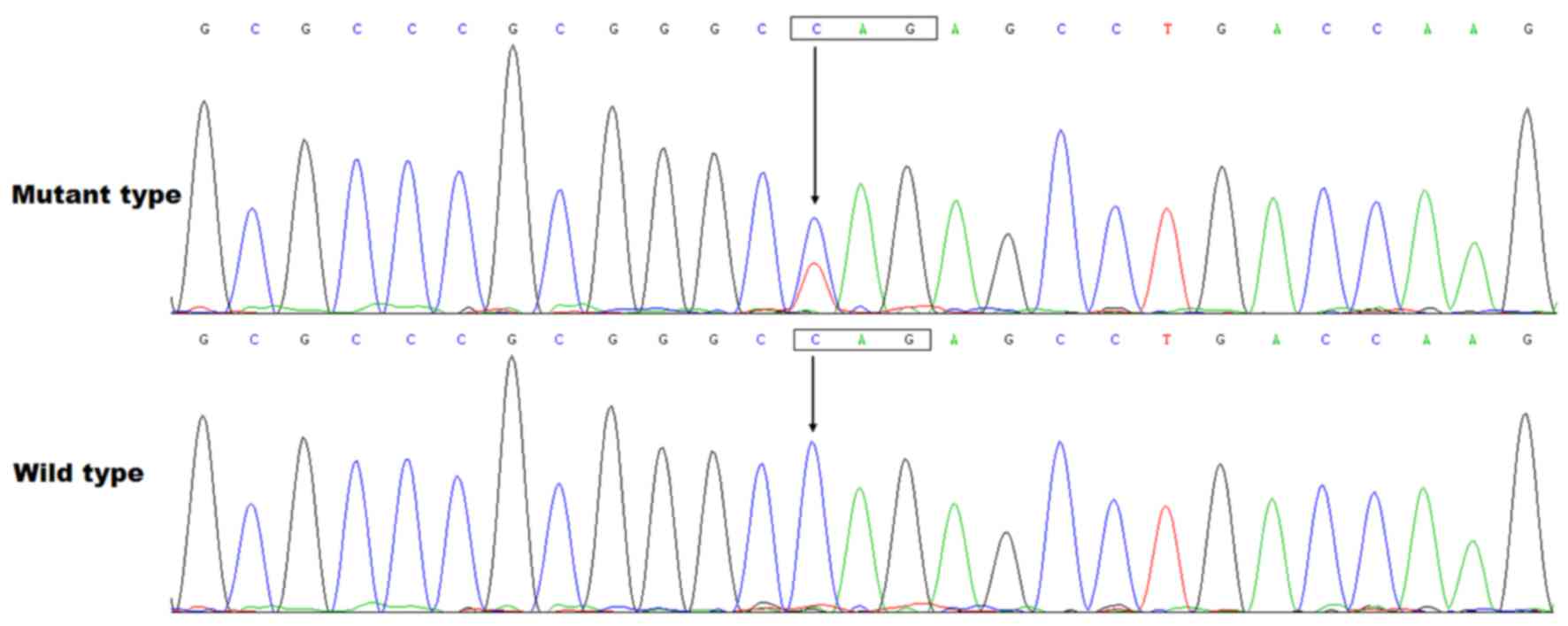
mutation in one patient, with a mutational prevalence of ~0.56%.
More specifically, a substitution of thymine for cytosine in the
first nucleotide of codon 118 (c.352C>T), predicting the
transition of the codon coding for glutamine into a stop codon at
amino acid position 118 (p.Q118X), was detected in a 3-month-old
female with DORV and VSD. The sequence chromatograms depicting the
heterozygous MESP1 mutation in addition to its control
sequence are presented in Fig. 1.
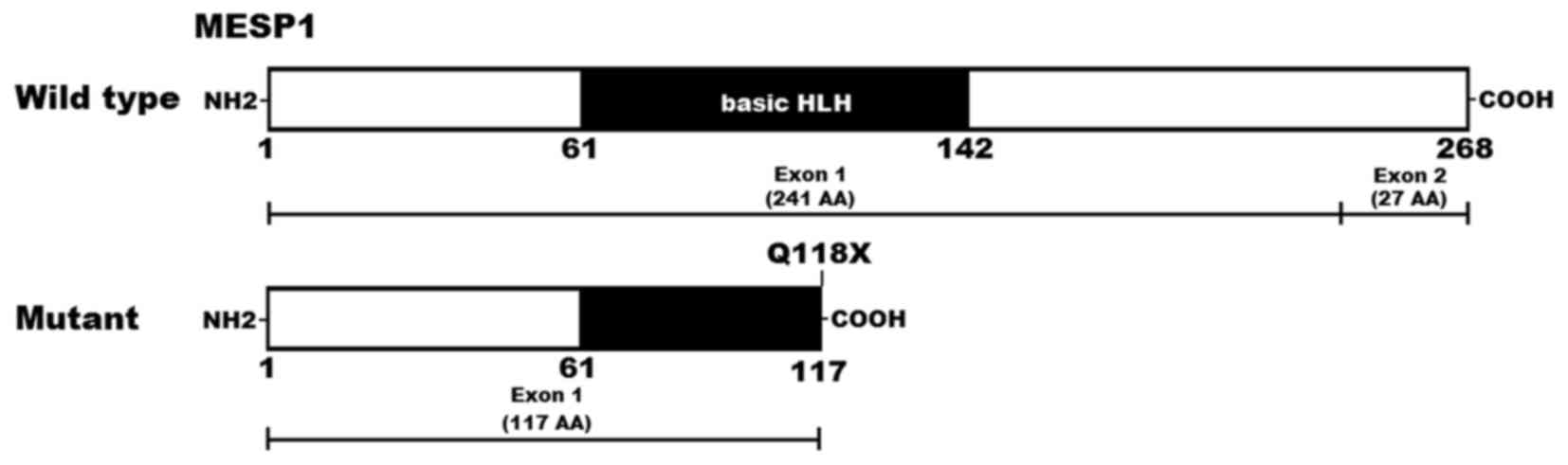
A schematic diagram displaying the location of the identified
mutation and the bHLH structural domain of the MESP1 protein is
presented in Fig. 2 (48). The nonsense mutation was not
identified in the 400 reference chromosomes or in the SNP, 1000 GP
and HGM databases (consulted July 5th, 2016). The mutation carrier
had no family history of CHD and her parents had no CHD. Genetic
screening of the mutation carrier's parents identified that the
mutation was absent in her parents, indicating that it is a de
novo mutation.
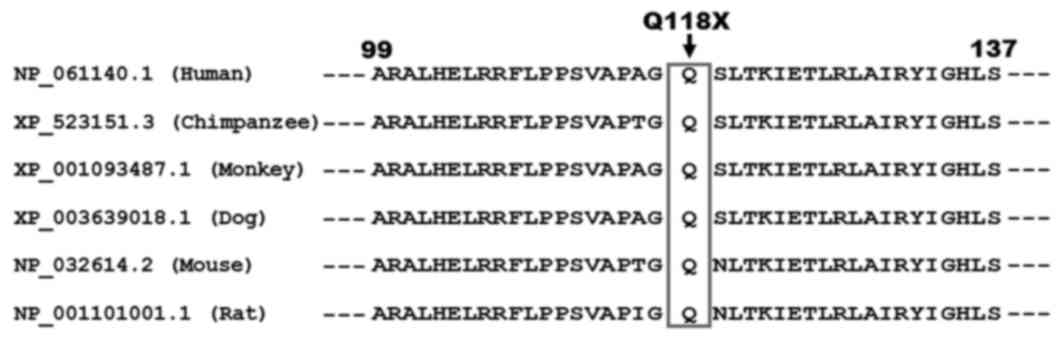
Alignment of multiple MESP1 sequences
across species
As presented in Fig.
3, a cross-species alignment of the MESP1 protein sequences
indicated that the altered amino acid glutamine at position 118 was
completely conserved evolutionarily.
Transcriptional activation of
Q118X-mutant MESP1
It has been demonstrated that MESP1 binds to the
promoter regions containing putative bHLH-binding sites (Eboxes) of
several transcription factors (41). However, MESP1 does not bind to
Eboxes alone, it forms heterodimers with E47 or E12 (E47 and E12
are two isoforms of TCF3, a member of the ubiquitous E-protein
family of bHLH transcription factors) in order to activate
transcription. In addition, it has been demonstrated that the
MESP1/E47 heterodimer activates transcription significantly more
than either one alone (41). Thus,
in order to assess the effect of a mutation on MESP1
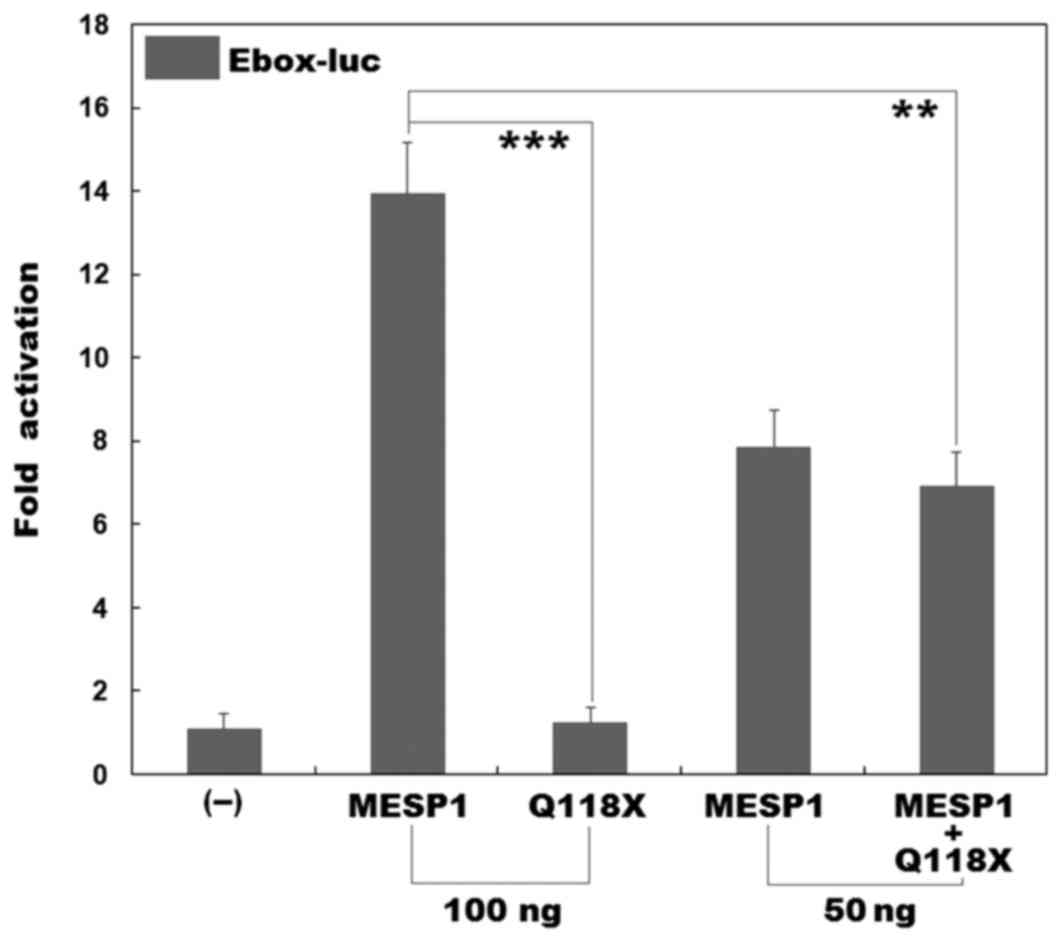
transcriptional activity, E47 was used in the present study. As
presented in Fig. 4, in the
presence of E47, the wild-type MESP1 and the Q118X-mutant MESP1
(Q118X) activated the Ebox-containing promoter by ~14-fold and
~1-fold, respectively. When wild-type MESP1 was co-expressed with
the same amount of Q118X-mutant MESP1, the induced transcriptional
activation was ~7-fold. These results indicate that mutant MESP1
does not have transcriptional activity or a dominant-negative
effect on its wild-type counterpart.
Discussion
In the present study, a novel heterozygous mutation
in MESP1, p.Q118X, was discovered in a patient with DORV and
VSD. The mutant allele was absent in the unaffected relatives
examined and in 400 control chromosomes. A cross-species alignment
of the MESP1 protein sequences indicated that the altered amino
acid was completely conserved evolutionarily. Biological assays
demonstrated that mutant MESP1 had no transcriptional activity.
Therefore, genetically compromised MESP1 may predispose an
individual to DORV and VSD.
The MESP1 gene maps on to human chromosome
15q26.1, which contains 2 exons and codes for a bHLH transcription
factor with 268 amino acids that is crucial for normal
cardiovascular development (38).
The bHLH domain is required for DNA sequence recognition and
binding to the consensus motif of CATATG (Ebox) within the
promoters of target genes; it is also responsible for
protein-protein interactions (38,41,48).
However, MESP1 do not bind to Eboxes alone and instead form
heterodimers with E47 or E12 in order to activate transcription
(41). E47 and E12 are two
isoforms of TCF3, a member of the ubiquitous E-protein family of
bHLH transcription factors (41).
It has been demonstrated that the MESP1/E47 heterodimer activates
transcription significantly more using a previously identified
MESP1 binding Ebox motif in the DKK1 enhancer when compared with
either one alone (41). In the
present study, the p.Q118X mutation detected in a patient with CHD
was located in the bHLH domain and is predicted to produce a
truncated protein with only a partial bHLH domain left. Thus it may
prevent the transcriptional activation of MESP1, which was verified
by functional analysis. Overall, these results together with
previous reports (41,45) strongly suggest that a MESP1
loss-of-function mutation is an alternative molecular mechanism
underpinning CHD.
An association between genetically defective
MESP1 and enhanced susceptibility to CHD has been reported
in mouse models. Genetic lineage tracing in mice indicated that
MESP1 was the earliest marker of cardiovascular progenitors,
tracing almost all of the cardiac cells including derivatives of
the primary and second heart fields, and serves a pivotal role in
cardiovascular morphogenesis, particularly during the early
specification and migration of cardiac precursors (37,38).
Homozygous MESP1-null mice suffered embryonic mortality due
to defects in heart tube formation and looping, resulting in
various degrees of cardia bifida from full to partial bifurcations,
which was most likely induced by the delay in mesodermal migration
and failure of ventral mesoderm fusion (37–40).
In addition, a number of studies have indicated that MESP1 resides
at the top of a large transcriptional hierarchy that regulates the
expression of 423 genes, representing 1.3% of the murine
transcriptome (38,41). Among the genes upregulated by
MESP1, there are a number of genes encoding cardiovascular core
transcription factors, including NKX2-5, GATA4,
TBX20 and HAND2 (41). In addition, targeted inactivation
of NKX2-5, GATA4, TBX20 or HAND2 in
mice has been demonstrated to cause embryonic lethality with
various cardiovascular developmental anomalies (49–56).
These experimental results suggest that the MESP1
loss-of-function mutation may contribute to CHD in humans.
In humans, MESP1 variations have been
causally linked to a number of CHDs. By high-resolution melting
curve analysis and sequencing, Werner et al (41) scanned the coding exons and flanking
introns of MESP1 in 647 unrelated patients with congenital
conotruncal and associated heart defects, and identified 6 rare,
nonsynonymous variants that were not seen in ethnically matched
controls, and 1 likely race-specific nonsynonymous variant.
Functional analyses identified that three of these variants reduced
the activation of transcription by MESP1. Lahm et al
(45) sequenced the coding regions
of MESP1 in 215 unrelated patients with congenital heart
disease, and identified two missense mutations in two patients that
were absent in the controls. Biological assays demonstrated that
one mutant had an enhanced transcriptional activity, however, the
other had no functional alteration. These well-established
CHD-associated MESP1 mutations are summarized in Table III. These results coupled with
those of the present study suggest that pathologic mutations in
MESP1 may predispose individuals to CHD.
| Table III.Mesoderm posterior 1 mutations
associated with congenital heart diseases in humans. |
Table III.
Mesoderm posterior 1 mutations
associated with congenital heart diseases in humans.
| Author, year | Nucleotide
change | Amino acid
change | Effect on protein
function | Cardiac structural
defects |
Familial/sporadic | Refs. |
|---|
| Werner et
al, 2016 | c.79_87 dup9 | p.P27_D29 dup | No significant
effect | VSD | Familial | (41) |
| Werner et
al, 2016 | c.209G>A | p.G70D | No significant
effect | VSD | n/a | (41) |
| Werner et
al, 2016 | c.310G>A | p.E104K | Loss of
function | TOF | Familial | (41) |
| Werner et
al, 2016 | c.359T>C | p.L120P | Loss of
function | TOF | Sporadic | (41) |
| Werner et
al, 2016 | c.436_437
delAG | p.L147PfsX9 | Loss of
function | VSD | n/a | (41) |
| Werner et
al, 2016 | c.503A>G | p.D168G | No significant
effect | VSD | n/a | (41) |
| Werner et
al, 2016 | c.804G>C | p.K268N | No significant
effect | VSD, CoA, AA | Familial | (41) |
| Lahm et al,
2013 | c.33G>C | p.E11D | Gain of
function | TOF | n/a | (45) |
| Lahm et al,
2013 | c.528A>T | p.T176S | No significant
effect | ASD | n/a | (45) |
| Zhang et al,
2017 | c.352C>T | p.Q118X | Loss of
function | DORV, VSD | Sporadic | Present study |
In conclusion, the results of the present study
indicate that there is an association between the MESP1
loss-of-function mutation and an increased susceptibility to DORV
in humans. This widens the known mutational spectrum of
MESP1 associated with CHD and highlights the potential
broader role of MESP1 in cardiovascular development and CHD.
Thus, the present study provides evidence for the potential
implications of genetic counseling and he development of
personalized therapeutic strategies for the treatment of patients
with CHD.
Acknowledgements
The present study was supported by the Key Program
for Basic Research of Shanghai, China (grant no. 14JC1405500), the
National Natural Science Fund of China (grant nos. 81470372 and
81641014), the Natural Science Fund of Shanghai, China (grant no.
16ZR1432500) and the Key Project for Basic Research of Shanghai
Chest Hospital, China (grant no. 2014YZDH10102).
References
|
1
|
Postma AV, Bezzina CR and Christoffels VM:
Genetics of congenital heart disease: The contribution of the
noncoding regulatory genome. J Hum Genet. 61:13–19. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Writing Group Members, . Mozaffarian D,
Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, Das SR, de
Ferranti S, Després JP, et al: Heart Disease and Stroke
Statistics-2016 Update: A Report From the American Heart
Association. Circulation. 133:e38–e360. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
van der Linde D, Konings EE, Slager MA,
Witsenburg M, Helbing WA, Takkenberg JJ and Roos-Hesselink JW:
Birth prevalence of congenital heart disease worldwide: A
systematic review and meta-analysis. J Am Coll Cardiol.
58:2241–2247. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhu H: Forkhead box transcription factors
in embryonic heart development and congenital heart disease. Life
Sci. 144:194–201. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bruneau BG: The developmental genetics of
congenital heart disease. Nature. 451:943–948. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Fahed AC, Gelb BD, Seidman JG and Seidman
CE: Genetics of congenital heart disease: The glass half empty.
Circ Res. 112:707–720. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Andersen TA, Troelsen Kde L and Larsen LA:
Of mice and men: Molecular genetics of congenital heart disease.
Cell Mol Life Sci. 71:1327–1352. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lalani SR and Belmont JW: Genetic basis of
congenital cardiovascular malformations. Eur J Med Genet.
57:402–413. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Patel SS and Burns TL: Nongenetic risk
factors and congenital heart defects. Pediatr Cardiol.
34:1535–1555. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zaidi S, Choi M, Wakimoto H, Ma L, Jiang
J, Overton JD, Romano-Adesman A, Bjornson RD, Breitbart RE, Brown
KK, et al: De novo mutations in histone-modifying genes in
congenital heart disease. Nature. 498:220–223. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Al Turki S, Manickaraj AK, Mercer CL,
Gerety SS, Hitz MP, Lindsay S, D'Alessandro LC, Swaminathan GJ,
Bentham J, Arndt AK, et al: Rare variants in NR2F2 cause congenital
heart defects in humans. Am J Hum Genet. 94:574–585. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Werner P, Paluru P, Simpson AM, Latney B,
Iyer R, Brodeur GM and Goldmuntz E: Mutations in NTRK3 suggest a
novel signaling pathway in human congenital heart disease. Hum
Mutat. 35:1459–1468. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xiang R, Fan LL, Huang H, Cao BB, Li XP,
Peng DQ and Xia K: A novel mutation of GATA4 (K319E) is responsible
for familial atrial septal defect and pulmonary valve stenosis.
Gene. 534:320–323. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Shi LM, Tao JW, Qiu XB, Wang J, Yuan F, Xu
L, Liu H, Li RG, Xu YJ, Wang Q, et al: GATA5 loss-of-function
mutations associated with congenital bicuspid aortic valve. Int J
Mol Med. 33:1219–1226. 2014.PubMed/NCBI
|
|
15
|
Huang RT, Xue S, Xu YJ, Zhou M and Yang
YQ: Somatic GATA5 mutations in sporadic tetralogy of Fallot. Int J
Mol Med. 33:1227–1235. 2014.PubMed/NCBI
|
|
16
|
Wang X, Ji W, Wang J, Zhao P, Guo Y, Xu R,
Chen S and Sun K: Identification of two novel GATA6 mutations in
patients with nonsyndromic conotruncal heart defects. Mol Med Rep.
10:743–748. 2014.PubMed/NCBI
|
|
17
|
Zhao L, Ni SH, Liu XY, Wei D, Yuan F, Xu
L, Xin-Li, Li RG, Qu XK, Xu YJ, et al: Prevalence and spectrum of
Nkx2.6 mutations in patients with congenital heart disease. Eur J
Med Genet. 57:579–586. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cowan J, Tariq M and Ware SM: Genetic and
functional analyses of ZIC3 variants in congenital heart disease.
Hum Mutat. 35:66–75. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Qu XK, Qiu XB, Yuan F, Wang J, Zhao CM,
Liu XY, Zhang XL, Li RG, Xu YJ, Hou XM, et al: A novel NKX2.5
loss-of-function mutation associated with congenital bicuspid
aortic valve. Am J Cardiol. 114:1891–1895. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wei D, Gong XH, Qiu G, Wang J and Yang YQ:
Novel PITX2c loss-of-function mutations associated with complex
congenital heart disease. Int J Mol Med. 33:1201–1208.
2014.PubMed/NCBI
|
|
21
|
Homsy J, Zaidi S, Shen Y, Ware JS, Samocha
KE, Karczewski KJ, DePalma SR, McKean D, Wakimoto H, Gorham J, et
al: De novo mutations in congenital heart disease with
neurodevelopmental and other congenital anomalies. Science.
350:1262–1266. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Li Y, Klena NT, Gabriel GC, Liu X, Kim AJ,
Lemke K, Chen Y, Chatterjee B, Devine W, Damerla RR, et al: Global
genetic analysis in mice unveils central role for cilia in
congenital heart disease. Nature. 521:520–524. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Guimier A, Gabriel GC, Bajolle F, Tsang M,
Liu H, Noll A, Schwartz M, El Malti R, Smith LD, Klena NT, et al:
MMP21 is mutated in human heterotaxy and is required for normal
left-right asymmetry in vertebrates. Nat Genet. 47:1260–1263. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Pan Y, Geng R, Zhou N, Zheng GF, Zhao H,
Wang J, Zhao CM, Qiu XB, Yang YQ and Liu XY: TBX20 loss-of-function
mutation contributes to double outlet right ventricle. Int J Mol
Med. 35:1058–1066. 2015.PubMed/NCBI
|
|
25
|
Li X, Wang G, An Y, Li H, Li Y and Wu C:
Association between sequence variations in RCAN1 promoter and the
risk of sporadic congenital heart disease in a Chinese population.
Pediatr Cardiol. 36:1393–1399. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Yang J, Zhu M, Wang Y, Hou X, Wu H, Wang
D, Shen H, Hu Z and Zou J: Whole-exome sequencing identify a new
mutation of MYH7 in a Chinese family with left ventricular
noncompaction. Gene. 58:138–142. 2015. View Article : Google Scholar
|
|
27
|
Zheng J, Li F, Liu J, Xu Z, Zhang H, Fu Q,
Wang J and Sun K: Investigation of somatic NKX2-5 mutations in
Chinese children with congenital heart disease. Int J Med Sci.
12:538–543. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Wang J, Mao JH, Ding KK, Xu WJ, Liu XY,
Qiu XB, Li RG, Qu XK, Xu YJ, Huang RT, et al: A novel NKX2.6
mutation associated with congenital ventricular septal defect.
Pediatr Cardiol. 36:646–656. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Pan Y, Wang ZG, Liu XY, Zhao H, Zhou N,
Zheng GF, Qiu XB, Li RG, Yuan F, Shi HY, et al: A novel TBX1
loss-of-function mutation associated with congenital heart disease.
Pediatr Cardiol. 36:1400–1410. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sun YM, Wang J, Qiu XB, Yuan F, Xu YJ, Li
RG, Qu XK, Huang RT, Xue S and Yang YQ: PITX2 loss-of-function
mutation contributes to tetralogy of Fallot. Gene. 577:258–264.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lu CX, Gong HR, Liu XY, Wang J, Zhao CM,
Huang RT, Xue S and Yang YQ: A novel HAND2 loss-of-function
mutation responsible for tetralogy of Fallot. Int J Mol Med.
37:445–451. 2016.PubMed/NCBI
|
|
32
|
Cao Y, Wang J, Wei C, Hou Z, Li Y, Zou H,
Meng M, Wang W and Jiang L: Genetic variations of NKX2-5 in
sporadic atrial septal defect and ventricular septal defect in
Chinese Yunnan population. Gene. 575:29–33. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Chen J, Qi B, Zhao J, Liu W, Duan R and
Zhang M: A novel mutation of GATA4 (K300T) associated with familial
atrial septal defect. Gene. 575:473–477. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Yoshida A, Morisaki H, Nakaji M, Kitano M,
Kim KS, Sagawa K, Ishikawa S, Satokata I, Mitani Y, Kato H, et al:
Genetic mutation analysis in Japanese patients with non-syndromic
congenital heart disease. J Hum Genet. 61:157–162. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Sun YM, Wang J, Qiu XB, Yuan F, Li RG, Xu
YJ, Qu XK, Shi HY, Hou XM, Huang RT, et al: A HAND2
loss-of-function mutation causes familial ventricular septal defect
and pulmonary stenosis. G3 (Bethesda). 6:987–992. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
McCulley DJ and Black BL: Transcription
factor pathways and congenital heart disease. Curr Top Dev Biol.
100:253–277. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Saga Y, Kitajima S and Miyagawa-Tomita S:
Mesp1 expression is the earliest sign of cardiovascular
development. Trends Cardiovasc Med. 10:345–352. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Bondue A and Blanpain C: Mesp1: A key
regulator of cardiovascular lineage commitment. Circ Res.
107:1414–1427. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Saga Y: Genetic rescue of segmentation
defect in MesP2-deficient mice by MesP1 gene replacement. Mech Dev.
75:53–66. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Saga Y, Miyagawa-Tomita S, Takagi A,
Kitajima S, Miyazaki JI and Inoue T: MesP1 is expressed in the
heart precursor cells and required for the formation of a single
heart tube. Development. 126:3437–3447. 1999.PubMed/NCBI
|
|
41
|
Werner P, Latney B, Deardorff MA and
Goldmuntz E: MESP1 mutations in patients with congenital heart
defects. Hum Mutat. 37:308–314. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schott JJ, Benson DW, Basson CT, Pease W,
Silberbach GM, Moak JP, Maron BJ, Seidman CE and Seidman JG:
Congenital heart disease caused by mutations in the transcription
factor NKX2-5. Science. 281:108–111. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Garg V, Kathiriya IS, Barnes R,
Schluterman MK, King IN, Butler CA, Rothrock CR, Eapen RS,
Hirayama-Yamada K, Joo K, et al: GATA4 mutations cause human
congenital heart defects and reveal an interaction with TBX5.
Nature. 424:443–447. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Kirk EP, Sunde M, Costa MW, Rankin SA,
Wolstein O, Castro ML, Butler TL, Hyun C, Guo G, Otway R, et al:
Mutations in cardiac T-box factor gene TBX20 are associated with
diverse cardiac pathologies, including defects of septation and
valvulogenesis and cardiomyopathy. Am J Hum Genet. 81:280–291.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lahm H, Deutsch MA, Dreßen M, Doppler S,
Werner A, Hörer J, Cleuziou J, Schreiber C, Böhm J, Laugwitz KL, et
al: Mutational analysis of the human MESP1 gene in patients with
congenital heart disease reveals a highly variable sequence in exon
1. Eur J Med Genet. 56:591–598. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Wang XH, Huang CX, Wang Q, Li RG, Xu YJ,
Liu X, Fang WY and Yang YQ: A novel GATA5 loss-of-function mutation
underlies lone atrial fibrillation. Int J Mol Med. 31:43–50.
2013.PubMed/NCBI
|
|
47
|
David R, Brenner C, Stieber J, Schwarz F,
Brunner S, Vollmer M, Mentele E, Müller-Höcker J, Kitajima S,
Lickert H, et al: MesP1 drives vertebrate cardiovascular
differentiation through Dkk-1-mediated blockade of Wnt-signalling.
Nat Cell Biol. 10:338–345. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Shi X, Zirbes KM, Rasmussen TL, Ferdous A,
Garry MG, Koyano-Nakagawa N and Garry DJ: The transcription factor
Mesp1 interacts with cAMP-responsive element binding protein 1
(Creb1) and coactivates Ets variant 2 (Etv2) gene expression. J
Biol Chem. 290:9614–9625. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Lyons I, Parsons LM, Hartley L, Li R,
Andrews JE, Robb L and Harvey RP: Myogenic and morphogenetic
defects in the heart tubes of murine embryos lacking the homeo box
gene Nkx2-5. Genes Dev. 9:1654–1666. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Terada R, Warren S, Lu JT, Chien KR,
Wessels A and Kasahara H: Ablation of Nkx2-5 at mid-embryonic stage
results in premature lethality and cardiac malformation. Cardiovasc
Res. 91:289–299. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Furtado MB, Wilmanns JC, Chandran A, Tonta
M, Biben C, Eichenlaub M, Coleman HA, Berger S, Bouveret R, Singh
R, et al: A novel conditional mouse model for Nkx2-5 reveals
transcriptional regulation of cardiac ion channels.
Differentiation. 91:29–41. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Molkentin JD, Lin Q, Duncan SA and Olson
EN: Requirement of the transcription factor GATA4 for heart tube
formation and ventral morphogenesis. Genes Dev. 11:1061–1072. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Kuo CT, Morrisey EE, Anandappa R, Sigrist
K, Lu MM, Parmacek MS, Soudais C and Leiden JM: GATA4 transcription
factor is required for ventral morphogenesis and heart tube
formation. Genes Dev. 11:1048–1060. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Singh MK, Christoffels VM, Dias JM, Trowe
MO, Petry M, Schuster-Gossler K, Bürger A, Ericson J and Kispert A:
Tbx20 is essential for cardiac chamber differentiation and
repression of Tbx2. Development. 132:2697–2707. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Srivastava D, Thomas T, Lin Q, Kirby ML,
Brown D and Olson EN: Regulation of cardiac mesodermal and neural
crest development by the bHLH transcription factor, dHAND. Nat
Genet. 16:154–160. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Yamagishi H, Olson EN and Srivastava D:
The basic helix-loop-helix transcription factor, dHAND, is required
for vascular development. J Clin Invest. 105:261–270. 2000.
View Article : Google Scholar : PubMed/NCBI
|