Introduction
Chemotherapy is an important cancer treatment and
numerous anti-cancer agents have been developed. However, the
adverse effects and systemic toxicity induced by these agents
limits their application (1,2).
Cisplatin (cisdiamminedichloroplatinum II; CP) is a
platinum-containing anticancer therapeutic that is used for the
treatment of various human carcinomas, including head, oral, lung
and neck cancer, metastatic tumors of testis and ovaries,
progressed bladder cancer and other solid tumors. The exact
mechanism underlying CP-induced toxicity remains to be fully
elucidated. Previous studies have demonstrated that the anticancer
behavior of CP originates from its capacity to attach to the N-7
position of purine bases of cellular DNA causing mono-adducts
formation, which are converted into inter- and intra-strand cross
links with a reaction at the secondary reactive site of drug
together with the second nucleobase (3–5).
Mitochondria have been revealed to be the cellular power plants
(6,7). A direct association between
mitochondrial dysfunction and the toxicity of chemotherapeutic
agents has been demonstrated (8),
and mitochondria are now considered anticancer drug targets.
Previous in vitro studies have revealed that CP-treated rat
hepatic cells undergo alterations to mitochondrial structure and
function (9,10). These alterations may be crucial in
strengthening various aspects of CP hepatotoxicity. Natural
antioxidants have been investigated as potential nutraceuticals to
minimize the adverse effects and increase the efficacy of
chemotherapeutic agents (11).
Quercetin (3,3′, 4′, 5,7-pentahydroxyflavone; QR) is a large class
of polyphenolic compounds ubiquitously present in plants and food
sources. It is primarily present in vegetables, fruits, red wine,
tea and other aromatic plants (12). QR has been investigated as a
therapeutic agent to ameliorate various toxicities, including
nephrotoxicity (13),
cardiotoxicity (14),
neurotoxicity (15) and
hepatotoxicity (16).
In addition, QR has been reported to contribute to
various pharmacological and biological activities, including
antimicrobial (17), antioxidant
(18), anti-inflammatory (19) and anticancer activities (20). It has been demonstrated to inhibit
oxidative stress-induced mitochondrial damage (21). The present study aimed to
investigate the effects of QR on CP-induced mitochondrial
dysfunction using an in vitro model.
Materials and methods
Chemicals
4-amino-3-hydroxy-1-naphtalenesulfonic acid (ANSA),
bovine serum albumin (BSA), butylated hydroxy toluene (BHT),
1-chloro-2, 4 dinitrobenzene (CDNB), 2,6, dichlorophenol
indophenols (DCIP), 2,4-dinitrophenyl hydrazine (DNPH),
5,5′-dithiobis (2-nitrobenzoicacid) (DTNB),
ethylenediaminetetraacetic acid (EDTA), ethylene glycol-O, -O'-bis,
(2-Aminoethyl) tetraacetic acid epinephrine, reduced glutathione
(GSH), hydrogen peroxide (H2O2), nicotinamide
adenine dinucleotide reduced (NADH), nicotinamide adenine
dinucleotide phosphate reduced tetra sodium salt (NADPH),
o-phoshoric acid (OPA), thiobarbituric acid (TBA) and trichloro
acetic acid (TCA) were purchased from Sigma-Aldrich; Merck
Millipore (Darmstadt, Germany). CP and QR were obtained from Dr
Reddy's Laboratories, Ltd. (Hyderabad, India) and HiMedia
Laboratories Pvt. Ltd. (Mumbai, India), respectively. General
chemicals were purchased from Sigma-Aldrich; Merck Millipore, Sisco
Research Laboratories Pvt. Ltd. (Mumbai, India) and Merck Ltd.
India (Mumbai, India).
Animals
Male Wistar rats (n=24; weight, 180–250 g) were
acquired from the Central Animal House of Jamia Hamdard (New Delhi,
India). The rats were acclimatized for a week prior to the
initiation of the experiments. Animals were housed at a temperature
of 22±2°C and a relative humidity of 65±10% under a 12-h light/dark
cycle, and had ad libitum access to standard rodent food and
deionized water. Experiments were performed according to the
standard guidelines of Institutional Animal Ethics Committee of
Jamia Hamdard (New Delhi, India). The study was approved by the
Institutional Animal Ethics Committee of Jamia Hamdard.
Mitochondrial preparation
Mitochondria were isolated by differential
centrifugation, as previously described (22). Briefly, liver from anaesthtized
(Nembutal, 150 mg/kg, i.p., Sigma-Aldrich) adult rats were
excised and homogenized using a mechanical Potter Elvehjem
homogenizer in an ice-cold isolation buffer containing 0.25 M
sucrose, 1 mM EDTA adjusted with Tris to pH 7.4, and centrifuged at
800 × g for 5 min at 4°C. The supernatant was centrifuged at 5,100
× g for 4 min at 4°C. Subsequently, the obtained pellet was
resuspended in a 0.25 M sucrose solution adjusted with Tris to pH
7.4, and centrifuged at 12,300 × g for 2 min at 4°C. Finally, the
pellet was resuspended in a 0.25 M sucrose solution adjusted with
Tris to pH 7.4, centrifuged at 12,300 × g for 10 min (4°C) and
resuspended in a buffer containing 0.25 M sucrose, 0.5 mM EDTA
adjusted with Tris to pH 7.4. The protein concentration of the
stock mitochondrial stock preparation was 4.5 mg/ml, as determined
by Waseem and Parvez (22).
Experimental design
(pre-incubation)
For in vitro investigations of CP-induced
hepatic mitotoxicity and its modulation by QR, mitochondrial
samples were analyzed as following: Group I (untreated control),
group II (QR), group III (CP) and group IV (CP with QR
pre-treatment). In group IV, liver mitochondria were pre-treated
with 50 µM QR at 37°C for 1 h prior to exposure to 100 µg/ml CP for
1 h (22). The concentration of QR
was selected according to previous in vitro studies on
hepatic and non-hepatic cells (17,21).
The schedule was designed so that the end point of all groups
occurred concurrently.
Evaluation of mitochondrial lipid
peroxidation (LPO)
LPO was quantified using the protocol described by
Waseem and Parvez (22). The
reaction mixture consisted of 0.01 M BHT, 6.7 mg/ml TBA, 1% chilled
OPA and 250 µl mitochondrial preparation. The rate of LPO was
determined as nmoles thiobarbituric acid reactive substances
formed/h/g of tissue using a molar extinction coefficient of
1.56×105 M−1cm−1.
Estimation of mitochondrial protein
oxidation (PC)
PC content was assessed using the protocol described
by Waseem and Parvez (22).
Mitochondria (2 mg/ml) were mixed with 0.01 M DNPH in 2 M HCl for 1
h at room temperature and precipitated with 60 mg/ml TCA. The
protein pellet was washed two or three times with a solution of
ethanol/ethyl acetate (1:1 ratio, v/v). Proteins were subsequently
solubilized in 6 M guanidine hydrochloride and 50% formic acid, and
centrifuged at 10,000 × g for 5 min at 4°C. The carbonyl level was
quantified spectrophotometrically at a wavelength of 360 nm.
Results were expressed as nmoles DNPH incorporated/mg protein using
a molar extinction coefficient of 21,000
M−1cm−1.
Determination of mitochondrial
GSH
The GSH level was assessed according to the
procedure of Tabassum et al (23). Mitochondria were primarily
precipitated with 40 mg/ml sulphosalicylic acid and were maintained
at 4°C for 1 h, followed by centrifugation at 1,500 × g for 15 min
at 4°C. The reaction mixture (total volume, 3 ml) consisted of 100
mM sodium phosphate buffer (pH 7.4), 0.01 M DTNB and 400 µl
mitochondria stock preparation. The absorbance of the reacted
product was measured at a wavelength of 412 nm on a dual-beam
spectrophotometer. The GSH content was expressed as µmoles GSH/g
tissue.
Estimation of mitochondrial
non-protein-bound thiols (NP-SH)
NP-SH levels were measured according to the protocol
of Waseem and Parvez (22), with
certain minor modifications. Mitochondria (1–2 mg/ml) were
precipitated with 400 mg/ml TCA and subsequently centrifuged at
3000 × g for 15 min at 4°C. Following this, 400 mM Tris buffer (pH
8.9) and 10 mM DTNB were added to the supernatant. Absorbance was
measured at a wavelength of 412 nm, and results were expressed as
µmoles NP-SH/g tissue using a molar extinction coefficient of
13,100 M−1 cm−1.
Activity of mitochondrial glutathione
S-transferase (GST)
The method of Waseem and Parvez (22) was used to evaluate GST activity,
with certain minor modifications. The reaction mixture consisted of
100 mM sodium phosphate buffer (pH 7.4), 10 mM GSH, 10 mM CDNB and
100 µl mitochondrial suspension. Absorbance was measured at a
wavelength of 340 nm at 30 sec intervals for 3 min, and results
were expressed as µmoles CDNB conjugate formed/min/mg protein using
a molar extinction coefficient of 9.6×103
M−1cm−1.
Kinetics of mitochondrial glutathione
peroxidase (GPx)
The method of Waseem and Parvez (22) was used to evaluate GPx activity,
with certain minor modifications. The reaction mixture consisted of
100 mM sodium phosphate buffer, 1 mM EDTA, 1 mM sodium azide, 10 mM
GSH, 2 mM NADPH, 10 µl H2O2 (10.32 M) and 4–5
mg/ml mitochondrial suspension, in a final volume of 2 ml. NADPH
oxidation was measured kinetically at a wavelength of 340 nm at 30
sec intervals for 3 min. The enzyme activity was calculated as
nmoles NADPH oxidized/min/mg protein, using a molar extinction
coefficient of 6.22×103
M−1cm−1.
Activity of mitochondrial
manganese-superoxide dismutase (Mn-SOD)
Mn-SOD activity was assessed according to the
procedure of Waseem and Parvez (23). Mitochondria (180 µl stock
preparation) were treated with 0.05 M glycine buffer (pH 10.4) and
20 mg/ml epinephrine. The enzymatic activity was measured
kinetically at a wavelength of 480 nm at 30 sec intervals for 3
min. The activity was expressed as nmoles epinephrine protected
from oxidation/min/mg protein using a molar extinction coefficient
of 4,020 M−1cm−1.
Activity of complex I
(NADH-dehydrogenase)
The procedure of King and Howard (24) was used to measure
NADH-dehydrogenase activity, with certain minor modifications. The
reaction mixture consisted of 600 µM DCIP, 2 mM glycyl glycine
buffer, 600 µM NADH and 100 µl mitochondrial stock preparation. The
absorbance was measured at a wavelength of 600 nm. The enzyme
activity was expressed as nmoles NADH oxidized/min/mg protein using
a molar extinction coefficient of 21,000 M
−1cm−1.
Activity of complex II (succinate
dehydrogenase)
Succinate dehydrogenase activity was assessed
according to the protocol of Waseem and Parvez (22). The reaction mixture consisted of
100 mM phosphate buffer (pH 7.4), 10 mg/ml BSA, 0.0015 M potassium
ferricynanide, 15 mM sodium succinate and 100 µl mitochondrial
stock preparation. The absorbance was measured for 3 min at a
wavelength of 420 nm. The enzyme activity was expressed as nmoles
succinate produced/min/mg protein using a molar extinction
coefficient of 1,000 M−1cm−1.
Activity of complex III (mitochondrial
dehydrogenase, MTT)
The MTT reduction rate was used to measure the
mitochondrial respiratory complex activity according to the method
of Kamboj et al (25), with
certain minor modifications. Briefly, 100 µg mitochondrial
preparation was suspended in 1.5 ml eppendorf tubes with ice cold
buffer C, and incubated at 37°C in the presence of 20 µl of MTT
(0.1 mg/ml). After 30 min incubation period, tubes were centrifuged
at 1,000 × g for 10 min, and the blue formazan crystals were
solubilised in 1 ml DMSO. The absorbance was measured at 595 nm.
The results were expressed as nmoles formazan formed/min/mg protein
using a molar extinction coefficient of 51,000
M−1cm−1.
Activity of complex V (total
ATPase)
Total ATPase activity was quantified by measuring
the hydrolysis rate of ATP to ADP and inorganic phosphate (Pi),
according to the protocol of Waseem and Parvez (22), with certain minor modifications.
Mitochondria (0.2 mg stock preparation) were incubated in ATPase
buffer (50 mM Tris and 5 mM MgCl2, pH 7.5) at 37°C with
5 mM ATP for 10 min. The reaction was terminated via the addition
of 100 mg/ml TCA. The suspension was subsequently centrifuged at
3,000 × g for 20 min at 4°C, and the supernatants were mixed with
0.5 ml distilled water. The reaction measuring Pi production was
initiated by adding a mixture containing 720 mM sodium bisulfite,
41.6 mM sodium sulfite and 10 mM ANSA. The enzyme activity was
measured at 660 nm and expressed as µg Pi liberated/min/mg
protein.
Protein content determination
The protein content of mitochondria was assessed
according to the protocol described by Lowry et al (26). BSA (1 mg/ml) served as the
standard.
Statistical analysis
Results are expressed as the mean ± standard error.
Data were analyzed using one-way analysis of variance followed by
Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference. Statistical analyses were
performed using GraphPad Prism software version 5 (GraphPad
Software, Inc., La Jolla, CA, USA).
Results
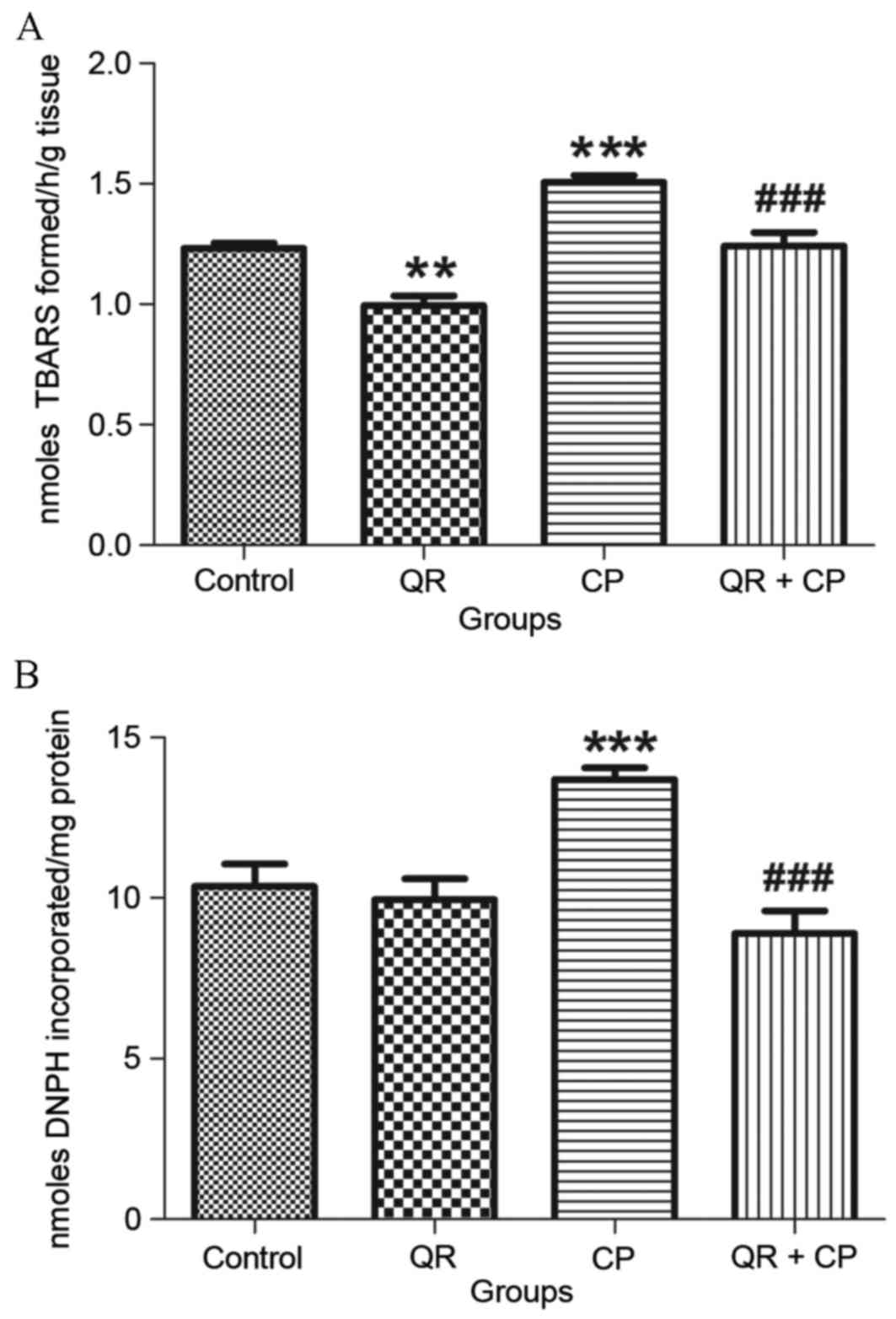
QR inhibits the CP-induced increase in
LPO and PC levels
CP treatment (group III) significantly increased LPO
(Fig. 1A) and PC (Fig. 1B) levels (P<0.001, respectively)
compared with the control group (group I). QR pre-treatment (group
IV) significantly decreased LPO and PC levels (P<0.001) compared
with CP treatment alone (group III). QR treatment alone (group II)
significantly decreased LPO levels compared with the control group
(P<0.01); however, it had no significant effect on PC
levels.
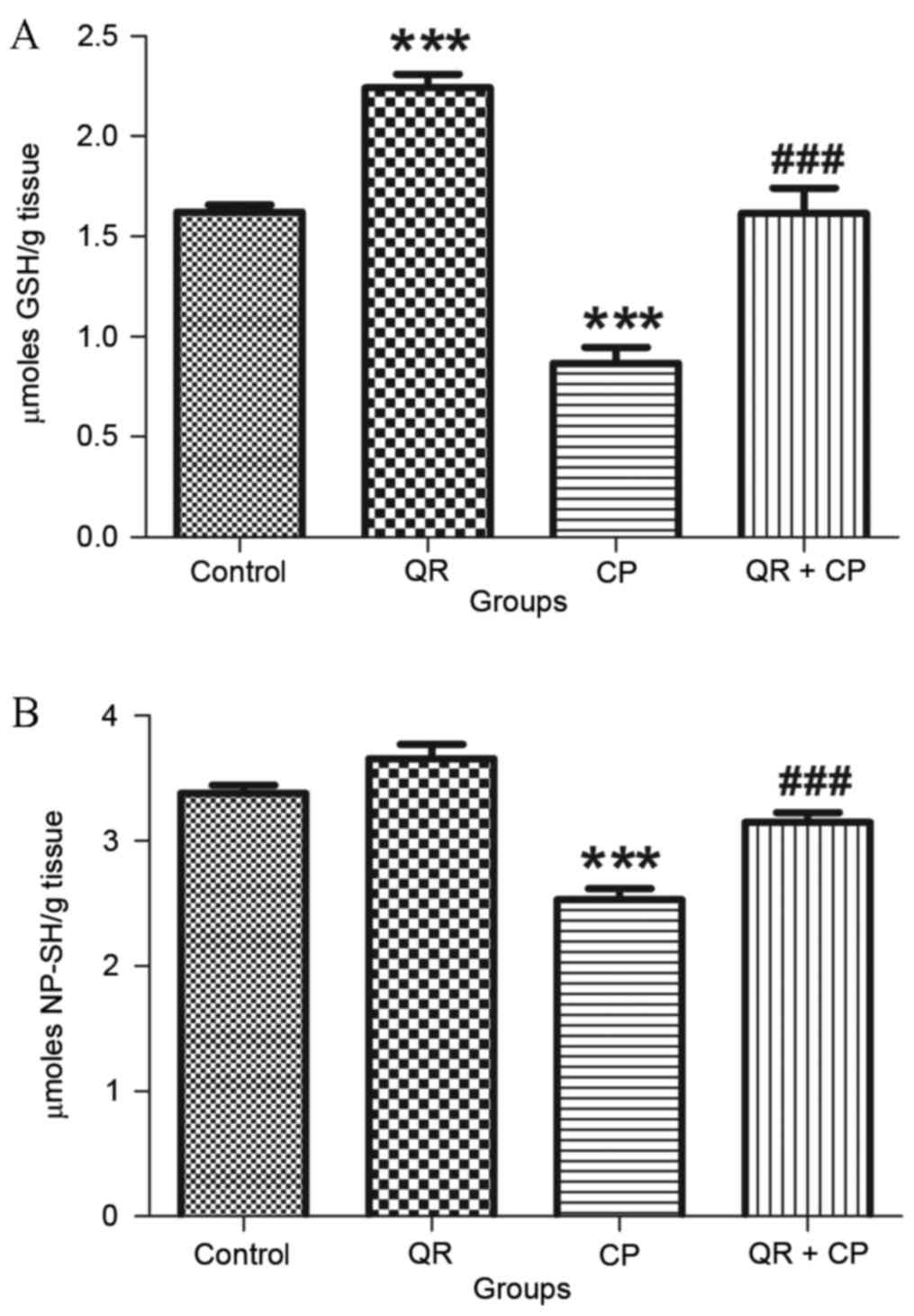
QR attenuates the CP-induced decrease
in GSH and NP-SH levels
CP treatment significantly decreased GSH (Fig. 2A) and NP-SH (Fig. 2B) levels in liver mitochondria
compared with the control group (P<0.001). QR pre-treatment
significantly attenuated these effects (P<0.001). QR alone
significantly increased GSH levels compared with the control group
(P<0.001); however, it had no significant effect on NP-SH
content.
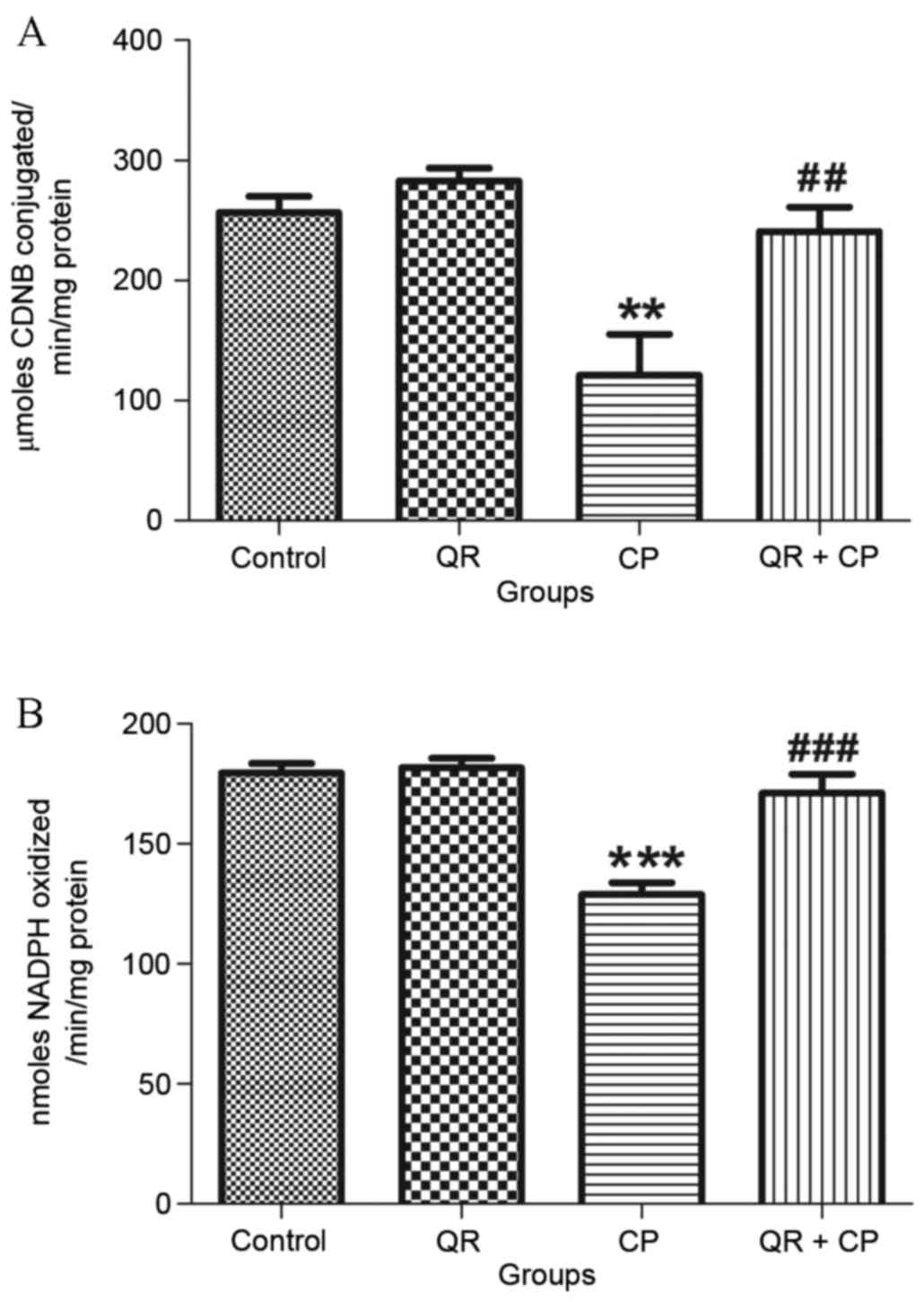
QR protects against the CP-induced
decrease in glutathione metabolizing enzyme levels
CP treatment significantly decreased GST (Fig. 3A) and GPx (Fig. 3B) activities compared with the
control group (P<0.01 and P<0.001, respectively). QR
pre-treatment significantly attenuated these effects. QR alone had
no significant effects on GST or GPx activities.
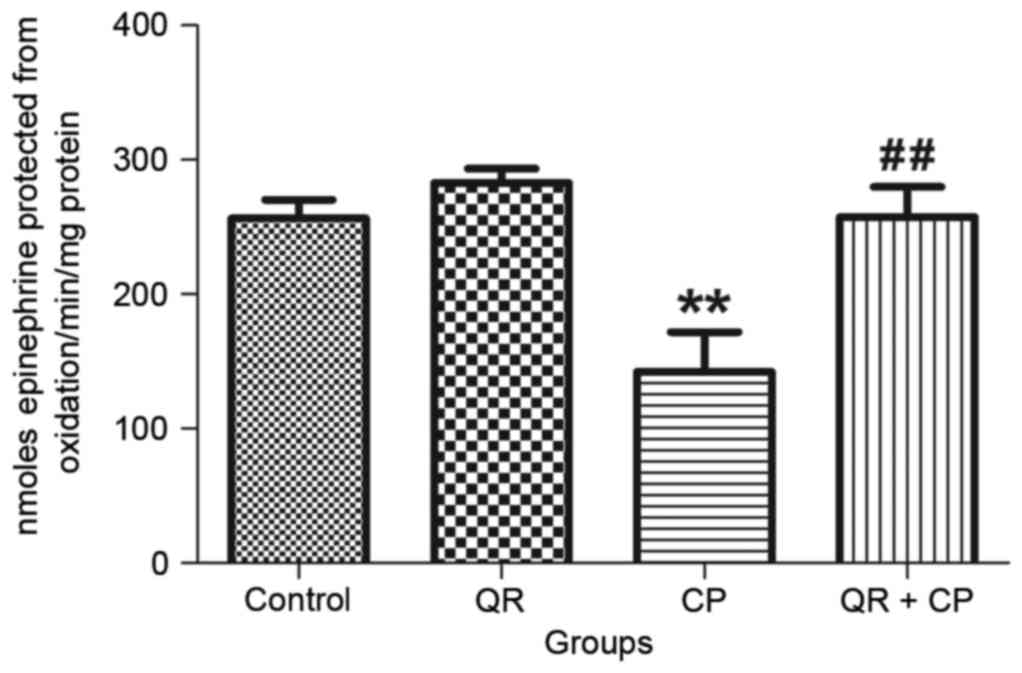
QR modulates the CP-induced decrease
in Mn-SOD activity
CP treatment significantly decreased Mn-SOD activity
(Fig. 4) in liver mitochondria
compared with the control group (P<0.01). QR pre-treatment
significantly abrogated this effect (P<0.01). QR alone had no
significant effect of Mn-SOD.
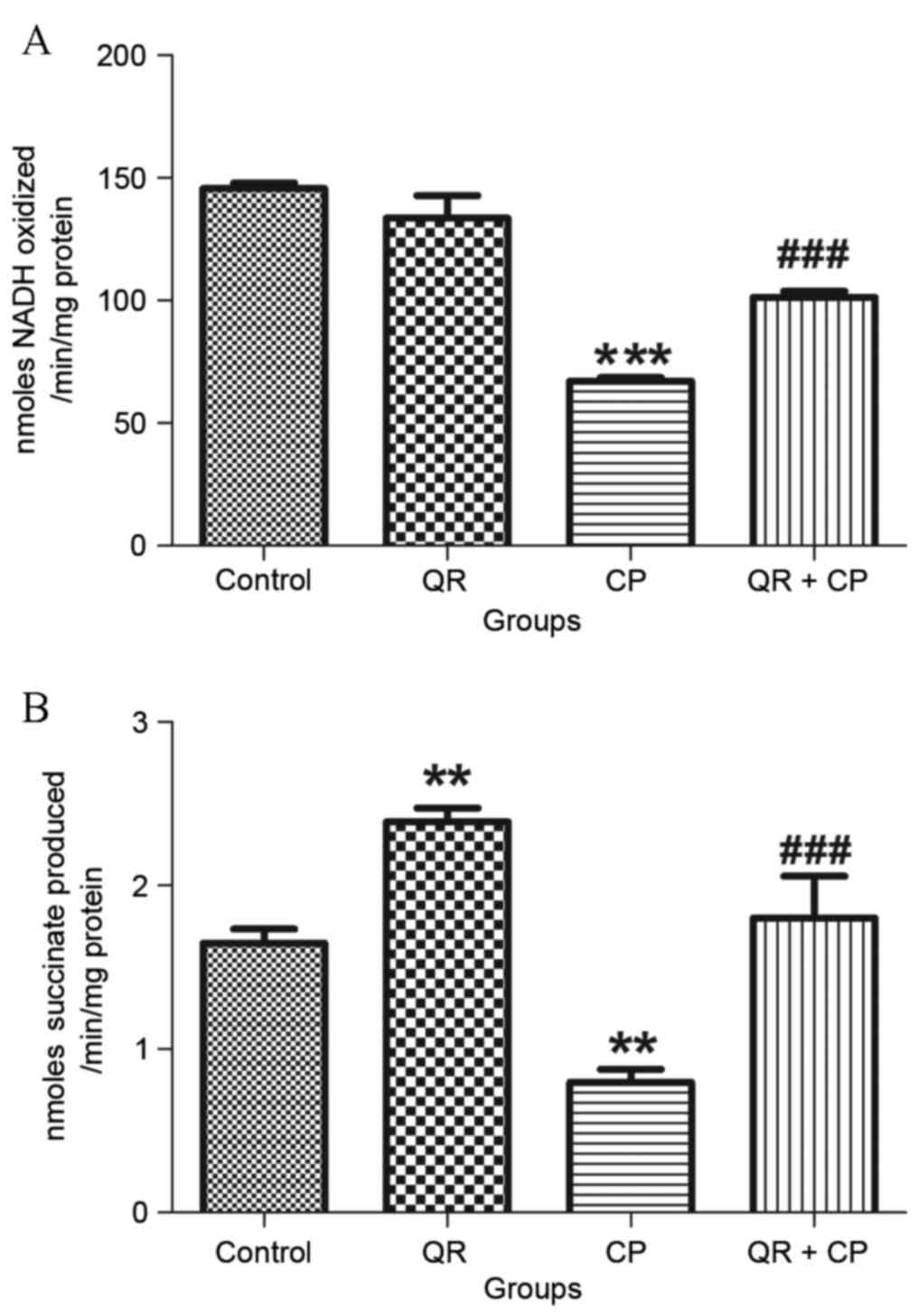
QR ameliorates the CP-induced decrease
in complex I and II enzyme activities
CP treatment significantly reduced NADH
dehydrogenase (Fig. 5A) and
succinate dehydrogenase (Fig. 5B)
activities compared with the control group (P<0.01 and
P<0.001, respectively). QR pre-treatment significantly
attenuated these effects (P<0.001). QR alone significantly
increased succinate dehydrogenase activity compared with the
control group (P<0.01); however, it had no significant effect on
NADH dehydrogenase activity.
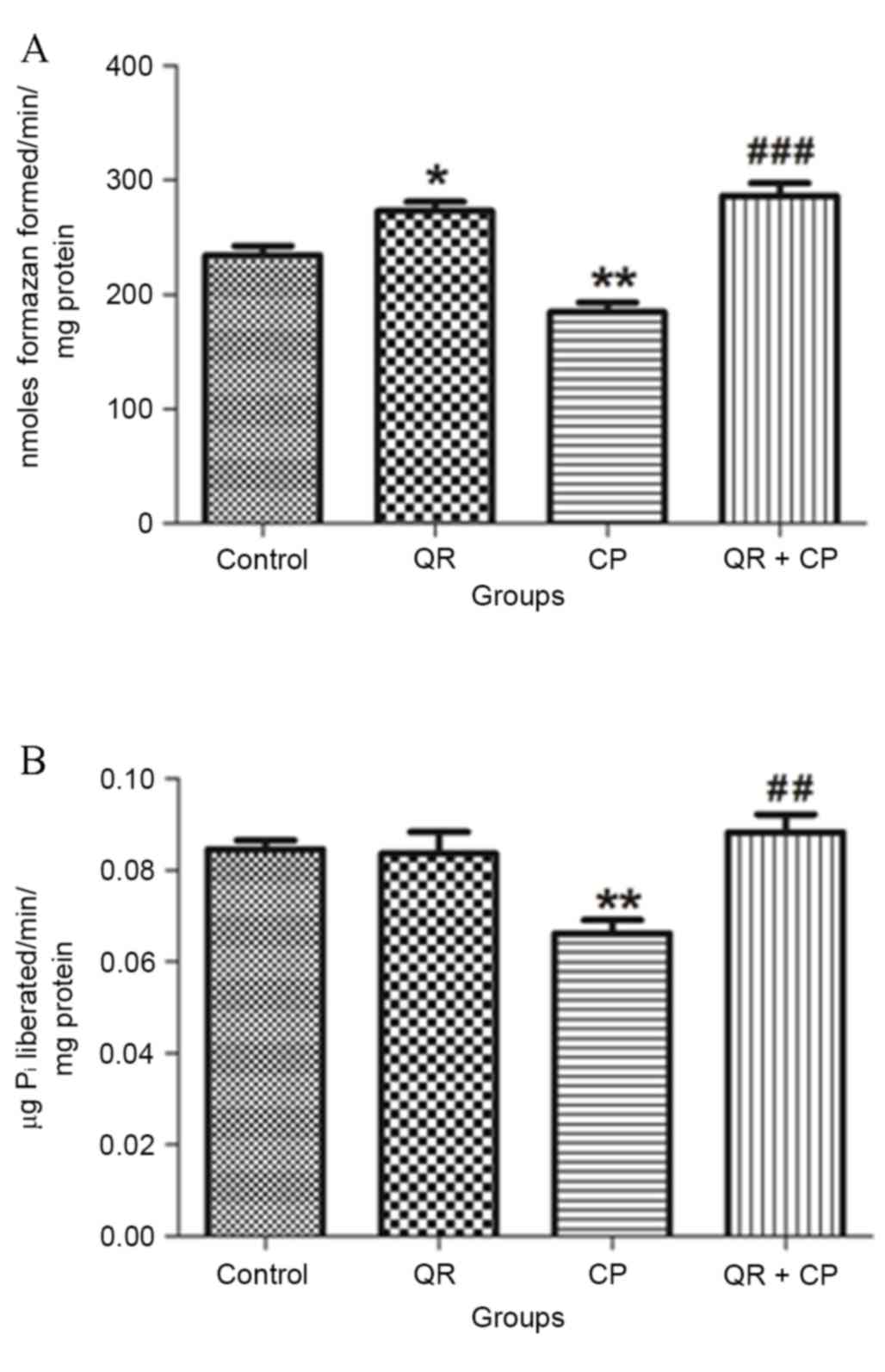
QR modulates the CP-induced inhibition
of complex III and V enzyme activities
CP treatment significantly decreased MTT ability
(Fig. 6A) and total ATPase
activity (Fig. 6B) compared with
the control group (P<0.01, respectively). QR pre-treatment
significantly abrogated these effects (P<0.01 and P<0.001,
respectively). QR alone significantly increased MTT ability
compared with the control group (P<0.05); however, it had no
significant effect on total ATPase activity.
Discussion
CP is a platinum-based heavy metal that is a
chemotherapeutic agent used for the treatment of various cancers
(22). However, the application of
CP is limited as a result of the hepatotoxicity that has been
demonstrated to develop following treatment in various animal
models. Our previous study (27)
was the first to observe that CP significantly elevated LPO levels
in isolated rat liver mitochondria. LPO has been suggested as a
primary underlying mechanism by which free radicals damage cells.
The significant increase in LPO levels may be due to its poor
antioxidant defenses or the non-stimulation of antioxidant enzymes,
due to oxidative stress. In addition, increased LPO levels or a
reduction in antioxidants have been associated with complex IV
activity decrease, which may ultimately result in
mitochondria-dependent apoptosis (28). In the present study, QR
pre-treatment was observed to significantly restore LPO levels and
alter antioxidant status. QR decreased the oxidative stress marker
LPO via reactive oxygen species (ROS) scavenging (29). In the present study, QR prevented
the CP-induced increase in LPO levels, which may sustain cellular
integrity and defense against damage due to free radicals.
PC is an extensively used biomarker and the
predominant indicator of protein carbonyl accumulation and protein
oxidation. The results of the present study support this, as PC
content increased in isolated liver mitochondria on exposure to CP.
The biomarker of oxidative protein damage is protein carbonylation,
as a result of xenobiotically-induced oxidative stress. QR
pre-treatment significantly restored PC contents in isolated liver
mitochondria. The modulatory role of natural compounds on protein
carbonylation has been demonstrated by previous studies (30).
GSH is the primary antioxidant molecule and
detoxifies various types of endogenous and exogenous toxicants,
including CP, via GSH adducts formation (22). Additionally, the redox cycle of
GSH, consisting of GSH, GPx and glutathione reductase, is crucial
in scavenging the ROS generated by CP, to protect cells from
potential toxicity and carcinogenesis (31). In the present study, CP
significantly reduced the levels of GSH in liver mitochondria. QR
treatment increased mitochondrial GSH levels and pre-treatment with
QR inhibited the CP-induced reduction in GSH levels. These results
suggested that cells protected by QR pre-treatment are less
susceptible to CP-induced mitochondrial oxidative stress.
In the present study, NP-SH levels were
significantly decreased in isolated liver mitochondria by CP
treatment, which is in accordance with the GSH results, as GSH
levels in liver tissue comprise >90% of the NP-SH pool. It is
recognized that the total cellular thiol pool is integral in
homeostasis and is additionally important in oxidative physiology.
Treatment with QR increased the mitochondrial NP-SH levels
(32) and QR pre-treatment
significantly attenuated the CP-induced reduction in NP-SH levels,
suggesting that cells protected by QR pre-treatment are less
susceptible to CP-induced mitochondrial oxidative stress.
GSTs are a group of enzymes that conjugate GSH to
structurally diverse electrophilic compounds. GST catalyzes the
conjugation of GSH via a sulfhydryl group to electrophilic centers
on a wide variety of substrates. GST is responsible for scavenging
organic peroxides, and endogenous and exogenous electrophiles.
Diverse forms of GST have been revealed to bind CP in vivo
and in vitro (33). In the
present study, it was observed that CP significantly decreased GST
activity in isolated liver mitochondria. Reduced mitochondrial GST
activity may be associated with an elevation in ROS generation
following tissue injury (22). In
the present study, QR pre-treatment restored the activity of GST.
This may provide protection from oxidative stress due to excess
O2 and H2O2. Mitochondrial
oxidative damage occurs as a results of the respiratory chain;
complex I and III are the foremost sources of superoxide anion
(O2−) (22).
Energy or ATP production by oxidative phosphorylation takes place
in mitochondria and is catalyzed by membrane-bound protein
complexes, namely NADH-dehydrogenase (complex I), succinate
dehydrogenase (complex II), cytochrome c oxidoreductase (complex
III) and total ATPases. Succinate dehydrogenase contributes only by
transferring electrons to the electron transport chain (ETC),
whereas NADH-dehydrogenase is associated with proton translocation
and electron transfer. The defect in any of the enzyme complexes
responsible for oxidative metabolism may lead to mitochondrial
cytopathy (22) and to the opening
of the mitochondrial permeability transition pore that allows the
membrane potential to dissipate resulting in uncoupling of
oxidative phosphorylation and therefore impaired cellular ATP
production. Previous studies have demonstrated that mitochondrial
dysfunctions are involved in mitochondrial toxicity induced by
platinum-based chemotherapeutic agents, including CP (2,22).
The results of the present study suggested that CP inactivated
mitochondrial complex enzymes, as indicated by a reduction in
NADH-dehydrogenase and succinate dehydrogenase activities, MTT
ability and ATPase activity. QR pre-treatment significantly
protected these complex enzymes. QR has been revealed to modulate
mitochondrial dysfunction in rodents and its property as an
antioxidant is hypothesized to be responsible for its protective
effects in mitochondria (5,9).
The alterations in the activities of mitochondrial
complex enzymes may be involved in hepatotoxicity. This may be as a
result of free radicals, as well as a reduction in mitochondrial
transcription and translation. Furthermore, it has been revealed
that mitochondrial activity interference is associated with effects
on complex enzymes, particularly complexes I and III of the ETC,
which result in increased mitochondrial electron leakage. QR
pre-treatment restored the activity of complex enzymes in liver
mitochondria. This may be due to the increase in the scavenging and
inactivation of H2O2 and hydroxyl radicals
caused by QR (34). Previous
studies have demonstrated that QR protects against mitochondrial
oxidative stress and actively increases biologically active
mitochondria in cells, following treatment with the concentration
used in the present study (5,9).
In conclusion, the results of the present study
suggested that CP exerts hepatotoxic effects through the induction
of oxidative stress, indicated by the alterations in the
concentrations of mitochondrial complex enzymes and non-enzymatic
antioxidants. The protective effect of QR was associated with its
antioxidant potential, as it potentially acts as a free radical
scavenger, LPO inhibitor and GSH activator. Further studies,
particularly molecular experiments, are required to investigate the
mitigatory effect of QR on mitochondria-mediated anticancer drug
toxicities.
Acknowledgements
The present study was supported by the University
Grants Commission Basic Science Research (grant no. F-7/91/2007, to
M.W.), the Department of Biotechnology, Government of India (DBT
BioCARe Program; grant no. BT/Bio-CARe/01/10219/2013-14, to H.T.),
the University Grants Commission, New Delhi, Government of India
[grant no. F. 41-1286/2012 (SR), to S.P.] and the Department of
Science and Technology for funding under PURSE program (to J.H.,
2016).
References
|
1
|
Kellokumpu-Lehtinen PL, Hjälm-Eriksson M,
Thellenberg-Karlsson C, Aström L, Franzen L, Marttila T, Seke M,
Taalikka M, Ginman C, et al: SPCG-13: Toxicity in patients
receiving adjuvant docetaxel + hormonal treatment after radical
radiotherapy for intermediate or high-risk prostate cancer: A
preplanned safety report of the SPCG-13 trial. Prostate Cancer
Prostatic Dis. 15:303–307. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kim JH, Jeong SJ, Kwon HY, Park SY, Lee
HJ, Lee HJ, Lieske JC and Kim SH: Decursin prevents
cisplatin-induced apoptosis via the enhancement of antioxidant
enzymes in human renal epithelial cells. Biol Pharm Bull.
33:1279–1284. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Saad JS, Benedetti M, Natile G and
Marzilli LG: Basic coordination chemistry relevant to DNA adducts
formed by the cisplatin anticancer drug. NMR studies on compounds
with sterically crowded chiral ligands. Inorg Chem. 49:5573–5583.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mantri Y, Lippard SJ and Baik MH:
Bifunctional binding of cisplatin to DNA: Why does cisplatin form
1,2-intrastrand cross-links with ag but not with GA? J Am Chem Soc.
129:5023–5030. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baruah H, Wright MW and Bierbach U:
Solution structural study of a DNA duplex containing the guanine-N7
adduct formed by a cytotoxic platinum-acridine hybrid agent.
Biochemistry. 44:6059–6070. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Khan N: Recent advancements in diagnostic
tools in mitochondrial energy metabolism diseases. Adv Med Sci.
61:244–248. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Iwata T, Nishiyama N, Nagano K, Izumi N,
Mizuguchi S, Tsukioka T, Morita R, Chung K, Hanada S and Inoue K:
Role of pulmonary resection in the diagnosis and treatment of
limited-stage small cell lung cancer: Revision of clinical
diagnosis based on findings of resected specimen and its influence
on survival. Gen Thorac Cardiovasc Surg. 60:43–52. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burgeiro A, Gajate C, Dakirel H,
Villa-Pulgarín JA, Oliveira PJ and Mollinedo F: Involvement of
mitochondrial and B-RAF/ERK signaling pathways in berberine-induced
apoptosis in human melanoma cells. Anticancer Drugs. 22:507–518.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zicca A, Cafaggi S, Mariggiò MA, Vannozzi
MO, Ottone M, Bocchini V, Caviglioli G and Viale M: Reduction of
cisplatin hepatotoxicity by procainamide hydrochloride in rats. Eur
J Pharmacol. 442:265–272. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Liu JJ, Galettis P, Farr A, Maharaj L,
Samarasinha H, McGechan AC, Baguley BC, Bowen RJ, Berners-Price SJ
and McKeage MJ: In vitro antitumour and hepatotoxicity profiles of
Au(I) and Ag(I) bidentate pyridyl phosphine complexes and
relationships to cellular uptake. J Inorg Biochem. 102:303–310.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang X, Yeung ED, Wang J, Panzhinskiy EE,
Tong C, Li W and Li J: Isoliquiritigenin, a natural anti-oxidant,
selectively inhibits the proliferation of prostate cancer cells.
Clin Exp Pharmacol Physiol. 37:841–847. 2010.PubMed/NCBI
|
|
12
|
Pandey KB and Rizvi SI: Plant polyphenols
as dietary antioxidants in human health and disease. Oxid Med Cell
Longev. 2:270–278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Ciftci O, Ozdemir I, Vardi N, Beytur A and
Oguz F: Ameliorating effects of quercetin and chrysin on
2,3,7,8-tetrachlorodibenzo- p-dioxin-induced nephrotoxicity in
rats. Toxicol Ind Health. 28:947–954. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Matouk AI, Taye A, Heeba GH and El-Moselhy
MA: Quercetin augments the protective effect of losartan against
chronic doxorubicin cardiotoxicity in rats. Environ Toxicol
Pharmacol. 36:443–450. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Haleagrahara N, Siew CJ and Ponnusamy K:
Effect of quercetin and desferrioxamine on 6-hydroxydopamine
(6-OHDA) induced neurotoxicity in striatum of rats. J Toxicol Sci.
38:25–33. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sekaran S, Kandaswamy S, Gunasekaran K,
Perumal E, Basha FY Afsar, Mohan BJ Madhan and Jagadeesan A:
Protective role of quercetin on polychlorinated biphenyls
(Aroclor-1254) induced oxidative stress and apoptosis in liver of
adult male rats. J Biochem Mol Toxicol. 26:522–532. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kallio J, Jaakkola M, Mäki M, Kilpeläinen
P and Virtanen V: Vitamin C inhibits staphylococcus aureus growth
and enhances the inhibitory effect of quercetin on growth of
Escherichia coli in vitro. Planta Med. 78:1824–1830. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Di Cesare Mannelli L, Zanardelli M, Failli
P and Ghelardini C: Oxaliplatin-induced neuropathy: Oxidative
stress as pathological mechanism. Protective effect of silibinin. J
Pain. 13:276–284. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Youn H, Jeong JC, Jeong YS, Kim EJ and Um
SJ: Quercetin potentiates apoptosis by inhibiting nuclear
factor-kappaB signaling in H460 lung cancer cells. Biol Pharm Bull.
36:944–951. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tang Y, Gao C, Xing M, Li Y, Zhu L, Wang
D, Yang X, Liu L and Yao P: Quercetin prevents ethanol-induced
dyslipidemia and mitochondrial oxidative damage. Food Chem Toxicol.
50:1194–1200. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jakubowicz-Gil J, Langner E, Wertel I,
Piersiak T and Rzeski W: Temozolomide, quercetin and cell death in
the MOGGCCM astrocytoma cell line. Chem Biol Interact. 188:190–203.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Waseem M and Parvez S: Neuroprotective
activities of curcumin and quercetin with potential relevance to
mitochondrial dysfunction induced by oxaliplatin. Protoplasma.
253:417–430. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Tabassum H, Parvez S, Pasha ST, Banerjee
BD and Raisuddin S: Protective effect of lipoic acid against
methotrexate-induced oxidative stress in liver mitochondria. Food
Chem Toxicol. 48:1973–1979. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
King TE and Howard RL: Preparations and
properties of soluble NADH dehydrogenases from cardiac muscle.
Methods Enzymol. 10:275–294. 1967. View Article : Google Scholar
|
|
25
|
Kamboj SS and Sandhir R: Protective effect
of N-acetylcysteine supplementation on mitochondrial oxidative
stress and mitochondrial enzymes in cerebral cortex of
streptozotocin-treated diabetic rats. Mitochondrion. 11:214–222.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lowry OH, Rosebrough NJ, Farr AL and
Randal RJ: Protein measurement with the Folin phenol reagent. J
Biol Chem. 193:265–275. 1951.PubMed/NCBI
|
|
27
|
Waseem M, Bhardwaj M, Tabassum H,
Raisuddin S and Parvez S: Cisplatin hepatotoxicity mediated by
mitochondrial stress. Drug Chem Toxicol. 38:452–459. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Badary OA, Abdel-Maksoud S, Ahmed WA and
Owieda GH: Naringenin attenuates cisplatin nephrotoxicity in rats.
Life Sci. 76:2125–2135. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tucci P, Cione E, Perri M and Genchi G:
All-trans-retinoic acid induces apoptosis in Leydig cells via
activation of the mitochondrial death pathway and antioxidant
enzyme regulation. J Bioenerg Biomembr. 40:315–323. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Singh R and Sharma P: Hepatoprotective
effect of curcumin on lindane-induced oxidative stress in male
Wistar rats. Toxicol Int. 18:124–129. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fedorova M, Bollineni RC and Hoffmann R:
Protein carbonylation as a major hallmark of oxidative damage:
Update of analytical strategies. Mass Spectrom Rev. 33:79–97. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Hanigan MH and Devarajan P: Cisplatin
nephrotoxicity: Molecular mechanisms. Cancer Ther. 1:47–61.
2003.PubMed/NCBI
|
|
33
|
Ognjanović BI, Djordjević NZ, Matić MM,
Obradović JM, Mladenović JM, Stajn AŠ and Saičić ZS: Lipid
peroxidative damage on Cisplatin exposure and alterations in
antioxidant defense system in rat kidneys: A possible protective
effect of selenium. Int J Mol Sci. 13:1790–1803. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Chen YR, Chen CL, Zhang L, Green-Church KB
and Zweier JL: Superoxide generation from mitochondrial NADH
dehydrogenase induces self-inactivation with specific protein
radical formation. J Biol Chem. 280:37339–37348. 2005. View Article : Google Scholar : PubMed/NCBI
|