Introduction
Glioblastoma multiforme (GBM) is one of the most
fatal forms of brain cancer, and patients with this disease develop
resistance to chemotherapy and radiotherapy, which is associated
with a poor patient prognosis (1,2).
Cisplatin is one of the first-line chemotherapeutic agents used to
treat a number of human tumors, including GBM. It targets tumor
cells by interacting with and damaging DNA, which induces
apoptosis-mediated cell death (3).
Despite an initial response, repeated clinical treatment with
cisplatin usually results in the development of chemoresistance in
tumor cells (4,5). Therefore, investigating the molecular
mechanisms underlying cisplatin-resistance and identifying
efficient combination treatments to eliminate cisplatin resistance,
are urgently required for the development of effective therapeutic
strategies for GBM.
The long non-coding RNA (lncRNA), maternally
expressed gene 3 (MEG3), is an imprinted gene that is part of the
Δ-like non-canonical notch ligand 1-MEG3 locus located on human
chromosome 14q32 (6). Although
MEG3 is expressed in a number of normal human tissues, particularly
those of the brain and pituitary, loss of MEG3 expression has been
identified in several human cancers, including bladder cancer,
glioma, hepatocellular carcinoma and additional cancers (7–10).
In addition, MEG3 was reported to be downregulated in human
meningioma when compared with normal tissues, and was associated
with tumor grade (8,11). A Previous study demonstrated that
loss of MEG3 activated autophagy in bladder cancer (9). However, the effects of MEG3 on
cisplatin resistance and autophagy in GBM remain elusive.
In the present study, MEG3 expression was induced by
cisplatin treatment of U87 cells, and increased MEG3 expression
contributed to the induction of apoptosis. In addition,
overexpression of MEG3 eliminated cisplatin-induced autophagy in
U87 cells. Furthermore, inhibition of autophagy by MEG3 enhanced
cisplatin-induced apoptosis in U87 cells. Therefore, the results
present a novel therapeutic strategy for the treatment of GBM.
Materials and methods
Cell culture
The human U87 cell line, which is likely to be a
glioblastoma cell line of unknown origin (12), was obtained from the American Type
Culture Collection (Manassas, VA, USA). U87 cells were cultured at
37°C and 95% humidity in minimum essential medium (cat. no. M2297;
Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) containing 10% fetal
bovine serum (FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA). The medium was refreshed every other day, and cells were
passaged prior to reaching 100% confluence.
Reagents
The following primary antibodies were employed in
the present study: Anti-caspase 3 (cat. no. 9662; dilution,
1:1,000; rabbit; Cell Signaling Technology, Inc., Danvers, MA,
USA); anti-GAPDH (cat. no. SC-25778; dilution, 1:2,000; rabbit;
Santa Cruz Biotechnology, Inc., Dallas, TX, USA); anti-poly (ADP)
ribose polymerase (PARP; cat. no. SC-8007; dilution, 1:1,000;
mouse; Santa Cruz Biotechnology, Inc.); anti-microtubule-associated
proteins 1A/1B light chain 3A (LC3; cat. no. L7543; dilution,
1:1,000; rabbit; Sigma-Aldrich; Merck KGaA); anti-autophagy protein
5 (ATG5; cat. no. sc-133158; dilution, 1:500; mouse; Santa Cruz
Biotechnology, Inc.) and anti-p62 (cat. no. ab109012; dilution,
1:1,000; rabbit; Abcam, Cambridge, UK). The PARP antibody was used
to detect PARP and cleaved PARP, the LC3 antibody was used to
detect LC3 I and LC3 II, and the caspase 3 antibody was used to
detect pro-caspase-3 and cleaved-caspase-3. Horseradish
peroxidase-conjugated goat anti-mouse IgG (cat. no. A21010;
dilution, 1:5,000) and anti-rabbit IgG (cat. no. A21020; dilution,
1:5,000) were purchased from Abbkine Scientific Co., Ltd. (Wuhan,
China). Cisplatin was purchased from Sigma-Aldrich; Merck KGaA
(cat. no. P4394), and the phosphoinositide 3-kinase (PI3K)
inhibitor, 3-methyladenine (3-MA), was purchased from R&D
Systems, Inc. (Minneapolis, MN, USA). 3-MA inhibits autophagy by
preventing autophagosome formation via the inhibition of PI3K
catalytic subunit type 3 (13).
3-MA was resuspended in water, as organic solvents, such as
dimethyl sulfoxide (DMSO), may be cytotoxic. Due to its instability
in aqueous solutions, 50 mM stocks of 3-MA were freshly prepared,
and cells were treated with 5 mM 3-MA to inhibit autophagy.
Lentivirus transfection and MEG3
knockdown
The lentiviral transduction particles for short
hairpin (sh)RNA-mediated knockdown of ATG5 were purchased from
Sigma-Aldrich; Merck KGaA. The shRNA sequence targeting ATG5 was
5′-CCTTTCATTCAGAAGCTGTTT-3′ and the pLKO.1 vector (Sigma-Aldrich;
Merck KGaA) was used as negative control. The shRNA was cloned
using the PLKO.1 vector. Stable knockdown of ATG5 in cells was
achieved as previously described (14). The primer sequences,
5′-AGACGGCGGAGAGCAGAG-3′ and 5′-CACATTTATTGAGAGCACAGTGG-3′, were
used to generate plasmids encoding the full-length MEG3 sequence.
RNA was extracted from U87 cells using RNAiso Plus (Takara
Biotechnology Co., Ltd., Dalian, China) MEG3 cDNA was then reverse
transcribed using 100 µg total RNA from U87 cells and the Advantage
RT-For PCR kit (Takara Biotechnology Co., Ltd.), and was amplified
for 40 cycles of 95°C for 1 min, 54°C for 1 min and 72°C for 30
sec. To generate lentiviral vectors expressing MEG3, HEK293T cells
(1×105/well) cultured on a 6-cm dish were transfected with 2 µg
pCDH-MEG3 or pCDH vector (System Biosciences, Inc., Palo Alto, CA,
USA), together with 1.5 µg psPax2 (System Biosciences, Inc.) and
0.5 µg pMD2 G (System Biosciences, Inc.) vectors using
Lipofectamine 3000 (Thermo Fisher Scientific, Inc.). At 24 h
following transfection, the medium was replaced with Dulbecco's
modified Eagle's medium containing 10% FBS, and cells were
incubated for a further 24 h. The culture medium containing
lentiviral particles was centrifuged at 1,000 × g for 5 min at room
temperature. Viral particles were collected in the supernatant and
the concentration was determined using the Lenti-Pac™ Lentivirus
Concentration Solution (GeneCopoeia, Inc., Rockville, MD, USA). The
virus titer was determined by calculating the multiplicity of
infection. For infection of U87 cells (1×105), 100 µl viral
particles (1×108/ml) were used for 12 h. In order to establish a
stable cell line, at 24 h after infection was performed, 1×105
cells that were infected by viral particles (1×108/ml) at 37°C were
seeded in a 6-well plate and puromycin (5 µM) was used as a
selection marker for the infected cells for 24 h at 37°C. The
expression efficiency was evaluated by western blot analysis.
For knockdown of MEG3 in U87 cells, small
interfering (si)RNA molecules were custom designed and synthesized
by Sigma-Aldrich; Merck KGaA. The sequences of the siRNAs were as
follows: Control, 5′-TACGTCCAAGGTCGGGCAGGAAGA-3′; and MEG3,
5′-GACUUAAACCAAUGCCCUA-3′. This was transfected into U87 cells
(2×105/well) for 24 h using Lipofectamine 3000 and, at
36 h after transfection, cells were treated with 10 µM cisplatin
for 0 or 36 h. For certain experiments, cells with or without
knockdown of MEG3 were pretreated with 5 mM 3-MA or mock treatment
(water) at 36 h after transfection for 6 h at 37°C to inhibit
autophagy, immediately prior to treatment with 10 µM cisplatin for
0 or 36 h.
Western blot analysis
U87 cells (1×105/well) were seeded into the six well
cell culture plate and subjected to various treatments as described
in the above paragraphs. Total protein extracted from U87 cells
using radioimmunoprecipitation assay lysis buffer (Shanghai Qcbio
Science and Technologies Co., Ltd., Shanghai, China) was used for
immunoblotting. Briefly, cell lysates were centrifuged at 9,000 × g
for 10 min at 4°C, and the supernatant was collected. The protein
concentration was determined using a BCA Protein assay kit (Pierce;
Thermo Fisher Scientific, Inc.). Total protein (30–60 µg) was
separated on a 12% SDS-PAGE mini-gel, followed by transfer to a
nitrocellulose membrane. The membranes were blocked with 5%
fat-free dry milk in a Tris-buffered saline plus Tween-20 solution,
consisting of 50 mM Tris-HCl, 0.15 M NaCl and 0.1% Tween-20 (pH
7.5), overnight at 4°C. Membranes were then incubated with primary
antibodies overnight at 4°C and incubated with the secondary
antibodies (1:5,000) at room temperature for 1 h. An enhanced
chemiluminescence detection system (GE Healthcare, Chicago, IL,
USA) was used to visualize the expression of target proteins. Three
samples from each group were analyzed, and the results were
quantified using the Gel-Pro analyzer software (version, 4.0, GE
Healthcare).
Cell viability
U87 cells (2×104/well) were seeded in 96-well plates
and incubated for 24 h. Cells were then treated with 10 µM
cisplatin for 36 h at 37°C. MTT reagent (0.5 mg/ml; Cayman Chemical
Company, Ann Arbor, MI, USA) was subsequently added, and cells were
incubated for 4 h before the medium was removed and 100 µl DMSO was
added. Cell viability was quantified by measuring the optical
density (OD) at 490 nm using a microplate reader (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). At least three independent
experiments were performed, and the final inhibitory rate of cell
viability was calculated using the following formula: Inhibitory
rate = [1 - (OD490compound/OD490solvent)] × 100.
Annexin V-fluorescein isothiocyanate
(FITC) staining and fluorescence-activated cell sorting (FACS)
The staining protocol was performed using a FITC
Annexin V Apoptosis Detection kit I (BD Biosciences, Franklin
Lakes, NJ, USA), according to the manufacturer's protocol. Briefly,
5×105 cells were transfected with 100 µl pLKO.1 (1×108/ml) as a
control or 100 µl shRNA ATG5 (1×108/ml) for 12 h at 37°C and,
immediately following infection, the cells were transfected with
control or MEG3 siRNA at 37°C. After transfection for 24 h, the
cells were treated with 10 µM cisplatin for 36 h at 37°C. Cells
were subsequently centrifuged at 1,000 × g for 5 min at 4°C and
resuspended in 195 µl binding buffer. This was followed by
incubation with 5 µl Annexin V-FITC for 10 min at room temperature
in the dark. The cells were centrifuged again at 1,000 × g for 5
min at 4°C, before they were resuspended in 190 µl binding buffer
and 10 µl propidium iodide stain and gently agitated. FACS analysis
was performed using the BD Accuri C6 Flow Cytometer (BD
Biosciences) in order to detect cell apoptosis and the apoptosis
was analyzed using CFlow Plus software (version, 1.0264.15; BD
Biosciences).
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
U87 cells (2×105/well) were treated with 5 or 10 µM
cisplatin for 0, 24 or 36 h, prior to RNA extraction using RNAiso
Plus. RNA quality was examined by gel electrophoresis, and only
high quality RNA was used for subsequent analyses. A total of 1 µg
total RNA was used to synthesize cDNA using the PrimeScript™ RT
reagent kit (Perfect Real Time; Takara Biotechnology Co., Ltd.)
according to the manufacturer's instructions. qPCR was performed
using SYBR premix ExTaq (Takara Biotechnology Co., Ltd.) and ROX,
and amplified with the Stratagene Mx3000P qPCR system (Agilent
Technologies, Inc., Santa Clara, CA, USA). The primer sequences
used for qPCR analysis were as follows: Actin forward,
5′-CTCCATCCTGGCCTCGCTGT-3′, and reverse,
5′-GCTGTCACCTTCACCGTTCC-3′; MEG3 forward,
5′-GCATTAAGCCCTGACCTTTG-3′, and reverse,
5′-TCCAGTTTGCTAGCAGGTGA-3′. The mRNA expression was normalized to
the expression of actin using the 2-∆∆Cq method (15).
Statistical analysis
Data are presented as the mean ± standard deviation,
and statistical analysis was performed using SPSS software
(version, 22.0; IBM Corp., Armonk, NY, USA). Statistical analysis
was performed using one-way analysis of variance followed by
Dunnett's test. P<0.05 was considered to indicate a
statistically significant difference.
Results
MEG3 lncRNA is induced by cisplatin
treatment of human U87 cells
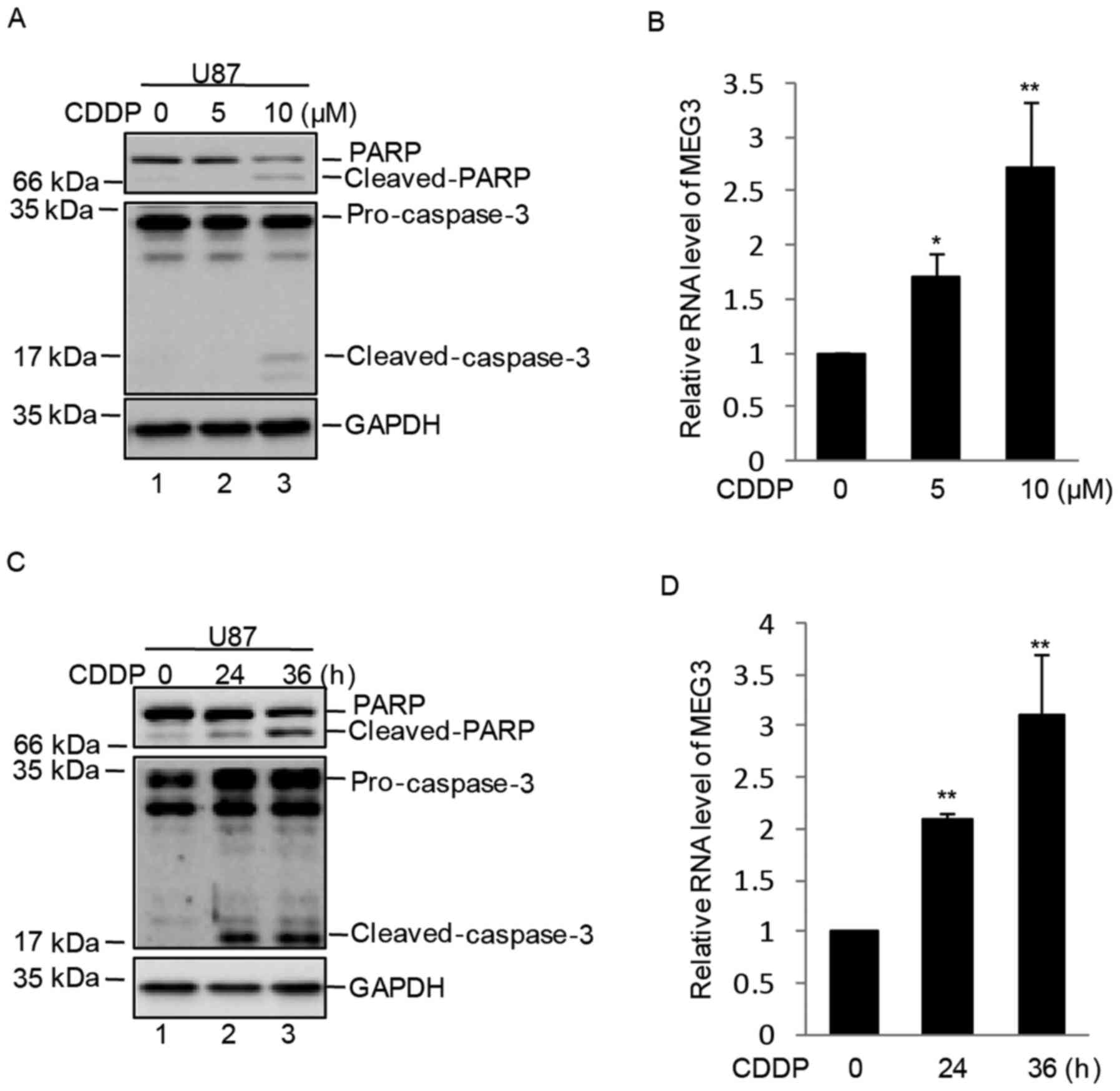
In order to evaluate MEG3 expression in response to
cisplatin treatment of GBM, human U87 cells were treated with 0, 5
or 10 µM cisplatin for 36 h. As shown in Fig. 1A, U87 cells were treated with 0, 5
or 10 µM cisplatin for 24 h and cell apoptosis was increased at 10
µM cisplatin as indicated by increased PARP and caspase-3 cleavage.
Subsequently, the mRNA expression of MEG3 was detected using
RT-qPCR and the results demonstrated that the mRNA expression
levels of MEG3 were increased in a dose-dependent manner following
treatment of U87 cells with cisplatin (Fig. 1B). The cells were subsequently
treated with 10 µM cisplatin for 0, 24 or 36 h. As shown in
Fig. 1C, cell apoptosis was
increased at 24 and 36 h, as indicated by increased PARP and
caspase-3 cleavage. In addition, the expression levels of MEG3 were
significantly upregulated in a time-dependent manner in U87 cells
(Fig. 1D).
MEG3 lncRNA promotes cisplatin-induced
apoptosis in U87 cells
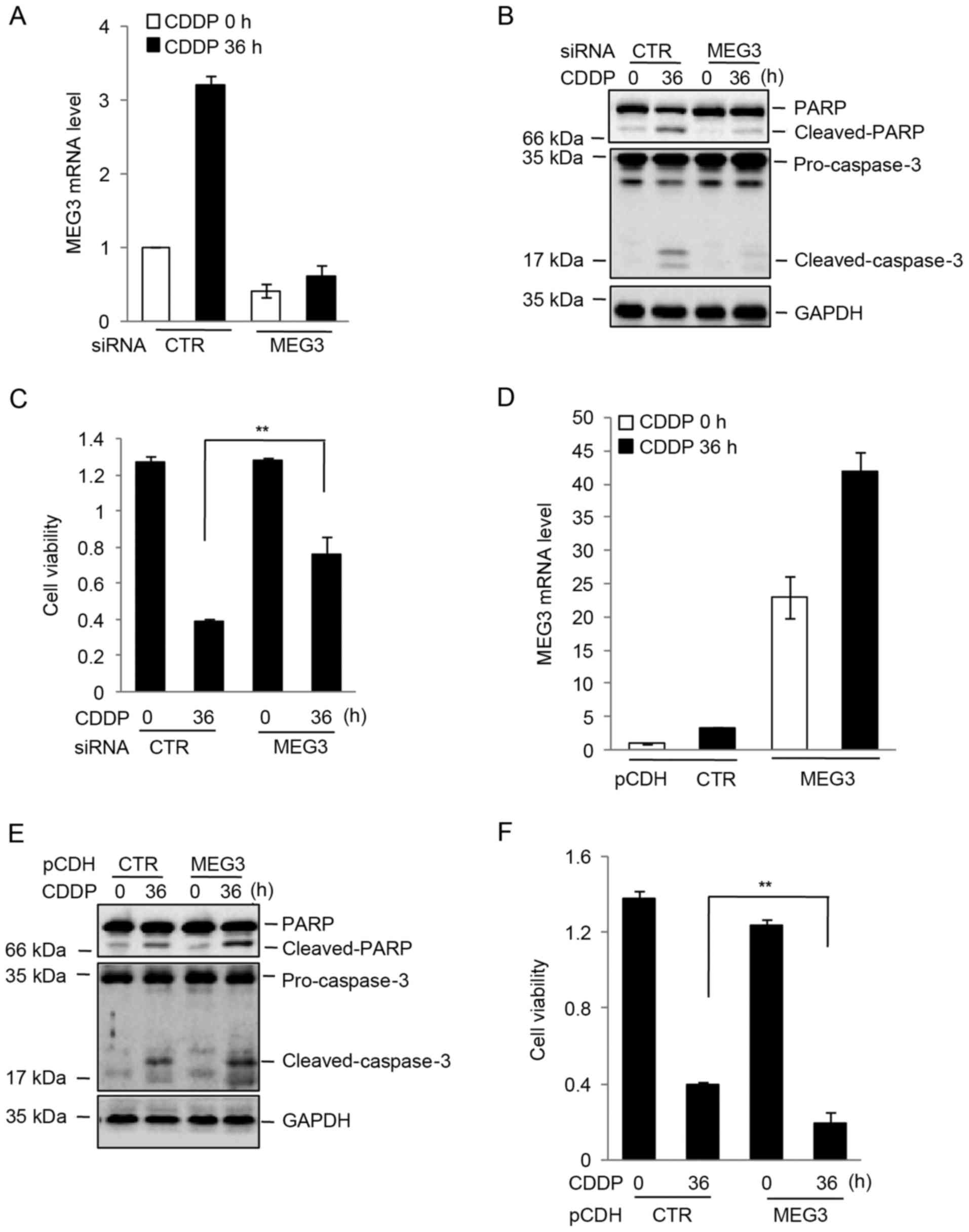
To determine whether elevated MEG3 affected cell
survival following cisplatin treatment, MEG3 expression was
inhibited by siRNA in U87 cells. The expression level of MEG3 was
markedly downregulated in MEG3-siRNA transfected U87 cells when
compared with those transfected with control siRNA (Fig. 2A). The cells were subsequently
treated with cisplatin for 0 or 36 h. When compared with cells
transfected with control siRNA, inhibition of MEG3 reduced the
protein expression levels of cleaved PARP (Fig. 2B). Similarly, decreased MEG3
expression attenuated the cisplatin-induced reduction in cell
viability (Fig. 2C). Conversely,
exogenous expression of MEG3 efficiently enhanced the effects of
cisplatin on the protein expression levels of cleaved PARP and cell
viability (Fig. 2D-F).
MEG3 lncRNA inhibits cisplatin-induced
autophagy
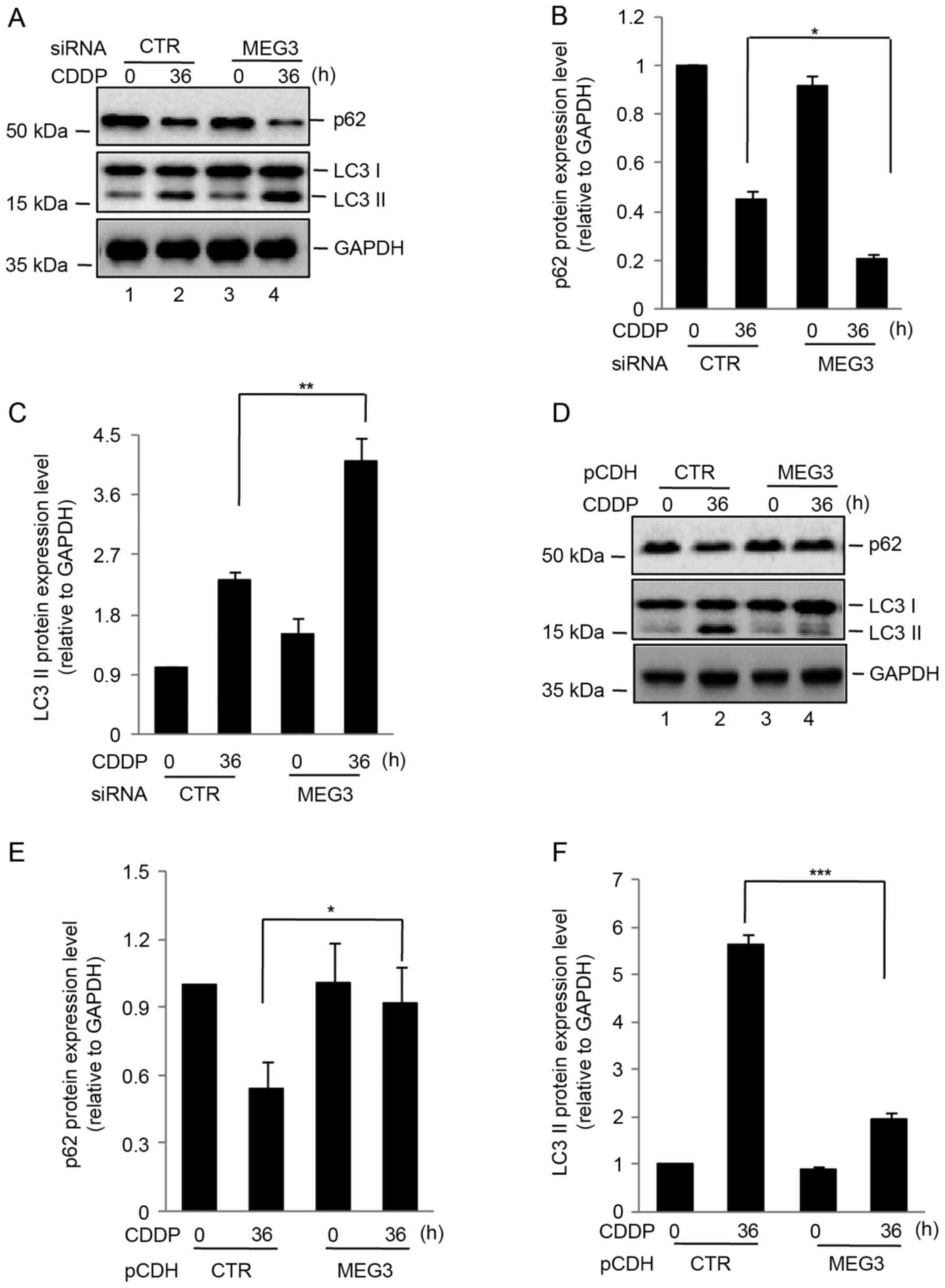
A previous study demonstrated that loss of MEG3
expression activated autophagy and increased the proliferation of
bladder cancer cells (9). In order
to investigate the effect of MEG3 on autophagy induced by
cisplatin, U87 cells transfected with control MEG3 siRNA were
treated with cisplatin for 0 or 36 h. Compared with the control
cells, silencing of MEG3 expression increased the protein
expression of LC3 II relative to GAPDH and promoted p62 degradation
(Fig. 3A-C). By contrast,
overexpression of MEG3 attenuated cisplatin-induced autophagy in
U87 cells (Fig. 3D-F).
LncRNA MEG3 enhances cisplatin-induced
apoptosis via suppression of autophagy
Autophagy has been demonstrated to be a key
mechanism that induces chemoresistance in cancer cells (9). Therefore, the present study
investigated whether the effects of MEG3 in promoting
cisplatin-induced apoptosis was dependent on the inhibition of
autophagy. U87 cells transfected with control or MEG3 siRNA were
pretreated with 3-MA (5 mM) for 6 h and exposed to 10 µM cisplatin
for 0 or 36 h. Cell apoptosis was analyzed. Compared with control
siRNA-transfected cells, inhibition of MEG3 markedly decreased the
expression of apoptosis-associated proteins, which was reversed
following treatment with the autophagy inhibitor, 3-MA (Fig. 4A and B). To confirm these
observations, cells were infected with lentiviruses expressing
shRNA targeting ATG5, which is a key molecule involved in the
formation of autophagic vacuoles and induces autophagy (16). The cells were treated with control
or MEG3 siRNA. When compared with control shRNA-transfected U87
cells that were transfected with MEG3 siRNA, knockdown of ATG5
significantly increased the level of apoptosis (Fig. 4C-F). These data indicated that
inhibition of autophagy by MEG3 promoted cisplatin-induced
apoptosis of U87 cells.
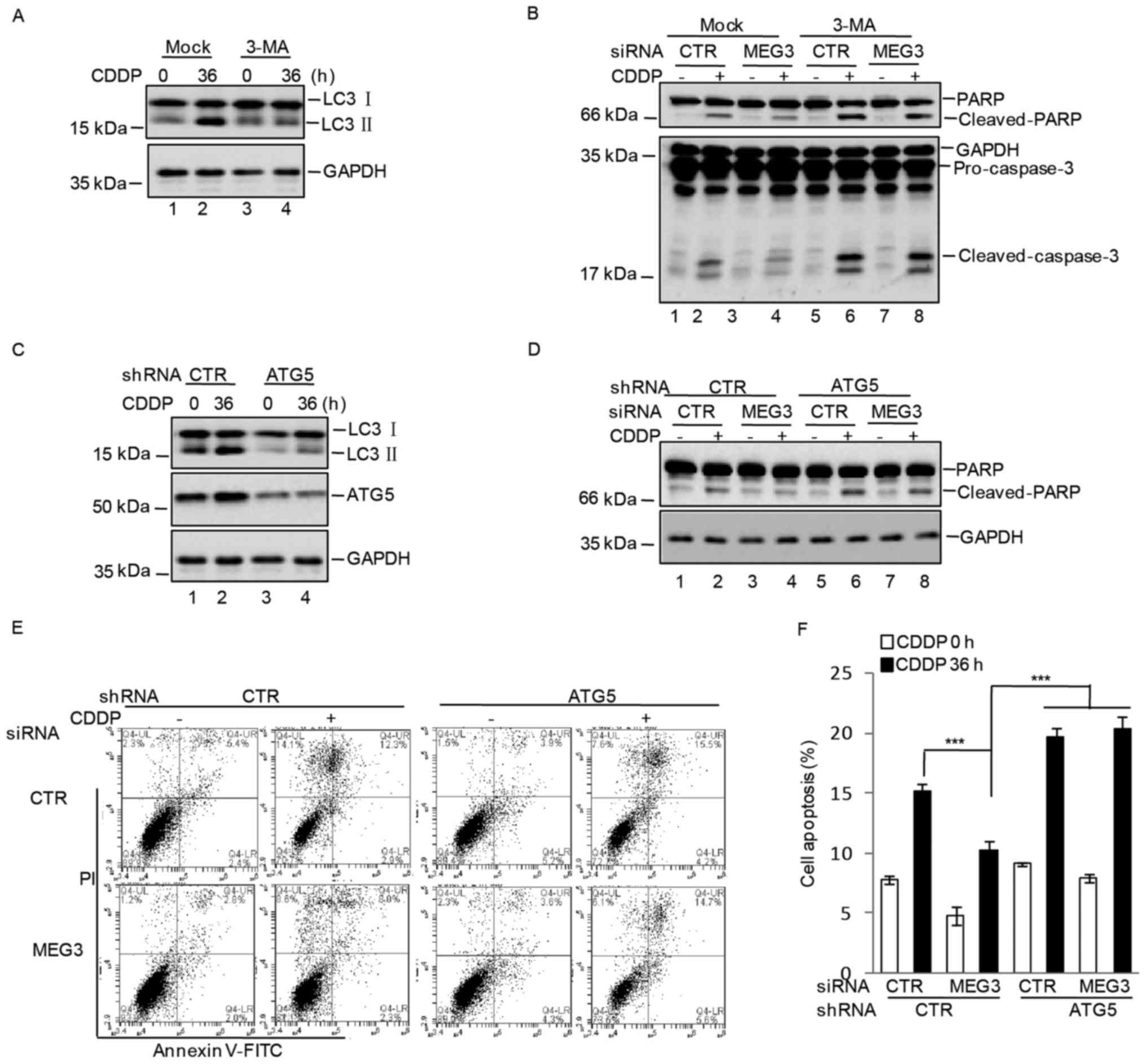 | Figure 4.MEG3 promoted CDDP-induced apoptosis
via inhibition of autophagy. (A) U87 cells were pretreated with or
without 3-MA (5 mM) for 6 h and were subsequently treated with 10
µM CDDP for 0 or 36 h, and the expression of autophagy-associated
proteins, LC3 I and II, were measured by western blotting. (B) U87
cells were transfected with CTR or MEG3 siRNA. At 36 h following
transfection, the cells were pretreated with or without 3-MA (5 mM)
for 6 h and were subsequently treated with 10 µM CDDP for 0 or 36
h. Cell lysates were separated by SDS-PAGE and the expression
levels of apoptosis-associated factors were analyzed by western
blotting. (C) Western blot analysis of the expression levels of
autophagy-associated proteins and ATG5 in U87 cells following
transfection with CTR or ATG5 shRNA and treatment with CDDP for 0
or 36 h. U87 cells were then transfected with CTR or ATG5 shRNA,
together with CTR or MEG3 siRNA. At 36 h following transfection,
cells were treated with or without 10 µM CDDP for 0 or 36 h. (D)
Cell lysates were separated by SDS-PAGE and the protein expression
levels of PARP and cleaved-PARP were analyzed by western blotting.
(E) The level of apoptosis in U87 cells transfected with CTR or
ATG5 shRNA, together with CTR or MEG3 siRNA and treated with or
without 10 µM CDDP for 0 or 36 h, was determined by flow cytometry
analysis and (F) the results were quantified. The results are
presented as the mean ± standard deviation (n=3). ***P<0.001 as
indicated. MEG3, maternally expressed gene 3; CDDP, cisplatin;
3-MA, 3-methyladenine; LC3, microtubule-associated proteins 1A/1B
light chain 3A; CTR, control; siRNA, small-interfering RNA; ATG5,
autophagy protein 5; shRNA, short-hairpin RNA; PARP, poly(ADP)
ribose polymerase; PI, propidium iodide; FITC, fluorescein
isothiocyanate. |
Discussion
LncRNAs, which were initially thought to be spurious
transcriptional ‘noise’, have been reported to participate in a
broad spectrum of biological processes, including maintenance of
genome integrity, cell differentiation and embryonic development
(17). In different types of
cancer cells, lncRNAs have been demonstrated to be involved in
tumorigenesis and chemoresistance (18).
The MEG3 lncRNA has been suggested to function as a
tumor suppressor in a number of human cancers, and the expression
level of MEG3 is significantly downregulated in several primary
cancers (6,8). A recent report indicated that MEG3
contributes to cisplatin-induced apoptosis in human lung cancer
cells (19). Similarly, the
results of the present study suggested that MEG3 expression was
induced by cisplatin treatment of human U87 cells in a time- and
dose-dependent manner. Silencing of MEG3 expression using siRNAs
inhibited cisplatin-induced apoptosis in U87 cells. By contrast,
exogenous MEG3 expression enhanced cisplatin-induced cell
apoptosis.
Previous studies have revealed that autophagy is
involved in mediating chemoresistance to cisplatin in several
cancer cell types (4,20). In the present study, MEG3 was
observed to regulate cisplatin-induced autophagy in U87 cells, as
inhibition of MEG3 expression promoted autophagy of cells treated
with cisplatin. However, the molecular mechanisms underlying these
effects remain unclear, and future studies will investigate this
further. The results of the current study demonstrated that
inhibition of autophagy by 3-MA or knockdown of ATG5 reversed the
decrease in cell apoptosis caused by MEG3 knockdown in U87 cells
treated with cisplatin. Therefore, the results suggest that MEG3
enhances cisplatin-induced apoptosis via inhibition of autophagy in
U87 cells and, therefore, MEG3 maybe considered a novel therapeutic
target to counteract cisplatin-resistant gliomas.
Acknowledgements
The present study was supported by the National
Nature Science Foundation of China (grant nos. 81372714, 81672480
and 81603448).
References
|
1
|
Van Meir EG, Hadjipanayis CG, Norden AD,
Shu HK, Wen PY and Olson JJ: Exciting new advances in
neuro-oncology: The avenue to a cure for malignant glioma. CA
Cancer J Clin. 60:166–193. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wen PY and Kesari S: Malignant gliomas in
adults. N Engl J Med. 359:492–507. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Kelland L: The resurgence of
platinum-based cancer chemotherapy. Nat Rev Cancer. 7:573–584.
2007. View
Article : Google Scholar : PubMed/NCBI
|
|
4
|
Liu X, Sun K, Wang H and Dai Y: Knockdown
of retinoblastoma protein may sensitize glioma cells to cisplatin
through inhibition of autophagy. Neurosci Lett. 620:137–142. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wainwright DA, Nigam P, Thaci B, Dey M and
Lesniak MS: Recent developments on immunotherapy for brain cancer.
Expert Opin Emerg Drugs. 17:181–202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Miyoshi N, Wagatsuma H, Wakana S,
Shiroishi T, Nomura M, Aisaka K, Kohda T, Surani MA, Kaneko-Ishino
T and Ishino F: Identification of an imprinted gene, Meg3/Gtl2 and
its human homologue MEG3, first mapped on mouse distal chromosome
12 and human chromosome 14q. Genes Cells. 5:211–220. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Anwar SL, Krech T, Hasemeier B, Schipper
E, Schweitzer N, Vogel A, Kreipe H and Lehmann U: Loss of
imprinting and allelic switching at the DLK1-MEG3 locus in human
hepatocellular carcinoma. PloS One. 7:e494622012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Balik V, Srovnal J, Sulla I, Kalita O,
Foltanova T, Vaverka M, Hrabalek L and Hajduch M: MEG3: A novel
long noncoding potentially tumour-suppressing RNA in meningiomas. J
Neurooncol. 112:1–8. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Ying L, Huang Y, Chen H, Wang Y, Xia L,
Chen Y, Liu Y and Qiu F: Downregulated MEG3 activates autophagy and
increases cell proliferation in bladder cancer. Mol Biosyst.
9:407–411. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zhou Y, Zhang X and Klibanski A: MEG3
noncoding RNA: A tumor suppressor. J Mol Endocrinol. 48:R45–R53.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhang X, Gejman R, Mahta A, Zhong Y, Rice
KA, Zhou Y, Cheunsuchon P, Louis DN and Klibanski A: Maternally
expressed gene 3, an imprinted noncoding RNA gene, is associated
with meningioma pathogenesis and progression. Cancer Res.
70:2350–2358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Allen M, Bjerke M, Edlund H, Nelander S
and Westermark B: Origin of the U87MG glioma cell line: Good news
and bad news. Sci Transl Med. 8:354re32016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Miller S, Oleksy A, Perisic O and Williams
RL: Finding a fitting shoe for Cinderella: Searching for an
autophagy inhibitor. Autophagy. 6:805–807. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han C, Gu H, Wang J, Lu W, Mei Y and Wu M:
Regulation of L-threonine dehydrogenase in somatic cell
reprogramming. Stem Cells. 31:953–965. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) methods. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Noda NN and Inagaki F: Mechanisms of
autophagy. Annu Rev Biophys. 44:101–122. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Schmitt AM and Chang HY: Long noncoding
RNAs in cancer pathways. Cancer Cell. 29:452–463. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Wu P, Zuo X, Deng H, Liu X, Liu L and Ji
A: Roles of long noncoding RNAs in brain development, functional
diversification and neurodegenerative diseases. Brain Res Bull.
97:69–80. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Liu J, Wan L, Lu K, Sun M, Pan X, Zhang P,
Lu B, Liu G and Wang Z: The long noncoding RNA MEG3 contributes to
cisplatin resistance of human lung adenocarcinoma. PLoS One.
10:e01145862015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yang X, Du T, Wang X, Zhang Y, Hu W, Du X,
Miao L and Han C: IDH1, a CHOP and C/EBPβ-responsive gene under ER
stress, sensitizes human melanoma cells to hypoxia-induced
apoptosis. Cancer Lett. 365:201–210. 2015. View Article : Google Scholar : PubMed/NCBI
|