Introduction
Gene transfer into normal cells serves an important
role in basic studies aiming to elucidate the fundamental processes
taking place in living organisms, as well as in gene therapy and
immunotherapy of human diseases. In the field of immunotherapy
there remains a requirement to develop and optimize gene transfer
into primary target cells. However, the majority of basic studies
are conducted with transformed cells, due to difficulties in
achieving stable and efficient levels of gene expression in normal
cells. Gene transfer into normal B cells is particularly difficult.
Notably, B cells induced to express the antigen of interest can
stimulate human antigen-specific cytotoxic T lymphocytes and are
useful and convenient source of autologous antigen-presenting cells
for examining human immune response. In addition, genetically
modified B cells may be used to induce anti-tumor immune response,
thus contribute to the development of novel strategies for
treatment of cancer. Following gene transfer, B cells have
additionally been successfully applied to induce tolerance and
prevent autoimmune responses in animal models. Unfortunately, the
use of nonviral methods for gene transfer into mature B cells is
particularly ineffective, and retroviral transduction of quiescent
mature B cells has not been reported in the literature. Therefore,
lentiviruses have attracted significant interest, considering their
ability to transduce nondividing cells (1,2).
Vectors based on murine leukemia virus (MLV) have previously been
used to transfer genes into transformed B cells or B-cell
precursors (3). However, only 1–4%
of B cells were transduced with these vectors, and they were
revealed to be ineffective in gene transduction of mature quiescent
B cells (4,5). Even with lentiviruses, quiescent B
cells require prior stimulation with cytokines, cell surface
molecules or other factors to stimulate entry from G0 to
G1 phase of the cell cycle, in order to become
transduced. This is achieved by various methods, including B-cell
activation via cluster of differentiation (CD)40, with either
antibodies or feeder cells expressing CD40 ligand (CD40L) (6); cytokines, such as interleukin (IL)-4
and IL-21; CpG oligonucleotides (5); anti-CD20 antibodies (7); infection with Epstein-Barr virus
(EBV); or a combination of these approaches (4). A major breakthrough was associated
with the generation of pseudotyped lentiviral vectors. Lentiviral
vectors pseudotyped with glycoprotein from the vesicular stomatitis
virus (VSVG) or envelope proteins from g-retroviruses, such as MLV,
have been reported to result in broad tropism in the majority of
human cells (8); however, they
were particularly ineffective in the case of B cells (6). Previous studies have revealed that
VSVG-lentiviruses require low-density lipoprotein-receptor, which
is inducible in numerous activated cells, but not in B cells
(9,10). Improved lentiviral pseudotypes have
been designed by incorporating measle virus (MV) glycoproteins F
and H into their surface (MV-lentiviruses) (11–13).
These vectors target normal B cells through MV receptors, such as
CD46 or signaling lymphocytic activation molecule (14,15).
Pseudotyped MV-lentiviruses have been reported to achieve ~50% gene
transfer efficiency in resting B cells (11); therefore, further improvements and
optimization strategies are required. Efficient delivery of genes
into primary human mature B cells may be used to investigate the
functions of genes-of-interest (GOIs) in these cells. In addition,
efficient gene delivery may result in the development of gene
therapies, as well as the development of novel tools for B-cell
immortalization, allowing for cloning of antigen-specific B-cell
populations, which may be used in the production of monoclonal
antibodies. Therefore, the aim of the present study was to further
optimize the methods for efficient B-cell transduction by using
various gene promoters to increase the levels of transgene
expression.
Materials and methods
Cell culture
The Raji human Burkitt lymphoma cell line (American
Type Culture Collection, Manassas, VA, USA) and human embryonic
kidney (HEK)-293T cell line (Leibniz Institute DSMZ-German
Collection of Microorganisms and Cell Cultures, Braunschweig,
Germany) were cultured in RPMI 1640 medium (Sigma-Aldrich; Merck
Millipore, Darmstadt, Germany). HT-1080 cells (American Type
Culture Collection) were cultured in Dulbecco's modified Eagle's
medium (DMEM; Sigma-Aldrich; Merck Millipore). The B95.8 human
EBV-producing cell line was kindly provided by Professor U. Wojda
(International Institute of Molecular and Cellular Biology, Warsaw,
Poland); these cells were cultured in RPMI 1640 medium until they
reached ≥2 mln/ml density. Subsequently, the supernatant was
collected and filtered through a 0.45 µm syringe filter (Promega
Corporation, Madison, Wisconsin, USA), aliquoted and stored at
−80°C until further use. All media were supplemented with 10%
heat-inactivated fetal bovine serum (FBS; HyClone; GE Healthcare
Life Sciences, Logan, UT, USA), 100 µg/ml streptomycin, 100 U/ml
penicillin and 250 ng/ml amphotericin B (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Cells were cultured at 37°C in
a humidified atmosphere containing 5% CO2, and were
passaged approximately every other day.
B-cell isolation from blood and in
vitro culture
Peripheral blood mononuclear cells (PBMCs) from
healthy donors (recruited between September and November 2013, 3
males and 3 females aged 25–45 years old, blood collected via
venipuncture from forearm) were isolated from full blood using
Histopaque-1077 (Sigma-Aldrich; Merck Millipore) according to the
manufacturer's protocol. B cells were isolated from PBMCs using the
magnetic EasySep™ Human B Cell Enrichment Kit (STEMCELL
Technologies Canada, Inc., Vancouver, BC, Canada) according to the
manufacturer's protocol. Cells were cultured in Iscove's modified
Dulbecco's medium (IMDM; Invitrogen; Thermo Fisher Scientific,
Inc.) supplemented with 10% heat-inactivated FBS, 100 µg/ml
streptomycin, 100 U/ml penicillin, and 250 ng/ml amphotericin B at
37°C in a humidified atmosphere containing 5% CO2. The
present study was approved by the Institutional Review Board of the
Medical University of Warsaw (Warsaw, Poland) and was conducted
according to the Declaration of Helsinki. Each patient provided
written informed consent for the procedures. For co-culture
experiments, HT-1080 pLVX-CD40L cells were incubated with mitomycin
C (10 µg/ml; Sigma Aldrich; Merck Millipore) for 3 h at 37°C, and
were reseeded into 6-well plates at a density of
4×105/well. A total of 5×106 B cells at a
density of 2.5×106/1 ml were then placed onto the
HT-1080 pLVX-CD40L cells in IMDM supplemented with 25 ng/ml IL-21
(PeproTech, Rocky Hill, NJ, USA).
Plasmids
A human CD40L coding sequence was amplified by
polymerase chain reaction (PCR) from cDNA obtained from healthy
donor PBMCs, and was cloned into a pLVX-internal ribosome entry
site (IRES)-Puro vector (Takara Bio Europe, Saint-Germain-en-Laye,
France) according to a standard protocol (16). Briefly, cells were washed with PBS,
pelleted, and resuspended in 1 ml TRIzol® reagent
(Invitrogen; Thermo Fisher Scientific, Inc.) to extract total RNA,
according to the manufacturer's protocol. First strand cDNA
synthesis was then performed: 0.5 µg total RNA was primed with
oligo(dT) using AMV reverse transcriptase (EURx Ltd., Gdansk,
Poland). PCR was performed using Mastercycler personal (Eppendorf
Instrumente GmbH, Hamburg, Germany) and Color Opti Taq polymerase
(EURx Ltd.) using the following PCR cycling conditions: Initial
denaturation at 94°C for 2 min, followed by 30 cycles of
denaturation at 94°C for 30 sec, annealing at 58°C for 30 sec,
elongation at 72°C for 60 sec, and a final elongation step at 72°C
for 10 min. Amplification products were analyzed by 1% agarose gel
electrophoresis. The following primers containing restriction sites
for XhoI and BamHI on the forward and reverse primer, respectively
were used: Forward,
5′-CCGACTCGAGACCATGATCGAAACATACAACCAAACTTCTCC-3′ and reverse,
5′-GCGGGGATCCTCAGAGTTTGAGTAAGCCAAAGGACG-3′. The sequence of the
construct (pLVX-CD40L-IRES-Puro) was confirmed by Sanger sequencing
using BigDyeTM Terminator Version 3.1 Ready Reaction Cycle
Sequencing kit (Thermo Fisher Scientific, Inc.), according to the
manufacturer's protocol. The human immunodeficiency virus
(HIV)-spleen focus-forming virus (SFFV) -monomeric red fluorescent
protein (mRFP)-woodchuck hepatitis virus posttranscriptional
regulatory element (WPRE) plasmid was a generous gift by Professor
Els Verhoeyen (Ecole Normale Supérieure de Lyon, Lyon, France)
(12). The sequences encoding
MCS-IRES-RFP, BiP-IRES-RFP, MCS-IRES-GFP and XBP1s-IRES-GFP were
synthesized by Epoch Life Science, Inc. (Missouri City, TX, USA)
and provided as inserts in pBluescript SK vectors. The inserts were
subsequently cloned by ligation into the BamHI and
XhoI sites of the HIV-SFFV-mRFP-WPRE plasmid. Sequences of
the constructs were confirmed by Sanger sequencing.
Lentiviral transduction of target
cells
For lentiviral production, HEK-293T cells were
seeded into 6-well plates (4×105/well) and were
co-transfected with 2 µg gene-of-interest (GOI)-containing vectors
and components of 2nd generation packaging vectors: 1.5 µg psPAX2
packaging vector and 1 µg pMD2.G envelope vector (both vectors were
obtained from Professor Didier Trono; École Polytechnique Fédérale
de Lausanne, Lausanne, Switzerland), using GeneJuice®
transfection reagent (EMD Millipore, Billerica, MA, USA), according
to the manufacturer's protocol. The following lentiviral vectors
were used in this study: pLVX–IRES-Puro and pLVX-IRES-ZsGreen1
(both from Clontech, Takara Bio Europe), pGIPZ (Open Biosystems; GE
Healthcare Life Sciences), pLVTHM (AddGene, Inc., Cambridge, MA,
USA). 72 h post-transfection, the lentivirus-containing medium was
collected, filtered and added to the culture of target HT-1080 or
human B cells with a multiplicity of infection (MOI) of 2. MOI was
determined using the Lenti-X p24 Rapid Titer kit (Clontech, Takara
Bio Europe), according to the manufacturer's protocol. To select
GOI-containing cells, either puromycin selection or sorting for
fluorescent-positive cells using a FACSAria III cell sorter (BD
Biosciences, La Jolla, CA, USA) was performed 7 days
post-transduction. HT-1080 cells modified with pLVX-CD40L-IRES-Puro
construct (HT-1080 pLVX-CD40L) were cultured in DMEM supplemented
with 1 µg/ml puromycin (Sigma Aldrich; Merck Millipore).
Staining of surface antigens
Cells were incubated with saturating amounts of
fluorochrome-conjugated antibodies (Table I) for 30 min at room temperature.
Prior to analysis cells were washed and resuspended in PBS. Cells
were analyzed using a FACScan (BD Biosciences).
 | Table I.Antibodies used for cell sorting. |
Table I.
Antibodies used for cell sorting.
| Antibody | Fluorochrome | Clone | Cat. no. | Dilution | Company |
|---|
| CD19 | FITC | 4G7 | 345776 | 1:10 | BD Biosciences |
| CD20 | FITC | L27 | 345792 | 1:10 | BD Biosciences |
| CD27 | PE | L128 | 340425 | 1:10 | BD Biosciences |
| CD138 | FITC | MI15 | 552723 | 1:10 | BD Biosciences |
| CD20 | PE | 2H7 | 555623 | 1:10 | BD Biosciences |
| IgD | FITC | Polyclonal | F0189 | 1:10 | Dako |
| IgM | FITC | Polyclonal | F0058 | 1:10 | Dako |
| IgG | FITC | Polyclonal | F0185 | 1:10 | Dako |
| IgA | FITC | Polyclonal | F0188 | 1:10 | Dako |
| CD40L | PE | 89–76 | 555700 | 1:10 | BD Biosciences |
EBV infection and generation of
lymphoblastoid cell lines (LCL)
The supernatant from B95.8 cells was collected and
stored as aforementioned, and was thawed on the day of infection.
Human B cells freshly isolated from buffy coats, as aforementioned,
were seeded at a density of 5 mln/ml in IMDM in a T25 flask.
Subsequently, 2 ml fresh RPMI 1460 medium was added, followed by 2
ml EBV-containing B95.8 supernatant. 24 h post-infection the cells
cultured at 37°C began to form clusters.
Fluorescent microscopy
Cell cultures were analyzed for the expression of
fluorescent proteins (GFP and RFP) using an inverted fluorescence
microscope (Eclipse TE-2000; Nikon Corporation, Tokyo, Japan)
equipped with Plan Fluor 10x/0.30 Ph1 DLL, S Plan Fluor ELWD
20x/0.45 Ph1 ADM and Plan Fluor ELWD 40x/0.60 Ph2 ADL objectives,
together with UV-2E/C, B-2E/C, G-2E/C and Y-2E/C fluorescence
filters. Images were captured using a QICAM FAST1394 12-bit digital
CCD camera (Q Imaging, Surrey, BC, Canada) and were analyzed using
Image-Pro Plus 7.0 software (Media Cybernetics, Inc., Rockville,
MD, USA).
ELISA
The concentration of immunoglobulin (Ig)G in cell
culture supernatants was evaluated using Human IgG ELISA kit (Koma
Biotech, Seoul, Korea), according to the manufacturer's protocol.
This assay allows for detection of IgG in a sample at the range of
1.95–125 ng/ml.
Western blotting
Cells were washed with PBS, pelleted, lysed in a
custom-made lysis buffer (50 mM HEPES pH 7.4, 1.0% Triton X-100,
150 mM NaCl, 10% glycerol, 5 mM EDTA) and separated by 10%
SDS-PAGE. Protein concentration prior to electrophoresis was
measured using the Bradford Protein Assay kit (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Total proteins (10–20 µg)
were loaded onto each well. Separated proteins were transferred
onto Protran nitrocellulose membranes (Schleicher & Schuell
BioScience GmbH, Dassel, USA). Membranes were blocked for 1 h at
room temperature with 5% non-fat milk in TBS-0.1% Tween (TBST; see
below). Membranes were then incubated with anti-XBP-1s (cat. no.
647502; 1:1,000; BioLegend, San Diego, CA, USA) overnight at 4°C in
the presence of 5% bovine serum albumin (Sigma Aldrich; Merck
Millipore). After washing with TBST, membranes were incubated with
anti-rabbit horseradish peroxidase (HRP)-conjugated secondary
antibody (cat.no. 111-035-144; Jackson ImmunoResearch Laboratories,
Inc., West Grove, PA, USA) for 1 h at room temperature. The
chemiluminescence reaction for HRP was developed using a
luminol-based chemiluminescence reagent SuperSignal West Pico
(Thermo Fisher Scientific, Inc.) and blots were visualized using
STELLA 8300 bioimager (Raytest, Straubenhardt, Germany). The blots
were then re-probed with anti-β-actin peroxidase-purified
immunoglobulin (clone AC-15; 1:50,000; Sigma Aldrich; Merck
Millipore) for 45 min at room temperature.
Results
Normal B cells co-cultured with
CD40L-expressing feeder cells start to proliferate after 7 days of
culture
The present study aimed to select optimal constructs
and promoters for genetic modification of B cells. As a terminal
cell type, B cells can usually only be cultured for a relatively
short time. Therefore, to prolong the culture period B cells were
co-cultured with feeder cells expressing human CD40L. Initially,
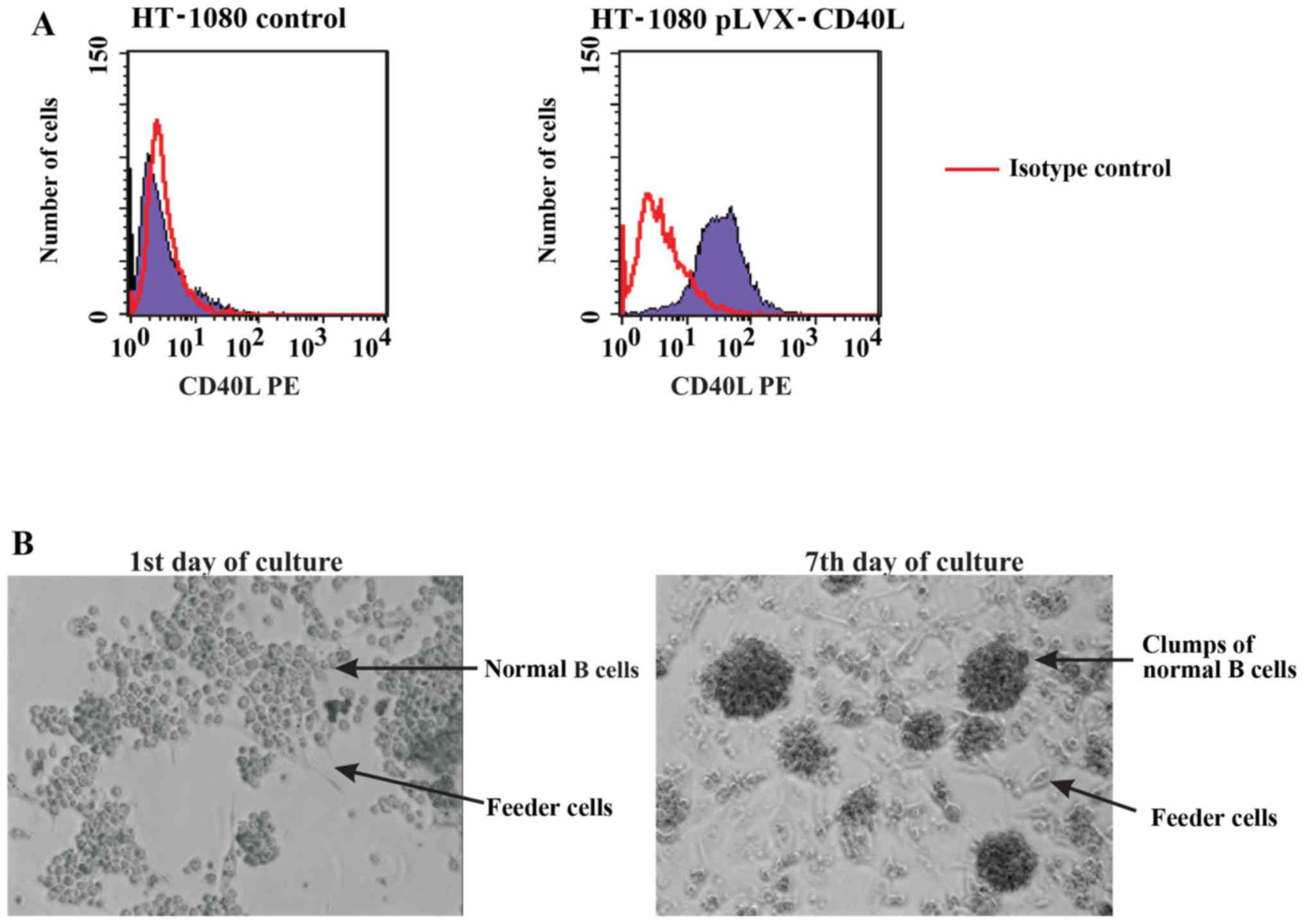
HT-1080 cells were modified with an expression vector encoding
CD40L (Fig. 1A). The CD40L coding
sequence was amplified by PCR from the cDNA of healthy donor PBMCs,
after which it was cloned into the pLVX-IRES-Puro vector and
subsequently used to modify HT-1080 cells. Feeder cells were
pretreated with mitomycin C to prevent their proliferation before
starting the co-cultures. B cells co-cultured with feeder cells
formed clumps of proliferating cells that were viable for ≥14
consecutive days (Fig. 1B).
B cells activated with CD40L and IL-21
are resistant to transduction
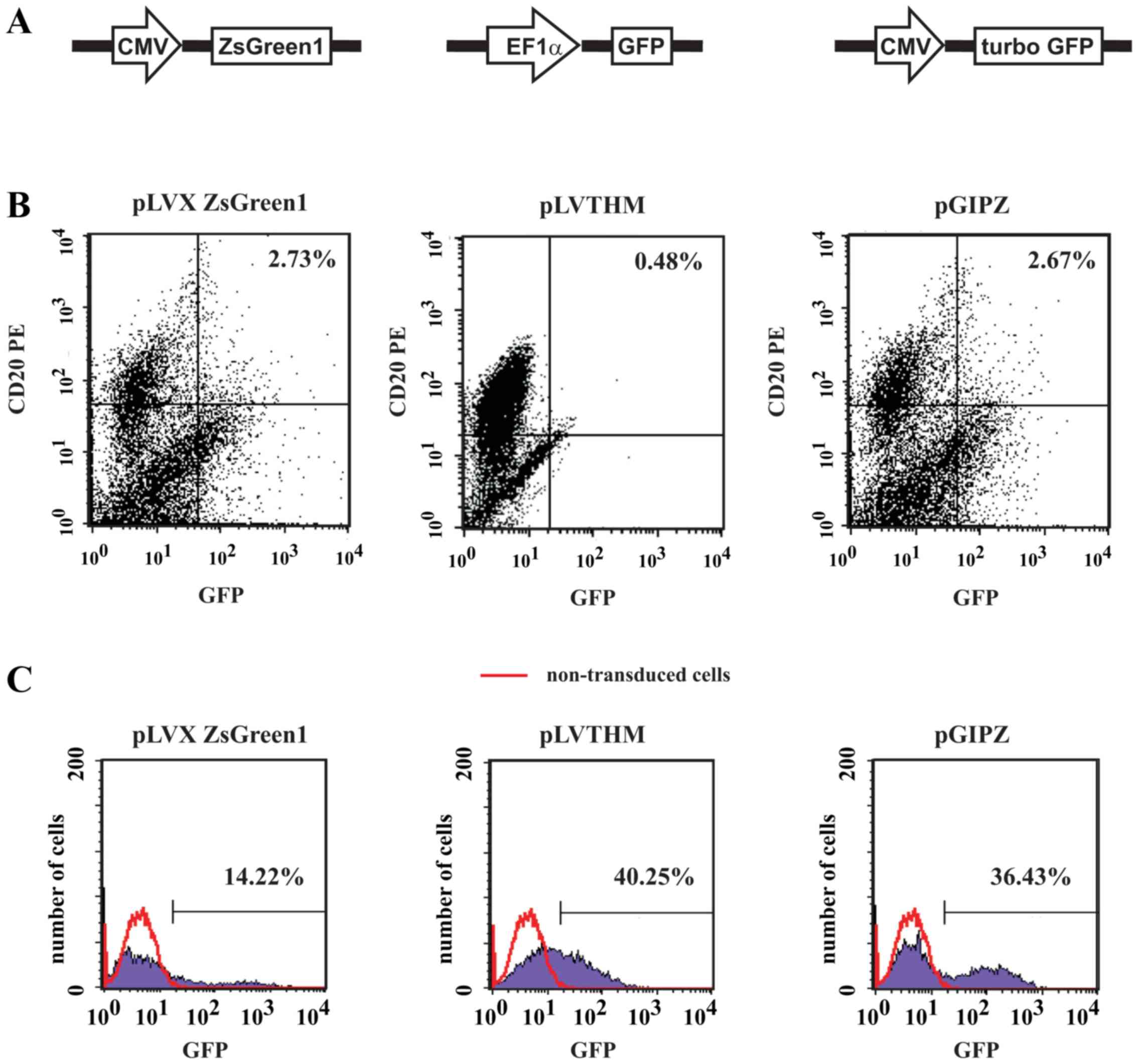
To determine the levels of transgene expression in B
cells three bicistronic plasmids were used that encode various GFPs
under the control of different promoters, namely pGIPZ
[cytomegalovirus (CMV) promoter; turbo GFP, which is an improved
variant of the green fluorescent protein CopGFP], pLVTHM
[elongation factor 1 alpha (EF1α) promoter; GFP) and
pLVX-IRES-ZsGreen1 (CMV promoter; ZsGreen1, which is a human
codon-optimized variant of ZsGreen) (Fig. 2A). Independent of the vectors used
in the present study, a very low level of transgene expression was
detected in B cells; expression did not exceed 10%, as assessed
with flow cytometry 7 days post-transduction (Fig. 2B). Notably, concomitant
transduction of Raji lymphoma cells with pLVX ZsGreen1, pGIPZ and
pLVTHM proved to be effective; the levels of transgene expression
ranged between 14.22 and 40.25% (Fig.
2C).
EBV infection of B cells generates
LCLs
Since Raji cells carry the latent EBV genome and are
positive for EBV nuclear antigen 1, and EBV infection has
previously been described to increase the sensitivity of B cells to
genetic modification (17), the
present study infected normal B cells with EBV to determine whether
this would allow for increased levels of transgene expression with
lentiviral vectors. Freshly isolated human B cells were mixed with
supernatants collected from B95.8 human EBV-producing cells, in
order to allow B cell infection and immortalization. Transformation
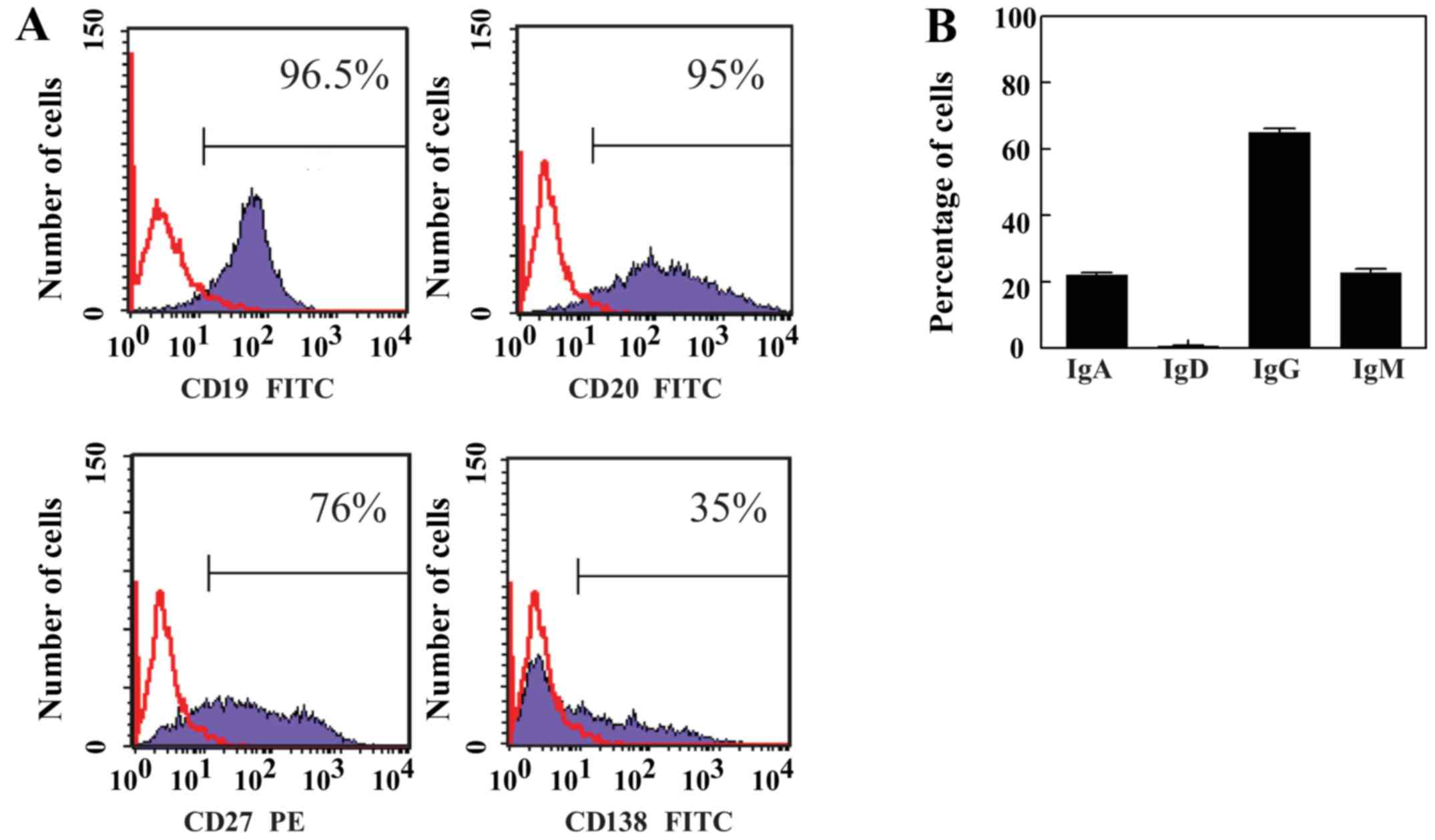
of B cells with EBV resulted in the generation of LCLs. These cells
were highly variable in shape and grew in clumps. The cells
expressed antigens characteristic for normal B lymphocytes, such as
CD19 and CD20 (Fig. 3A).
Furthermore, EBV-infected B cells displayed memory phenotype, as
they were positive for CD27. The expression of CD138, which is an
antigen that is present during activation and differentiation of B
cells and is specific for terminally differentiated B cells, was
variable in tested LCLs (Fig. 3A).
In addition, the membrane expression of Igs was determined, and the
results indicated that LCLs do not express membrane IgD. However,
~60% of LCLs expressed membrane IgG, whereas ~40% expressed
membrane IgA and membrane IgM (Fig.
3B). These results confirmed the findings of previous studies
(18,19), and suggested that EBV infection
expands the CD27+ memory cell pool, and the majority of
these cells exhibited a switch in isotype to IgG or IgA. In
addition, LCLs were tested for the secretion of Igs; the mean
output was 0.004 ng IgG per cell per day (data not shown).
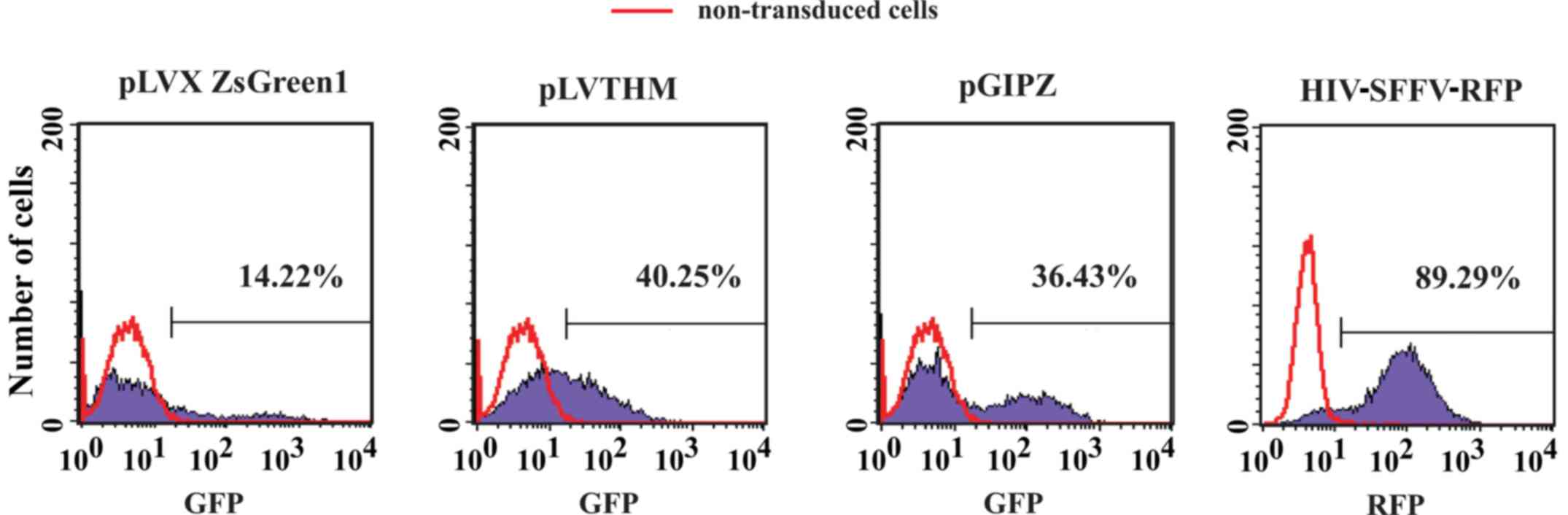
SFFV promoter provides the best level
of transgene expression in Raji cells and LCLs
Since CMV and EF1α promoters allowed for ≤40.25%
transgene expression in Raji cells further experiments were
conducted using the SFFV promoter. The results demonstrated that
SFFV was much stronger in driving transgene expression in human
hematopoietic cells compared with other promoters, including CMV or
EF1α (12). Notably, the level of
transgene expression in Raji cells was >89% when the SFFV
promoter was used (Fig. 4).
Subsequently, the present study analyzed the levels of transgene
expression in Raji cells, LCLs and normal CD40L-stimulated B cells
using the HIV-SFFV-mRFP-WPRE vector. The SFFV promoter resulted in
high levels of transgene expression in Raji and LCL cells, as
determined by fluorescent microscopy; however, normal
CD40L-stimulated B cells were still resistant to modification
(Fig. 5).
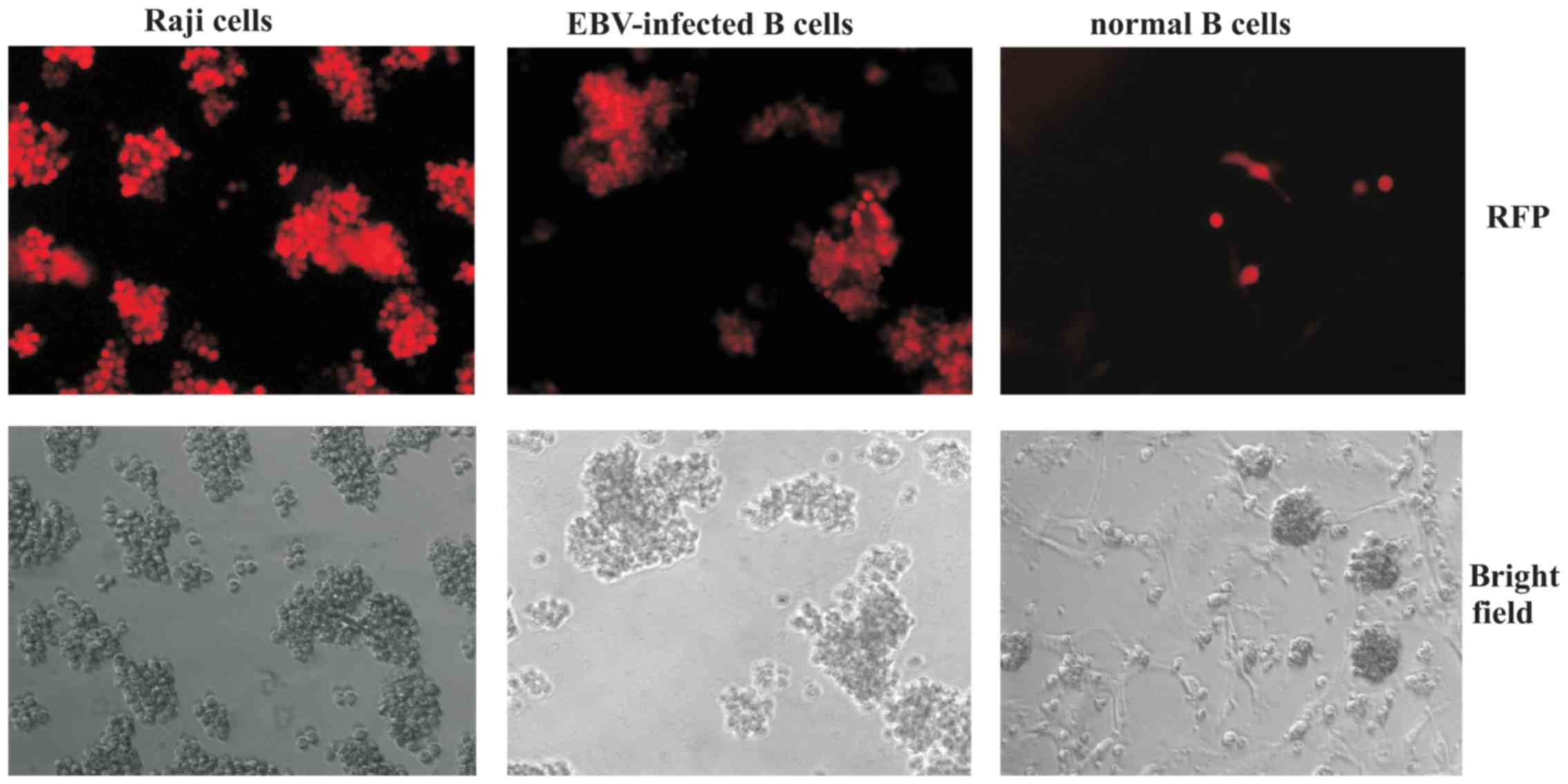 | Figure 5.Transduction of Raji cells, LCLs and
normal B cells with HIV-SFFV-mRFP-WPRE vector. Raji cells, LCLs and
normal B cells were modified with a HIV-SFFV-mRFP-WPRE vector.
After 7 days, cells were analyzed for RFP expression using
fluorescent microscopy (Nikon Eclipse TE2000-E microscope;
magnification, ×200). mRFP, monomeric red fluorescent protein;
LCLs, lymphoblastoid cell lines; HIV, human immunodeficiency virus;
SFFV, spleen focus-forming virus; WPRE, woodchuck hepatitis virus
posttranscriptional regulatory element; EBV, Epstein-Barr
virus. |
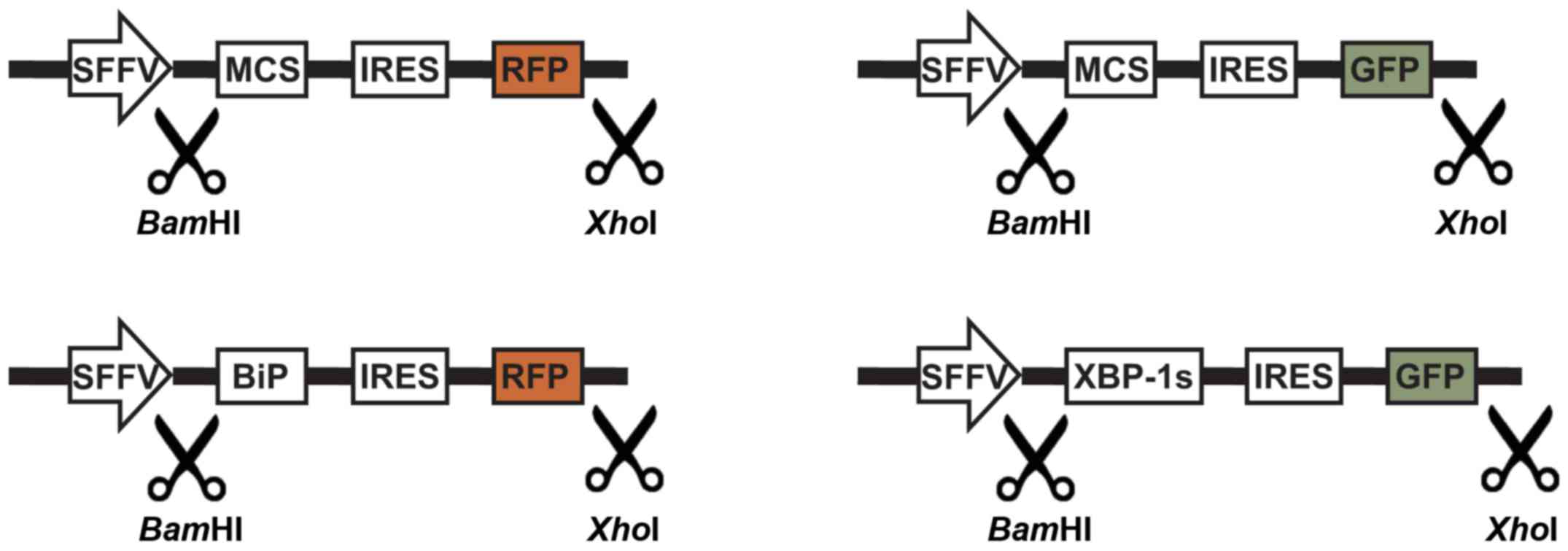
Constructs with IRES and fluorescent
markers enable the selection of transduced LCLs
Based on the HIV-SFFV-mRFP-WPRE vector, the present
study constructed vectors to allow expression of one transgene of
interest, and in addition, a fluorescent marker protein for
identification and selection of transduced cells. In order to
enable the coordinated and efficient expression of a transgene and
fluorescent marker directly from the SFFV promoter, attenuated IRES
was introduced in front of the fluorescent marker. Since a gene
located behind the IRES is expected to be translated with lower
efficiency than the one in front, such conformation ensures
high-level expression of the GOI, rather than the fluorescent
marker gene. As transgenes, the present study introduced genes
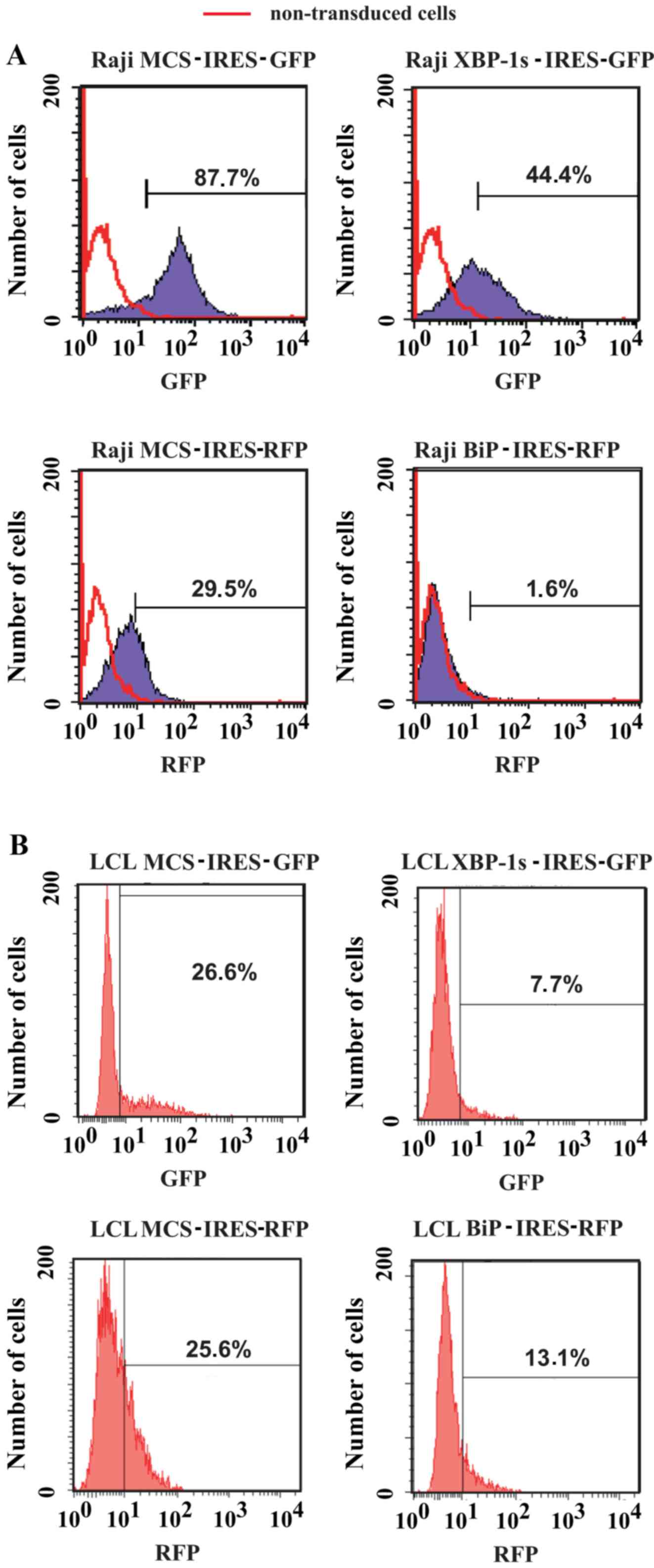
encoding XBP-1 s and BiP proteins (Fig. 6). Flow cytometric analysis and
sorting of Raji cells (Fig. 7A) or
LCLs (Fig. 7B) was conducted 10
days post-transduction. Notably, the levels of transgene expression
with GFP-encoding vectors was higher in Raji cells compared with in
LCLs, whereas the levels of transgene expression with RFP-encoding
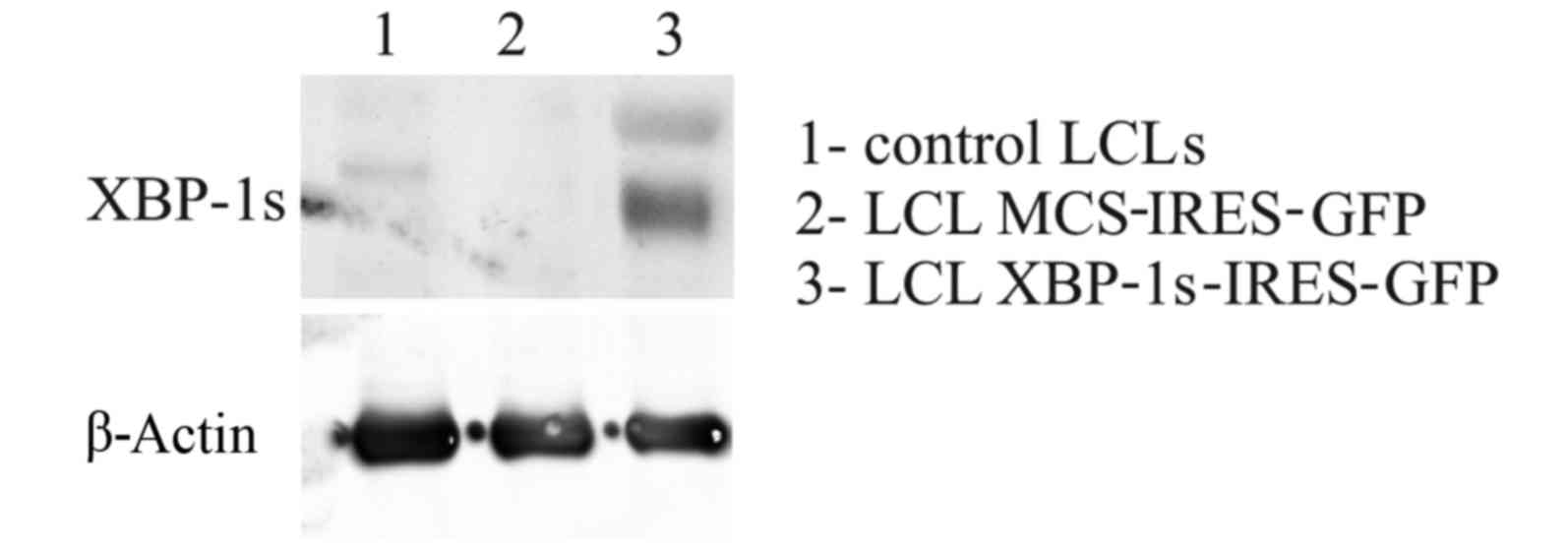
vectors were comparable between the cell lines (Fig. 7). Finally, after sorting, it was
demonstrated that LCLs were successfully modified to overexpress
the XBP-1 s protein, as confirmed with western blotting (Fig. 8).
Discussion
The present study aimed to identify a eukaryotic
promoter allowing for efficient and stable transduction of normal
human B cells with GOI. Quiescent cells are notoriously resistant
to various methods of gene transfer. Normal human B cells are
particularly difficult to transduce. Nonviral transfection
approaches are completely ineffective, and the use of viral vectors
requires complex genetic engineering to generate pseudotyped
lentiviruses. The present study aimed to evaluate the levels of
transgene expression with lentiviral vectors containing various
promoters, including CMV, EF1α and SFFV. However, despite
satisfactory (CMV and EF1α) and notable (SFFV) levels of transgene
expression (Fig. 4), allowing for
modification of ~90% of Raji cells, <10% of normal human B cells
were transduced with vectors containing these promoters (Fig 2B). Since Raji cells are immortalized
B cell-derived lymphoma cells carrying the latent EBV genome, the
present study aimed to investigate whether EBV infection of normal
B cells will make them more sensitive to lentiviral transduction.
Notably, infection of B cells with EBV resulted in the generation
of LCLs that could be effectively transduced with lentiviruses
carrying the SFFV promoter (Fig.
5). Therefore, these results confirmed the findings of a
previous study, which indicated that B cells stimulated with EBV
are easily modified with lentiviruses, whereas B cells activated
with CD40L and cytokines are resistant to transduction (17). To produce lentiviral particles the
2nd generation packaging vector psPAX2 and the VSVG-expressing
envelope vector pMD2.G were used. Using VSVG-lentiviruses the
present study managed to modify lymphoma cells and LCLs with high
efficacy; however, the present study failed to transduce quiescent
normal B cells. It has previously been reported that lentiviruses
pseudotyped with the Edmonston MV hemagglutinin and fusion
glycoproteins (Hgps and Fgps) allowed for efficient transduction of
quiescent human B and T cells (12). These findings strongly suggested
that the transduction efficiency may be determined at the level of
vector entry. It may be hypothesized that bicistronic vectors, when
used with lentiviral vectors pseudotyped with the Edmonston MV Hgps
and Fgps can serve as a potent transduction tool for normal B
cells.
As proof-of-concept, the present study used
bicistronic vectors containing genes encoding BiP or XBP-1, as well
as fluorescent reporter proteins mRFP or GFP, and demonstrated that
SFFV promoter-based lentiviruses may be used for stable
transduction of LCLs with GOI. In conclusion, the present study
demonstrated that it is possible to achieve an efficient and stable
lentiviral transduction of LCLs using SFFV promoter constructs.
This approach may be used in future studies aimed at studying
various processes associated with B-cell physiology. Such
bicistronic vectors expressing fluorescent proteins (GFP or mRFP)
also allow for tracking of modified cells in ex vivo or
in vivo studies.
Acknowledgements
The present study was supported by the National
Centre for Research and Development (grant no.
INNOTECH-K1/IN1/51/159542/NCBR/12).
References
|
1
|
Kay MA, Glorioso JC and Naldini L: Viral
vectors for gene therapy: The art of turning infectious agents into
vehicles of therapeutics. Nat Med. 7:33–40. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Naldini L, Blömer U, Gallay P, Ory D,
Mulligan R, Gage FH, Verma IM and Trono D: In vivo gene delivery
and stable transduction of nondividing cells by a lentiviral
vector. Science. 272:263–267. 1996. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jaleco AC, Stegmann AP, Heemskerk MH,
Couwenberg F, Bakker AQ, Weijer K and Spits H: Genetic modification
of human B-cell development: B-cell development is inhibited by the
dominant negative helix loop helix factor Id3. Blood. 94:2637–2646.
1999.PubMed/NCBI
|
|
4
|
Serafini M, Naldini L and Introna M:
Molecular evidence of inefficient transduction of proliferating
human B lymphocytes by VSV-pseudotyped HIV-1-derived lentivectors.
Virology. 325:413–424. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kvell K, Nguyen TH, Salmon P, Glauser F,
Werner-Favre C, Barnet M, Schneider P, Trono D and Zubler RH:
Transduction of CpG DNA-stimulated primary human B cells with
bicistronic lentivectors. Mol Ther. 12:892–899. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Janssens W, Chuah MK, Naldini L, Follenzi
A, Collen D, Saint-Remy JM and VandenDriessche T: Efficiency of
onco-retroviral and lentiviral gene transfer into primary mouse and
human B-lymphocytes is pseudotype dependent. Hum Gene Ther.
14:263–276. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Yang L, Bailey L, Baltimore D and Wang P:
Targeting lentiviral vectors to specific cell types in vivo. Proc
Natl Acad Sci USA. 103:11479–11484. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Burns JC, Friedmann T, Driever W,
Burrascano M and Yee JK: Vesicular stomatitis virus G glycoprotein
pseudotyped retroviral vectors: Concentration to very high titer
and efficient gene transfer into mammalian and nonmammalian cells.
Proc Natl Acad Sci USA. 90:8033–8037. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Finkelshtein D, Werman A, Novick D, Barak
S and Rubinstein M: LDL receptor and its family members serve as
the cellular receptors for vesicular stomatitis virus. Proc Natl
Acad Sci USA. 110:7306–7311. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Amirache F, Lévy C, Costa C, Mangeot PE,
Torbett BE, Wang CX, Nègre D, Cosset FL and Verhoeyen E: Mystery
solved: VSV-G-LVs do not allow efficient gene transfer into
unstimulated T cells, B cells, and HSCs because they lack the LDL
receptor. Blood. 123:1422–1424. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Frecha C, Costa C, Lévy C, Nègre D,
Russell SJ, Maisner A, Salles G, Peng KW, Cosset FL and Verhoeyen
E: Efficient and stable transduction of resting B lymphocytes and
primary chronic lymphocyte leukemia cells using measles virus gp
displaying lentiviral vectors. Blood. 114:3173–3180. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Frecha C, Lévy C, Costa C, Nègre D,
Amirache F, Buckland R, Russell SJ, Cosset FL and Verhoeyen E:
Measles virus glycoprotein-pseudotyped lentiviral vector-mediated
gene transfer into quiescent lymphocytes requires binding to both
SLAM and CD46 entry receptors. J Virol. 85:5975–5985. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Funke S, Maisner A, Mühlebach MD, Koehl U,
Grez M, Cattaneo R, Cichutek K and Buchholz CJ: Targeted cell entry
of lentiviral vectors. Mol Ther. 16:1427–1436. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dörig RE, Marcil A, Chopra A and
Richardson CD: The human CD46 molecule is a receptor for measles
virus (Edmonston strain). Cell. 75:295–305. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tatsuo H, Ono N, Tanaka K and Yanagi Y:
SLAM (CDw150) is a cellular receptor for measles virus. Nature.
406:893–897. 2000. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Sambrook J, Fritsch EF and Maniatis T:
Molecular Cloning: A laboratory manual. 2nd. Cold Spring Harbor
Laboratory Press; Cold Spring Harbor, NY: pp. 1.63–1.70. 1989
|
|
17
|
Bovia F, Salmon P, Matthes T, Kvell K,
Nguyen TH, Werner-Favre C, Barnet M, Nagy M, Leuba F, Arrighi JF,
et al: Efficient transduction of primary human B lymphocytes and
nondividing myeloma B cells with HIV-1-derived lentiviral vectors.
Blood. 101:1727–1733. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Heath E, Begue-Pastor N, Chaganti S,
Croom-Carter D, Shannon-Lowe C, Kube D, Feederle R, Delecluse HJ,
Rickinson AB and Bell AI: Epstein-Barr virus infection of naïve B
cells in vitro frequently selects clones with mutated
immunoglobulin genotypes: Implications for virus biology. PLoS
Pathog. 8:e10026972012. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Joseph AM, Babcock GJ and Thorley-Lawson
DA: EBV persistence involves strict selection of latently infected
B cells. J Immunol. 165:2975–2981. 2000. View Article : Google Scholar : PubMed/NCBI
|