Introduction
Cardiac fibrosis is an important pathological
feature of cardiac remodeling in heart diseases (1) and remains a major contributor to
morbidity and mortality rates in a variety of cardiovascular
diseases, including myocardial infarction, cardiac hypertrophy,
heart failure and severe arrhythmia (2). Although significant therapeutic
progress has been made in previous decades (3–5), the
molecular mechanisms underlying the development of cardiac fibrosis
remain to be elucidated. Cardiac fibroblasts (CFs), the most
prevalent cell type in the heart, are key in the regulation of
normal myocardial function. The proliferation of CFs and excessive
deposition of extracellular matrix (ECM) are the primary
pathological characteristics of cardiac fibrosis. It is also known
that transforming growth factor-β (TGF-β) is pivotal in mediating
CF function and cardiac fibrosis (6). CFs differentiate into cardiac
myofibroblasts (CMFs) via TGF-β1, and these differentiated cells
are actively involved in cardiac fibrosis.
Nucleotide-binding domain and leucine-rich repeat
(NLR) proteins are important in innate immune responses as
pattern-recognition receptors. NLRC5, the largest member of the NLR
protein family, contains three structural domains, including the
N-terminal atypical caspase activation and recruitment domain, the
centrally located NACHT (named after neuronal apoptosis inhibitory
protein, class II major histocompatibility complex transactivator,
HET-E and transition protein 1 proteins) and 27 leucine-rich
repeats at the C-terminal. Increasing evidence indicates that NLRC5
is important in regulating immune responses (7–9). For
example, Staehli et al reported that NLRC5 is expressed at
high levels and required for the regulating the expression of major
histocompatibility complex I in lymphocytes (10). Another previous study showed that
the knockdown of NLRC5 significantly suppressed TGF-β1-induced
proliferation, but increased apoptosis, and inhibited the
expression levels of collagen 1 and α-smooth muscle actin (α-SMA)
in hepatic stellate cells (11).
However, whether NLRC5 is involved in the pathogenesis of cardiac
fibrosis remains to be elucidated. The aim of the present study was
to examine the role of NLRC5 and its mechanisms in regulating
cardiac fibrosis.
Materials and methods
Cell culture
A total of 6 female Sprague-Dawley rats (age, 6
weeks; weight, 180–200 g) were obtained from the Animal Breeding
Center of Tianjin Medical University Metabolic Diseases Hospital
(Tianjin, China). They were housed in barrier facilities under a
12-h light/dark cycle at a temperature of 22±2°C and had free
access to laboratory chow and tap water. Rats were used to harvest
CFs. Briefly, rats were anesthetized intraperitoneally with sodium
pentobarbital (50 mg/kg). Heart ventricles were removed under
sterile conditions, placed in cold sterile calcium-free PBS, minced
into ~2-mm cubes, and treated with 1 mg/ml type II collagenase
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Dissociated cells
were cultured in Dulbecco's modified Eagle's medium (DMEM;
Invitrogen; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (FBS; Sigma-Aldrich; Merck
KGaA), 100 U/ml penicillin sulfate and 100 U/ml streptomycin
(Sigma-Aldrich; Merck KGaA) at pH 7.4, in an incubator with a
humidified atmosphere of 5% CO2 at 37°C. All
experimental procedures were approved by the guidelines of the
Animal Care and Use Committee of Tianjin Medical University
Metabolic Diseases Hospital (Tianjin, China).
Small interfering RNA (siRNA)
transfection
CFs at a density of 1×104 cells/well were
cultured to 80% confluence and transfected with small interfering
(si)RNA (2.5 µg) targeting NLRC5 (forward,
5′-GGGACTGAGAGCTTTGTAT-3′ and reverse, 5′-CGCACCCTAGACTGAAA-3′) or
with a non-targeting scrambled siRNA (forward,
5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′) at room temperature for 24 h, using
Lipofectamine™ RNAiMAX (Invitrogen; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. The siRNAs targeting rat
NLRC5 and scrambled siRNA were from GenePharma (Shanghai,
China).
Cell proliferation assay
An MTT assay was used to measure cell proliferation.
Briefly, the CFs were seeded at a density of 1×104
cells/well into 24-well plates and transfected with siRNA-NLRC5 or
scramble siRNA for 24 h. The cells were then treated with TGF-β1
(10 ng/ml; Sigma-Aldrich; Merck KGaA) for another 24 h.
Subsequently, 20 µl MTT (5 mg/ml) solution was added to each well
and incubation continued at 37°C for 4 h, followed by removal of
the culture medium and the addition of 100 ml of dimethyl sulfoxide
(Sigma-Aldrich; Merck KGaA). The absorbance at 450 nm was measured
using an ELISA microplate reader (Invitrogen; Thermo Fisher
Scientific, Inc.).
Transwell migration assay
Cell migration was analyzed using a Transwell
chamber (Corning Costar, Cambridge, MA, USA) assay. Briefly, the
CFs (1×104 cells/ml) transfected with siRNA-NLRC5 or
scramble siRNA were resuspended in 0.1 ml serum-free DMEM and
placed in the upper chambers. The lower chambers were filled with
500 µl DMEM containing 10% FBS as a chemoattractant. After 24 h
incubation at 37°C, the cells on the surface of upper chamber were
removed by scraping with a cotton swab. The migrated cells on the
lower surface of the filter were washed with TBS containing 0.1%
Tween-20, fixed with 100% methanol at 37°C for 15 min, stained with
0.1% hematoxylin and eosin at 37°C for 20 min, and counted under an
optical microscope (Olympus Corporation, Tokyo, Japan) in 5
randomly selected fields of view.
RNA isolation and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
analysis
Total RNA was extracted from the CFs using TRIzol
reagent (Invitrogen; Thermo Fisher Scientific, Inc.), according to
the manufacturer's protocol. Total RNA (1 µg) was reverse
transcribed into cDNA using the Transcriptor First Strand cDNA
Synthesis kit (Invitrogen; Thermo Fisher Scientific, Inc.),
according to the manufacturer's protocol. qPCR was performed on
cDNA using the SYBR green detection system (Bio SYBR-Green Master
mix; Takara Bio, Inc., Otsu, Japan). The reaction mixture contained
cDNA templates (1 µl), primers (2 µl of each forward and reverse
primer) and SYBR-Green qPCR Master mix (5 µl). The specific primers
were as follows: NLRC5 forward, 5′-CAGATGGTGGAAACTTTTAGCC-3′ and
reverse, 5′-AACTTCCTTAGCACCTGGATCA-3′; α-SMA forward,
5′-CTATTCCTTCGTGACTACT-3′ and reverse, 5′-ATGCTGTTATAGGTGGTGGTT-3′;
collagen I forward, 5′-TGGTGAACAGCCTGTACCCT-3′ and reverse,
5′-CACGGTAGTGCCCATCATTC-3′; connective tissue growth factor (CTGF)
forward, 5′-CAGGCTGGAGAAGCAGAGTCGT-3′ and reverse,
5′-CTGGTGCAGCCAGAAAGCTCAA-3′; and β-actin forward,
5′-GAGGCACTCTTCCAGCCTTC-3′ and reverse, 5′-GGATGTCCACGTCACACTTC-3′.
The protocol comprised 35 cycles at 94°C for 5 sec, at 59°C for 30
sec, and at 72°C for 1 min. The ratio of the relative expression of
target genes to β-actin was calculated using the 2−ΔΔCq
method from the quantification cycle numbers (12).
Western blot analysis
The CFs were lysed in radioimmunoprecipitation assay
lysis buffer (Beyotime Institute of Biotechnology, Nantong, China)
containing a phosphatase inhibitor and a protease inhibitor
cocktail (Sigma-Aldrich; Merck KGaA) on ice for 10 min. Protein
concentrations were determined using a bicinchoninic acid protein
assay kit (Pierce; Thermo Fisher Scientific, Inc.). Equal
quantities of protein (40 µg/lane) were loaded and separated by 12%
SDS-PAGE, and transferred onto nitrocellulose membranes (Bio-Rad
Laboratories, Inc., Hercules, CA, USA). Non-specific binding was
blocked by incubation with 5% non-fat milk in PBS containing 0.1%
Tween-20 at room temperature for 1 h. The membranes were then
incubated with primary antibodies overnight at 4°C, followed by
incubation with the appropriate horseradish peroxidase-conjugated
secondary antibody (1:2,500; cat. no. sc-516087; Santa Cruz
Biotechnology Inc., Dallas, TX, USA) at room temperature for 1 h.
The proteins were visualized using an enhanced chemiluminescence
detection system (Invitrogen; Thermo Fisher Scientific, Inc.). The
following antibodies were used: Goat anti-NLRC5 (1:3,000; cat. no.
sc-248094; Santa Cruz Biotechnology Inc.), rabbit anti-α-SMA
(1:3,000; cat. no. PA5-19465; Invitrogen; Thermo Fisher Scientific,
Inc.), rabbit anti-collagen I (1:3,000; cat. no. sc-28657; Santa
Cruz Biotechnology Inc.), rabbit anti-CTGF (1:2,500; cat. no.
sc-25440; Santa Cruz Biotechnology Inc.), rabbit anti-Smad3
(1:2,500; cat. no. PA5-34774; Invitrogen; Thermo Fisher Scientific,
Inc.), rabbit anti-phosphorylated (p-)Smad3 (1:3,000; cat. no.
44-246G; Invitrogen; Thermo Fisher Scientific, Inc.) and rabbit
anti-GAPDH (1:3,000; cat. no. sc-25778; Santa Cruz Biotechnology
Inc.) antibodies. Densitometry was performed using Gel-Pro Analyzer
software version 4.0 (Media Cybernetics, Inc., Rockville, MD,
USA).
Statistical analysis
Statistical analysis was performed using SPSS
software version 13.0 (SPSS, Inc., Chicago, IL, USA). Data are
expressed as the mean ± standard deviation of triplicate
independent samples. Comparisons between two groups and among
multiple groups were conducted using Student t-test and one-way
analysis of variance followed by Tukey's post hoc test,
respectively. P<0.05 was considered to indicate a statistically
significant difference.
Results
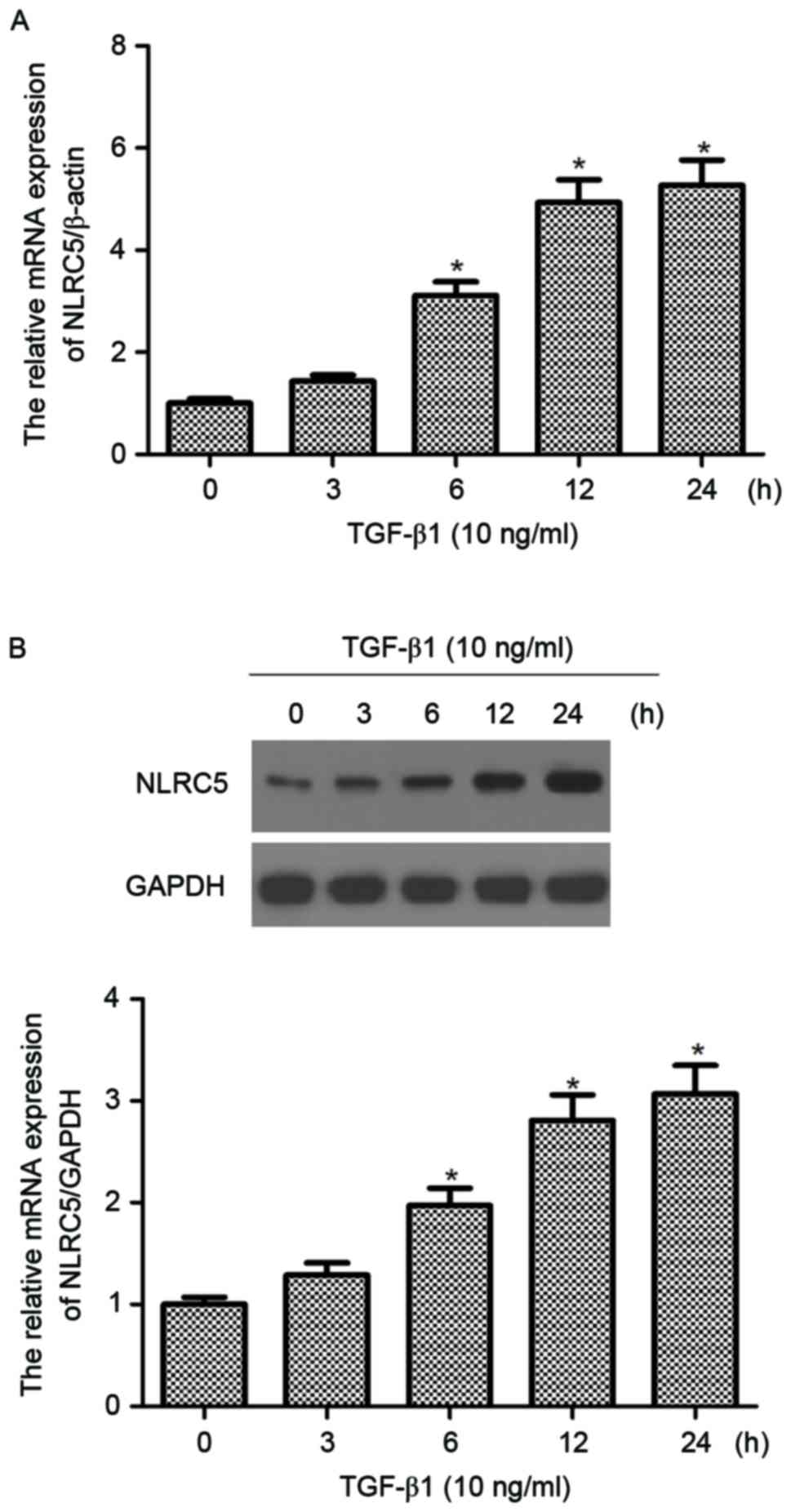
NLRC5 is upregulated in TGF-β1-induced
CFs
The present study investigated the expression of
NLRC5 in TGF-β1-induced CFs. As shown in Fig. 1A, compared with the untreated
group, the mRNA expression of NLRC5 was significantly increased by
TGF-β1 in CFs and increased in a time-dependent manner. The western
blot analysis demonstrated that the protein expression of NLRC5 was
also increased when incubated with TGF-β1 (Fig. 1B).
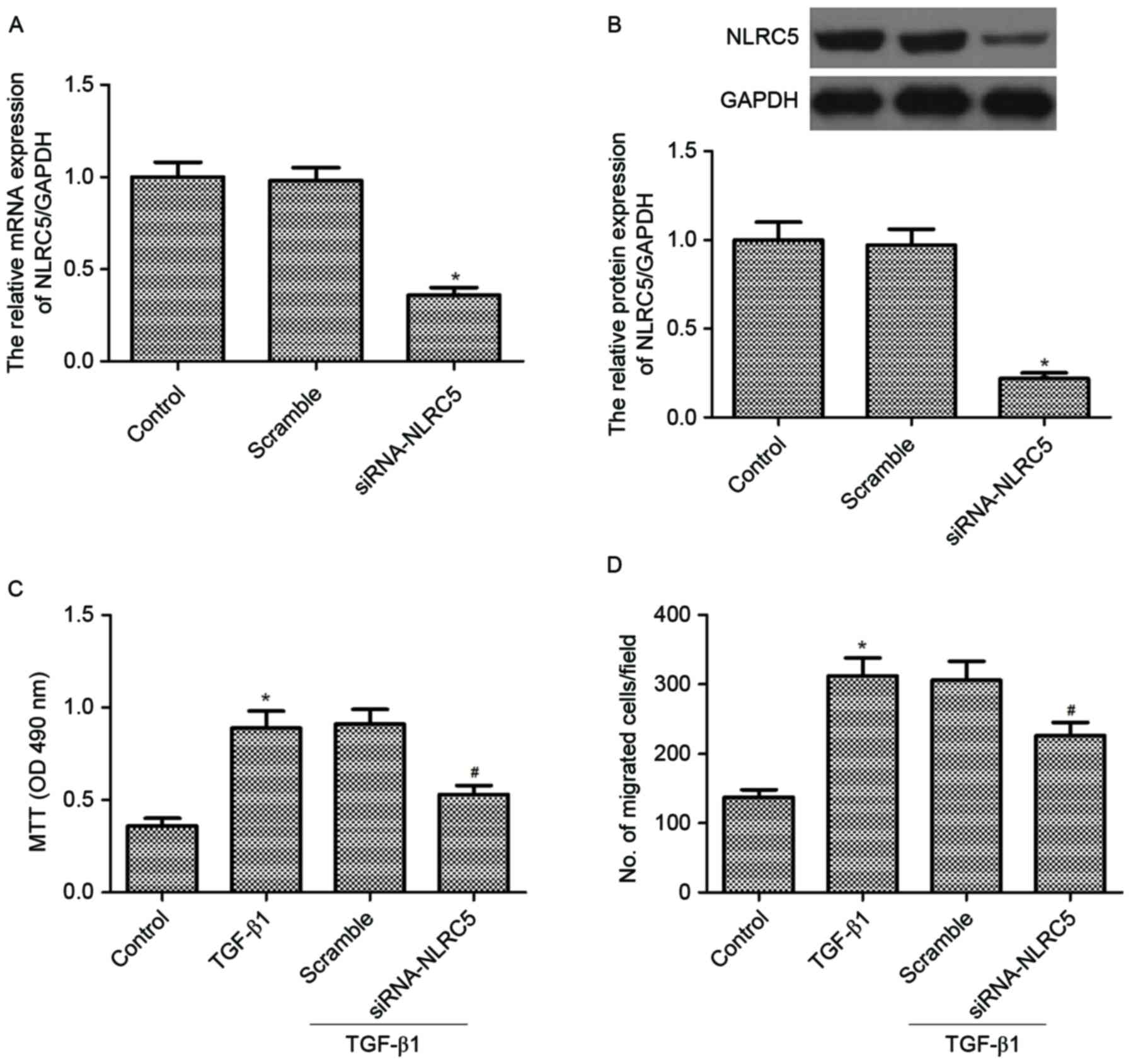
Silencing NLRC5 inhibits cell
proliferation and migration induced by TGF-β1 in CFs
To characterize the biological effect of NLRC5 on
cell proliferation and migration in CFs, NLRC5 was first knocked
down in the CFs using siRNA. The decreased expression levels of
NLRC5 were confirmed using RT-qPCR and western blot analyses. The
results demonstrated that the downregulation of NLRC5 significantly
decreased the mRNA and protein expression levels of NLRC5,
respectively (Fig. 2A and B).
The present study then examined the effect of NLRC5
on cell proliferation and migration in CFs induced by TGF-β1. The
results of the MTT assays showed that TGF-β1 significantly
increased the proliferation of CFs, compared with the control
group. However, silencing NLRC5 markedly inhibited TGF-β1-induced
CF proliferation (Fig. 2C).
Similarly, it was found that silencing NLRC5 markedly inhibited
TGF-β1-induced CF migration (Fig.
2D).
Silencing NLRC5 inhibits the
expression of α-SMA and pro-fibrotic molecules induced by TGF-β1 in
CFs
As the expression of α-SMA is a hallmark of
myofibroblast differentiation, the effects of NLRC5 on
TGF-β1-induced mRNA and protein levels of α-SMA were measured using
RT-qPCR and western blot analyses, respectively. As shown in
Fig. 3, compared with the control
group, TGF-β1 treatment markedly induced the expression of α-SMA at
the mRNA and protein levels. However, silencing NLRC5 inhibited the
TGF-β1-induced expression of α-SMA at the mRNA and protein levels.
Similarly, silencing NLRC5 suppressed the TGF-β1-induced expression
levels of collagen I and CTGF.
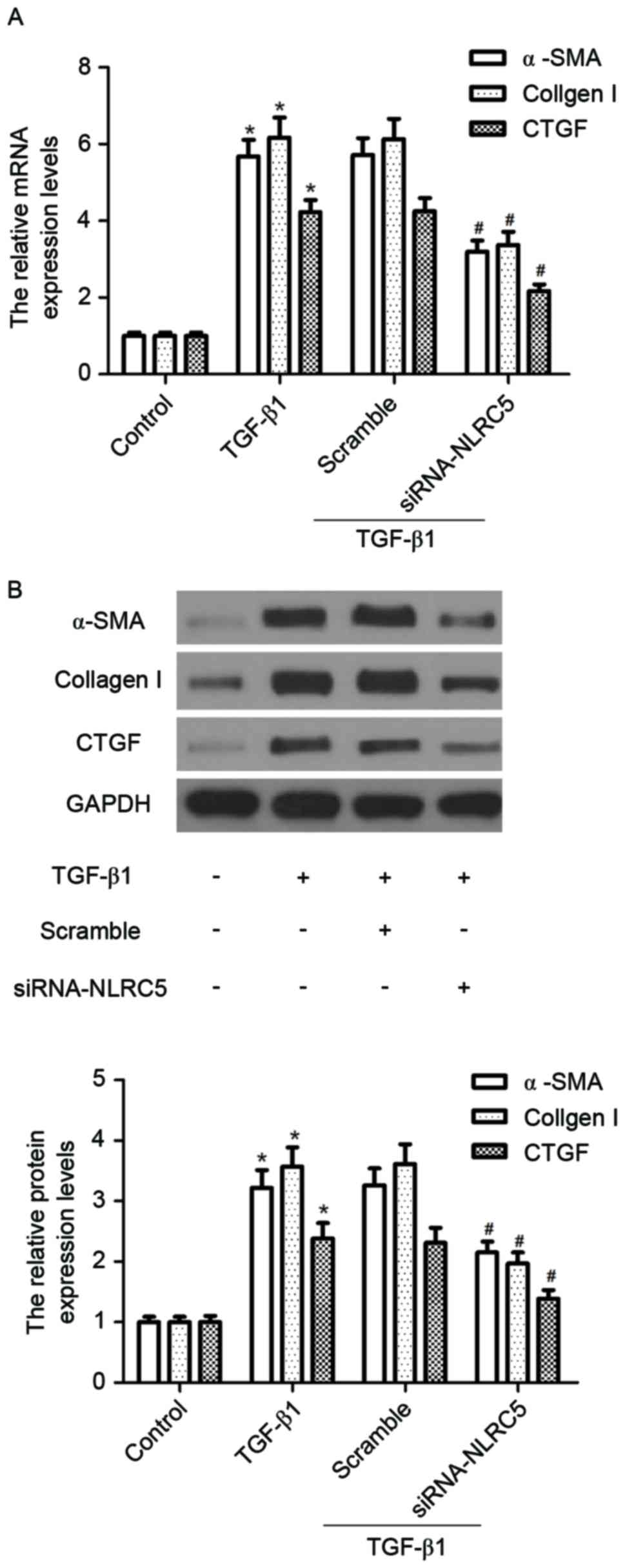 | Figure 3.Silencing NLRC5 inhibits the
expression of α-SMA and pro-fibrotic molecules induced by TGF-β1 in
CFs. CFs were seeded at a density of 1×104 cells/well
into 24-well plates and transfected with siRNA-NLRC5 or scramble
for 24 h, then stimulated with 10 ng/ml TGF-β1 for 24 h. (A) mRNA
expression levels of α-SMA, collagen I and CTGF were detected using
reverse transcription-quantitative polymerase chain reaction
analysis; (B) protein expression levels of α-SMA, collagen I and
CTGF were measured using western blot analysis. Relative
quantitative analyses of protein levels of α-SMA, collagen I and
CTGF were normalized to GAPDH. The results are expressed as the
mean ± standard deviation of three independent experiments.
*P<0.05, vs. control group; #P<0.05, vs. scramble
siRNA+TGF-β1 group. CFs, cardiac fibroblasts; siRNA, small
interfering RNA; α-SMA, α-smooth muscle actin; TGF-β1, transforming
growth factor-β1; CTGF, connective tissue growth factor. |
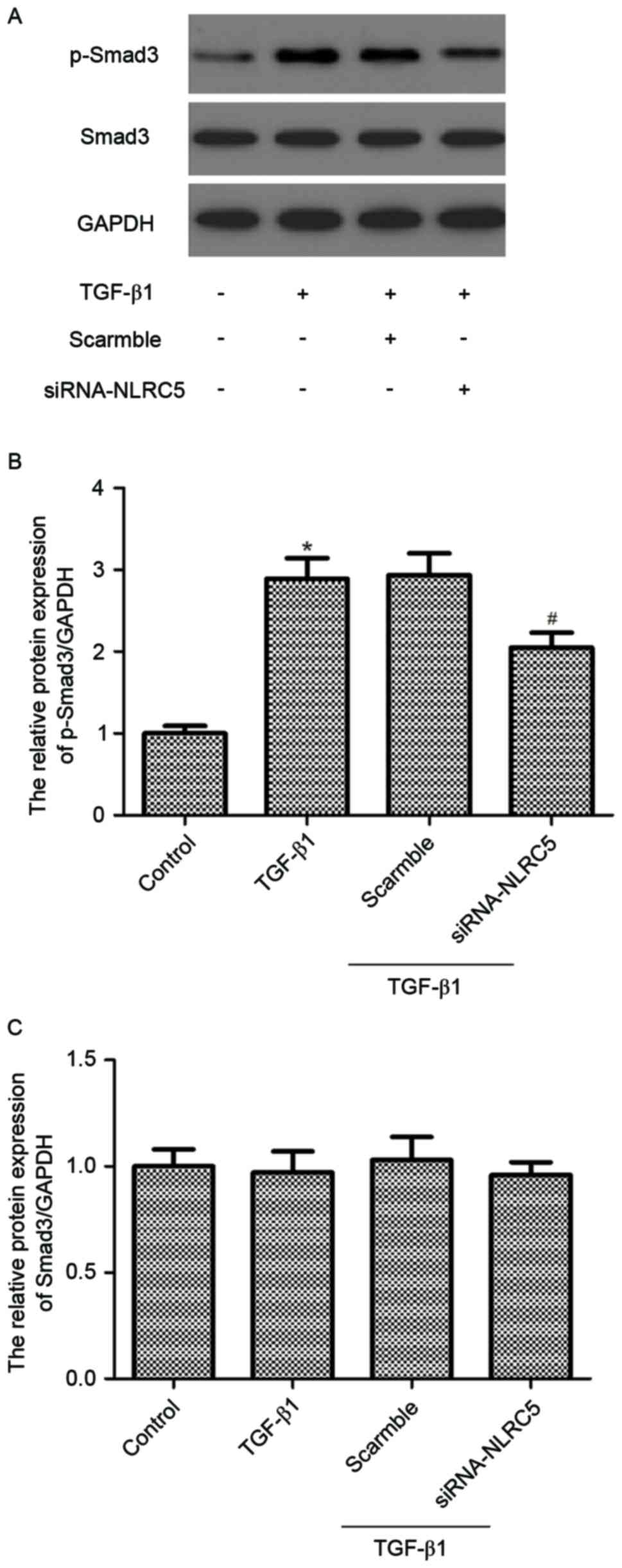
Silencing NLRC5 attenuates
TGF-β1-induced phosphorylation of Smad3 in CFs
It has been reported that activation of TGF-β1/Smad3
signaling is important in the development and progression of
cardiac fibrosis (13). Therefore,
the present study examined the effect of siRNA-NLRC5 on
TGF-β1/Smad3 signaling in CFs. The results showed that TGF-β1
treatment increased the phosphorylation of Smad3 in the cultured
rat CFs. However, silencing NLRC5 significantly inhibited the
phosphorylation of Smad3 induced by TGF-β1 (Fig. 4).
Discussion
In the present study, it was demonstrated that NLRC5
was upregulated in TGF-β1-induced CFs. The knockdown of NLRC5
inhibited cell proliferation and migration, it also suppressed
myofibroblast differentiation and the expression of pro-fibrotic
molecules in the TGF-β1-treated CFs. Furthermore, the knockdown of
NLRC5 attenuated the TGF-β1-induced phosphorylation of Smad3 in the
CFs.
NLRC5 was previously shown to be a critical
modulator in liver fibrogenesis, in which NLRC5 was significantly
upregulated in human liver fibrotic tissues (11). Consistent with the results of this
previous study, the present study observed that NLRC5 was
upregulated in TGF-β1-induced CFs, indicating that NLRC5 might be
in the development of cardiac fibrosis.
The proliferation of CFs is the primary pathological
characteristic of cardiac fibrosis (14). It has been reported that
myofibroblasts originate from resident fibroblasts, and invade and
repair injured tissues by secreting and organizing the ECM
(15). In the present study, it
was found that the knockdown of NLRC5 inhibited cell proliferation
and migration. These results suggested that siRNA-NLRC5 exerted an
anti-fibrotic effect through inhibiting the proliferation and
migration of CF.
The differentiation and activation of fibroblasts
into myofibroblasts, which express α-SMA, are essential in cardiac
fibrosis (16). Excessive collagen
deposition in the heart contributes to cardiac fibrosis (17). CTGF, a crucial pro-fibrotic factor,
also contributes to myofibroblast differentiation and activation,
and is a marker for activated fibroblasts in cardiac fibrosis
(18). Previous studies have shown
that TGF-β1 can stimulate collagen synthesis and inhibit the
degradation of collagen (19,20).
In the present study, it was found that TGF-β1 treatment induced
the expression levels of α-SMA, collagen I and CTGF. However,
silencing NLRC5 inhibited the expression of pro-fibrotic molecules
in the TGF-β1-treated CFs. These results suggested that siRNA-NLRC5
exerted an anti-fibrotic effect through inhibiting myofibroblast
differentiation and the expression of ECM in CFs.
Previous evidence indicates that the TGF-β1/Smad
signaling pathway is crucial in the myocardial remodeling process,
particularly in cardiac fibrosis (21–24).
As a primary downstream signal transducer of TGF-β1, Smad3 can be
phosphorylated by the activated type I receptor of TGF-β1, followed
by the formation of a complex with Smad4 and translocation into the
nucleus, where it acts as a transcription factor and regulates the
expression of target genes, including type I, type III collagen,
α-SMA and CTGF (25,26). It has been shown in several
experiments that TGF-β1 activates cardiac fibrosis, predominantly
through the TGF-β1/Smad signaling pathway. Bujak et al
confirmed that the TGF-β1-mediated induction of procollagen type
III and tenascin-C in isolated CFs is dependent on Smad3 (26). Another previous study reported that
Smad3 null fibroblasts showed impaired myofibroblast
transdifferentiation, reduced migratory potential and reduced
capacity to contract collagen pads upon TGF-β1 stimulation
(27). In the present study, it
was found that the knockdown of NLRC5 attenuated the TGF-β1-induced
phosphorylation of Smad3 in CFs. These results suggested that NLRC5
silencing ameliorated cardiac fibrosis by inhibiting the
TGF-β1/Smad3 signaling pathway in the rat CFs.
The results of the present study indicated that
NLRC5 acted as a key regulator of pathological cardiac fibrosis,
and that NLRC5 silencing ameliorated cardiac fibrosis by inhibiting
the TGF-β1/Smad3 signaling pathway. These results suggested that
NLRC5 may be a novel target for attenuating cardiac fibrosis.
References
|
1
|
Krenning G, Zeisberg EM and Kalluri R: The
origin of fibroblasts and mechanism of cardiac fibrosis. J Cell
Physiol. 225:631–637. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Burlew BS and Weber KT: Cardiac fibrosis
as a cause of diastolic dysfunction. Herz. 27:92–98. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Leask A: Potential therapeutic targets for
cardiac fibrosis: TGFbeta, angiotensin, endothelin, CCN2, and PDGF,
partners in fibroblast activation. Circ Res. 106:1675–1680. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Spinale FG, Coker ML, Bond BR and Zellner
JL: Myocardial matrix degradation and metalloproteinase activation
in the failing heart: A potential therapeutic target. Cardiovasc
Res. 46:225–238. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Brilla CG, Funck RC and Rupp H:
Lisinopril-mediated regression of myocardial fibrosis in patients
with hypertensive heart disease. Circulation. 102:1388–1393. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lijnen PJ, Petrov VV and Fagard RH:
Induction of cardiac fibrosis by transforming growth
factor-beta(1). Mol Genet Metab. 71:418–435. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Davis BK, Roberts RA, Huang MT, Willingham
SB, Conti BJ, Brickey WJ, Barker BR, Kwan M, Taxman DJ,
Accavitti-Loper MA, et al: Cutting edge: NLRC5-dependent activation
of the inflammasome. J Immunol 186. 186:1333–1337. 2011. View Article : Google Scholar
|
|
8
|
Kuenzel S, Till A, Winkler M, Häsler R,
Lipinski S, Jung S, Grötzinger J, Fickenscher H, Schreiber S and
Rosenstiel P: The nucleotide-binding oligomerization domain-like
receptor NLRC5 is involved in IFN-dependent antiviral immune
responses. J Immunol. 184:1990–2000. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Meissner TB, Li A, Biswas A, Lee KH, Liu
YJ, Bayir E, Iliopoulos D, van den Elsen PJ and Kobayashi KS: NLR
family member NLRC5 is a transcriptional regulator of MHC class I
genes. Proc Natl Acad Sci USA. 107:13794–13799. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Staehli F, Ludigs K, Heinz LX,
Seguín-Estévez Q, Ferrero I, Braun M, Schroder K, Rebsamen M,
Tardivel A, Mattmann C, et al: NLRC5 deficiency selectively impairs
MHC class I-dependent lymphocyte killing by cytotoxic T cells. J
Immunol. 188:3820–3828. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Xu T, Ni MM, Xing-Li, Li XF, Meng XM,
Huang C and Li J: NLRC5 regulates TGF-β1-induced proliferation and
activation of hepatic stellate cells during hepatic fibrosis. Int J
Biochem Cell Biol. 70:92–104. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(−Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Y, Huang XR, Wei LH, Chung AC, Yu CM
and Lan HY: miR-29b as a therapeutic agent for angiotensin
II-induced cardiac fibrosis by targeting TGF-β/Smad3 signaling. Mol
Ther. 22:974–985. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Porter KE and Turner NA: Cardiac
fibroblasts: At the heart of myocardial remodeling. Pharmacol Ther.
123:255–278. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
van Nieuwenhoven FA and Turner NA: The
role of cardiac fibroblasts in the transition from inflammation to
fibrosis following myocardial infarction. Vasc Pharmacol.
58:182–188. 2013. View Article : Google Scholar
|
|
16
|
Lijnen P, Petrov V and Fagard R:
Transforming growth factor-beta 1-mediated collagen gel contraction
by cardiac fibroblasts. J Renin Angiotensin Aldosterone Syst.
4:113–118. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Carver W, Nagpal ML, Nachtigal M, Borg TK
and Terracio L: Collagen expression in mechanically stimulated
cardiac fibroblasts. Circ Res. 69:116–122. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Chen MM, Lam A, Abraham JA, Schreiner GF
and Joly AH: CTGF expression is induced by TGF- beta in cardiac
fibroblasts and cardiac myocytes: a potential role in heart
fibrosis. J Mol Cell Cardiol. 32:1805–1819. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bujak M and Frangogiannis NG: The role of
TGF-beta signaling in myocardial infarction and cardiac remodeling.
Cardiovasc Res. 74:184–195. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Seeland U, Haeuseler C, Hinrichs R,
Rosenkranz S, Pfitzner T, Scharffetter-Kochanek K and Böhm M:
Myocardial fibrosis in transforming growth factor-beta(1)
(TGF-beta(1)) transgenic mice is associated with inhibition of
interstitial collagenase. Eur J Clin Invest. 32:295–303. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhao XY, Zhao LY, Zheng QS, Su JL, Guan H,
Shang FJ, Niu XL, He YP and Lu XL: Chymase induces profibrotic
response via transforming growth factor-beta 1/Smad activation in
rat cardiac fibroblasts. Mol Cell Biochem. 310:159–166. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Araújo-Jorge TC, Waghabi MC,
Hasslocher-Moreno AM, Xavier SS, Higuchi Mde L, Keramidas M, Bailly
S and Feige JJ: Implication of transforming growth factor-beta1 in
Chagas disease myocardiopathy. J Infect Dis. 186:1823–1828. 2002.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shyu KG, Wang BW, Chen WJ, Kuan P and Hung
CR: Mechanism of the inhibitory effect of atorvastatin on endoglin
expression induced by transforming growth factor-beta1 in cultured
cardiac fibroblasts. Eur J Heart Fail. 12:219–226. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Sassoli C, Chellini F, Pini A, Tani A,
Nistri S, Nosi D, Zecchi-Orlandini S, Bani D and Formigli L:
Relaxin prevents cardiac fibroblast-myofibroblast transition via
notch-1-mediated inhibition of TGF-β/Smad3 signaling. PLoS One.
8:e638962013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Leask A: TGFbeta, cardiac fibroblasts, and
the fibrotic response. Cardiovasc Res. 74:207–212. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Bujak M, Ren G, Kweon HJ, Dobaczewski M,
Reddy A, Taffet G, Wang X-F and Frangogiannis NG: Essential role of
Smad3 in infarct healing and in the pathogenesis of cardiac
remodeling. Circulation. 116:2127–2138. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Dobaczewski M, Bujak M, Li N,
Gonzalez-Quesada C, Mendoza LH, Wang XF and Frangogiannis NG: Smad3
signaling critically regulates fibroblast phenotype and function in
healing myocardial infarction. Circ Res. 107:418–428. 2010.
View Article : Google Scholar : PubMed/NCBI
|