Introduction
Craniosynostosis is characterized by premature
fusion of one or more cranial sutures, resulting in an abnormal
growth pattern of the skull (1).
Craniosynostosis can be clinically manifested as Crouzon syndrome,
Jackson-Weiss syndrome, or Pfeiffer syndrome. Crouzon syndrome,
first reported by Louis Edouard Octave (2–5) in
1912, is recognized as one of the most common craniosynostosis
syndromes (6). The prevalence of
Crouzon syndrome is between 1/60,000 to 1/1,000 live birth,
depending on race, region, and ethnicity (7–9).
Crouzon syndrome is typically characterized by craniosynostosis,
exorbitism, hypertelorism, midface hypoplasia, hooked nose, thin
vermilion of the upper lip, and mandibular prognathism (10,11).
Unlike Pfeiffer syndrome that can present hand abnormalities, such
as wide and deviated thumbs, or Jackson-Weiss syndrome that can
present broad great toes with medial deviation and
tarsal-metatarsal coalescence, Crouzon syndrome usually does not
present limb abnormalities (1,9).
Most patients with Crouzon syndrome present altered ocular
appearance such as ocular proptosis, and initially seek medical
care from neurosurgeons or ophthalmologists, rather than
orthopedists (8,12,13).
Since Crouzon syndrome is a relatively rare syndrome, and is
usually not easy to diagnose, molecular diagnosis will provide
useful information for the disease diagnosis and genetic
counseling.
Craniosynostosis is generally associated with
abnormal function of fibroblast growth factor receptors (FGFRs)
(14,15). To date, more than 50 distinct
mutations in the FGFR2 gene have been linked to Crouzon
syndrome. Approximately 95% of patients have a mutation in either 8
(IIIa) or exon 10 (IIIc), which encode the extracellular
immunoglobulin-like III (IgIII) domain of the receptor (14,16).
Growth factors, such as FGF and TGF, play pivotal roles for
controlling cell growth and differentiation (17–23).
Mutations in FGFR2 can lead to increased ligand affinity and
altered ligand specificity, disrupting the differentiation of
mesenchymal stem cells, and therefore causing developmental defects
(24,25). Although Crouzon syndrome is often
inherited as an autosomal dominant trait, de novo mutations
at FGFR2 can also result in sporadic cases (3,5,26).
Here, we report the results of a mutational analysis of two
sporadic patients with Crouzon syndrome from two unrelated Chinese
families.
Materials and methods
Patient recruitment and clinical
evaluations
All experimental protocols and methods which were
carried out in accordance with the guidelines were approved by the
Ethics Committee of Zhongshan Ophthalmic Center. Informed consents
were obtained from all participating subjects in accordance with
the Declaration of Helsinki. The following series of ophthalmic
tests were performed in patients and their family members. Visual
acuity was examined using the Early Treatment Diabetic Retinopathy
Study (EDTRS) chart (Precision Vision, LaSalle, IL, USA). Anterior
segment photographs were captured by a BX 900 slit lamp
(Haag-Streit AG, Köniz, Switzerland). Anterior segment measurements
were obtained by a Pentacam HR version 70700 (OCULUS Optikgeräte
GmbH, Wetzlar, Germany). Optical coherence tomography (OCT) was
carried out by Cirrus HD-OCT (Carl Zeiss Meditec, Dublin, CA, USA).
Computed tomography (CT) and physical examinations, including blood
examination, urinalysis, electrocardiogram, chest X-ray, blood
biochemistry, blood lipid, and blood coagulation tests, were
conducted to exclude systemic diseases.
Sample collection and mutational
screening
Genomic DNA samples were extracted from peripheral
blood leucocytes of the patients and their relatives with the
Qiagen kit (Qiagen Inc., Chatsworth, CA, USA) according to the
manufacturer's instructions. DNA concentration and purity were
measured by NanoDrop™ ND-1000 spectrophotometer (Thermo Fisher
Scientific Inc., Wilmington, DE, USA). In addition, DNA samples
collected from 200 subjects from the same population without
diagnostic features of Crouzon syndrome were used as controls.
Exons 8 and 10 in the FGFR2 gene were amplified using
polymerase chain reaction (PCR) as described previously (24,25,27).
Primers were obtained from the Beijing Genomics Institute
(Guangzhou, China). The sequences of the primers are listed in
Table I. All reagents used for the
PCR reactions were purchased from Takara Bio Inc. (Tokyo, Japan).
The amplification included a single 5-min step at 94°C; followed by
40 cycles of 94°C for 45 sec, 61°C for 45 sec, and 72°C for 45 sec;
and finally a 10-min step at 72°C. The PCR products were sequenced
in both directions using an ABI3730 Automated Sequencer (PE
Biosystems, Foster City, CA, USA). The sequencing results were
analyzed using SeqMan (version 2.3; Technelysium Pty, Ltd.,
Brisbane, QLD, Australia), and compared against reference sequences
obtained from the National Center for Biotechnology Information
(NCBI) database (accession no. NC_000010) (28).
 | Table I.Summary of the primers and product
length used for the amplification of the exons of FGFR 2. |
Table I.
Summary of the primers and product
length used for the amplification of the exons of FGFR 2.
| Exon | Forward
(5′-3′) | Reverse
(5′-3′) | Product size
(bp) | Annealing
temperature (°C) |
|---|
| FGFR2-8
(IIIa) |
GGTCTCTCATTCTCCCATCCC |
CCAACAGGAAATCAAAGAACC | 325 | 61 |
| FGFR2-10
(IIIc) |
CCTCCACAATCATTCCTGTGTC |
ATAGCAGTCAACCAAGAAAAGGG | 257 | 61 |
Results
Clinical presentations
We diagnosed two patients of two unrelated families
from the southern region of China. Systemic diseases were excluded
upon examination.
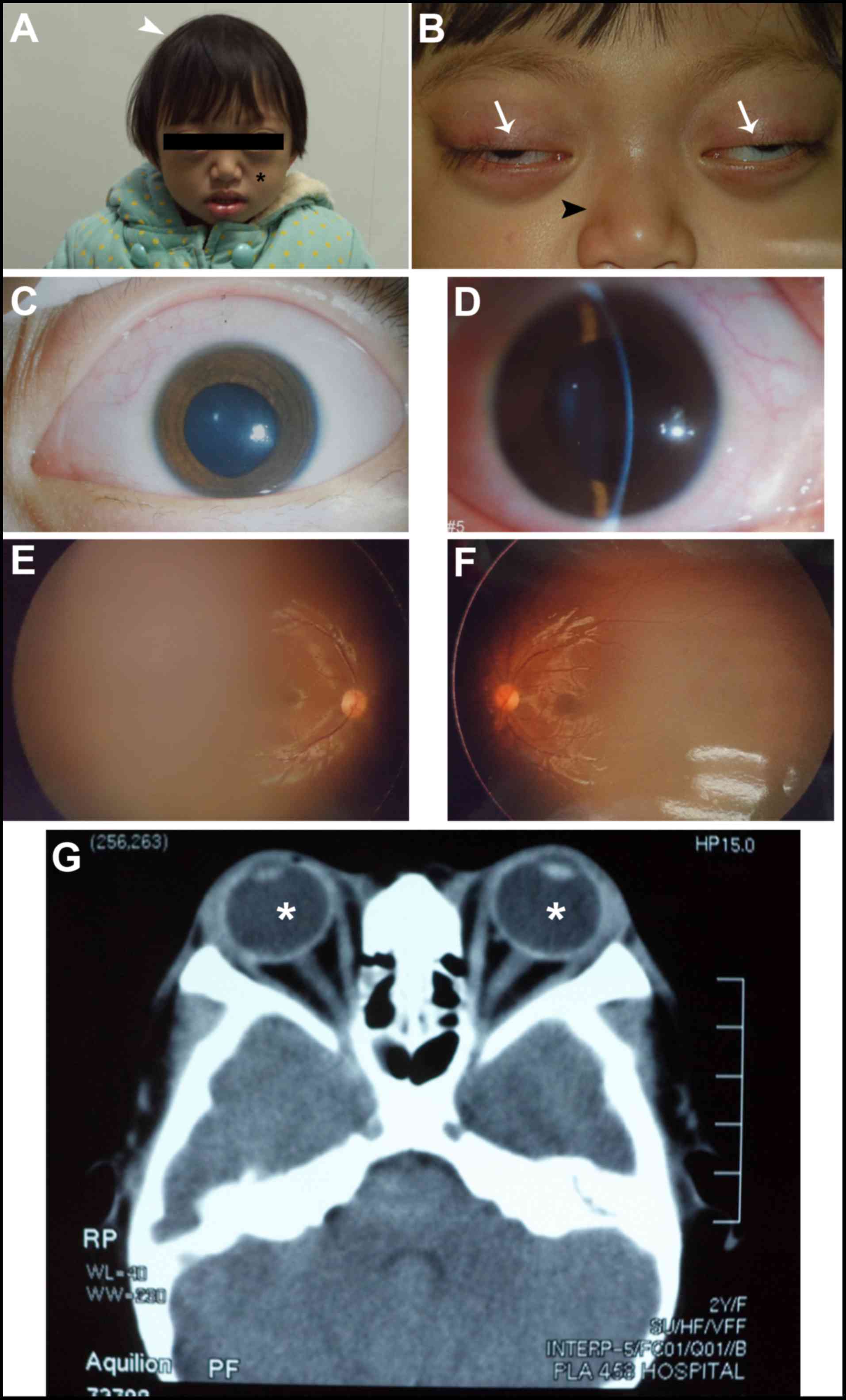
Patient #1 was a two-year-old girl and was the only
child of two healthy parents (Fig.
1). She was referred by her local pediatrician at two months of
age due to concerns about an elongated head shape and the possible
diagnosis of sagittal synostosis. Until this point, the patient's
development was otherwise unremarkable, with normal feeding and
steady weight gain after birth. Examination of this patient
revealed shallow orbits and ocular proptosis, accompanied by
midface hypoplasia, craniosynostosis, a curved beak-like nose
(Fig. 1A), and clinically normal
hands and feet. An approximately 2 mm gap was observed when she
attempted to close her eyelids (Fig.
1B). The patient presented with exotropia in both eyes, but the
corneas were transparent with normal size. Also, the lenses were
transparent and normally positioned (Fig. 1C and D). Fundus examination showed
normal retinas (Fig. 1E and F).
Because of the patient was young, we were unable to measure visual
acuity, but the child had normal visual tracking and the results of
the optometry were +3.0 D (OD) and +3.25 D (OS). CT scan revealed
shallow orbits and exotropia in both eyes (Fig. 1G). Both parents had normal visual
acuity and unremarkable eye examinations, and all family members
had no known history of learning difficulties or genetic
problems.
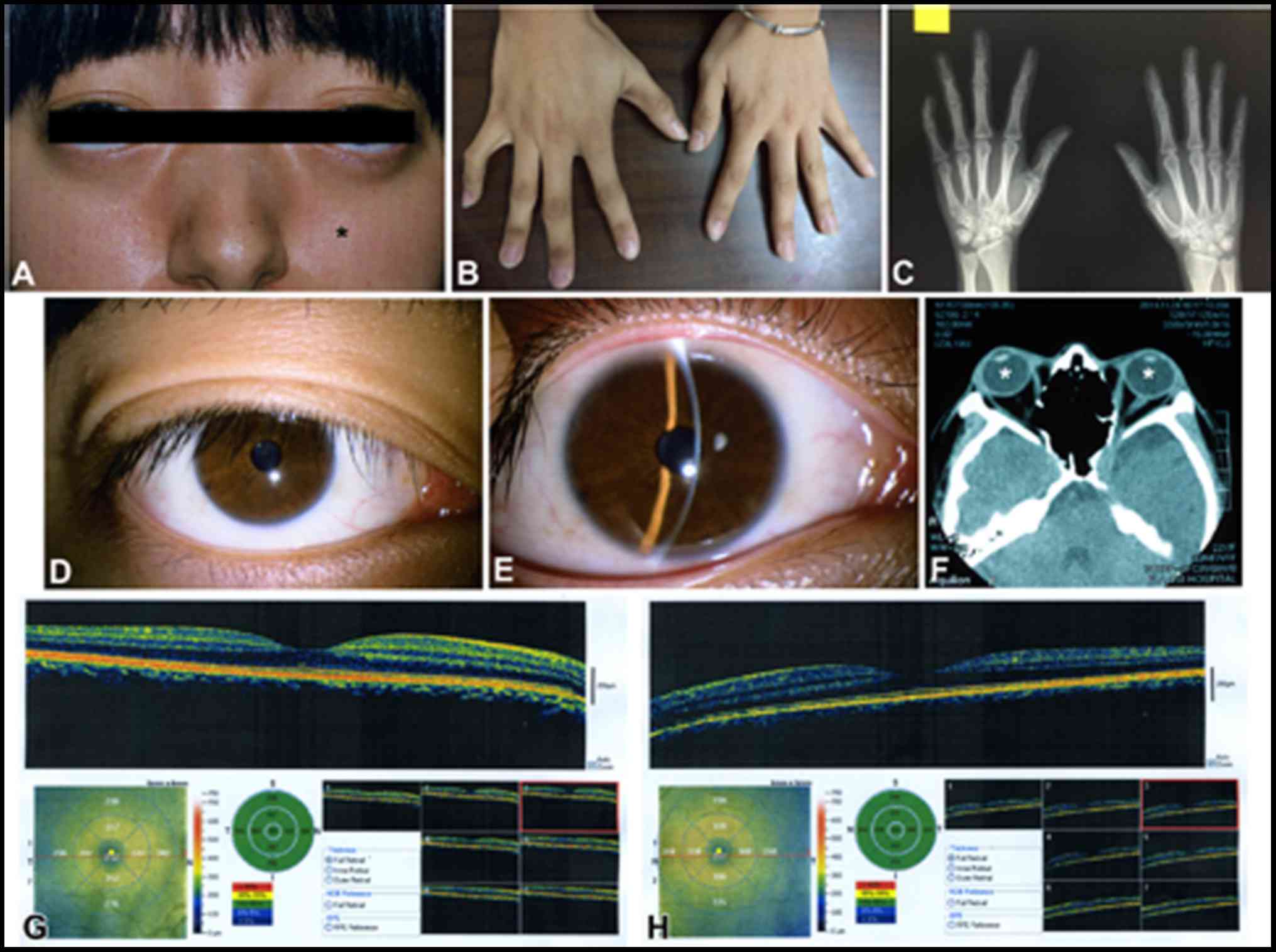
Patient #2 was a 21-year-old woman and was also the
only child of two healthy parents (Fig. 2). She presented with midface
hypoplasia and craniosynostosis (Fig.
2A). She had normal visual acuity. Her hands and feet had
normal flexibility. Radiography showed no obvious carpal fusion
(Fig. 2B and C). No abnormalities
were detected in the cornea or lens (Fig. 2D and E). CT scan revealed shallow
orbits (Fig. 2F). OCT revealed
normal retina in both eyes (Fig. 2G
and H). In general, the clinical manifestations of this patient
were less severe than patient #1.
Mutational screening
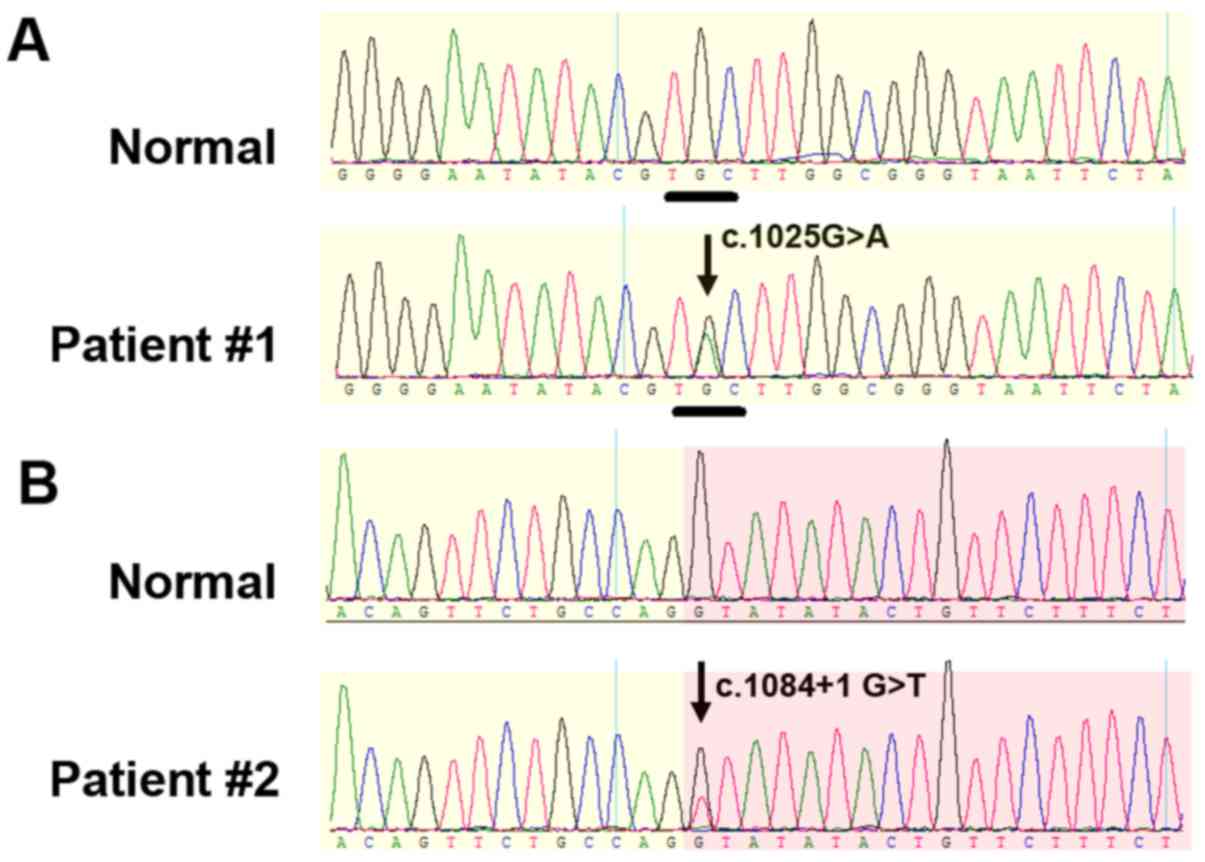
Patient #1 carried a heterozygous missense mutation
(c.1025G>A; p.C342Y) in exon 10 of the FGFR2 gene (Fig. 3A). Patient #2 carried a
heterozygous mutation (IVS10+1G>T; c.1084+1 G>T) in intron 10
of the FGFR2 gene (Fig. 3B). This
mutation is located at a splicing site. Both mutations were not
presented in any of the unaffected family members or unrelated
controls, therefore are considered de novo mutations.
Clinical manifestations and mutational screening
results of the two patients in this study are summarized in
Table II.
| Table II.Summary of clinical manifestations
and mutational screening results of the two patients. |
Table II.
Summary of clinical manifestations
and mutational screening results of the two patients.
|
|
|
| Clinical
manifestations |
|
|---|
|
|
|
|
|
|
|---|
| Patient (#) | Gender | Age | Facial
characteristics | Limbs | Lens/cornea | Fundus | CT scan | Mutation |
|---|
| 1 | Female | 2 | Midface hypoplasia,
craniosynostosis, curved beak-like nose, lagophthalmus | Normal | Normal | Normal | Shallow orbits,
proptosis, exotropia | c.1025G>A |
| 2 | Female | 21 | Midface hypoplasia,
craniosynostosis | Normal | Normal | Normal | Shallow orbits | c.1084+1 G>T,
IVS10 +1G>T; |
Discussion
Crouzon syndrome is a common autosomal dominant form
of craniofacial complexes, characterized by premature
craniosynostosis, orbital proptosis, and midface hypoplasia
(7). Both patients we reported
here do not present limb malformations, which differentiates
Crouzon syndrome from other types of craniosynostosis (1,9).
In patient #1, the c.1025G>A mutation causes a
cysteine-to-tyrosine substitution at amino acid 342 in FGFR2. The
loss of this cysteine residue is one of the most frequent mutations
in Crouzon syndrome patients and has been reported in French,
British, and German populations (5,6,29–32).
Therefore, the amino acid C342 in FGFR2 is considered as a mutation
‘hotspot’. Several identified mutations at this position are C342R
(c.1024T>C), C342Y (c.1025G>A), C342S (c.1025G>C), C342F
(c.1025G>T), and C342W (c.1026C>G) (30,32,33).
Mutations at C342 can cause Crouzon syndrome as well as Pfeiffer
syndrome (34). Studies have shown
that C342 is part of the disulfide bridge that stabilizes the IgIII
loop in all FGFR proteins and is the most conserved extracellular
amino acid in the Ig superfamily. The loss of C342 leaves an
unbridged C278, which may cause the ligand-independent dimerization
of receptor molecules, leading to constitutive receptor activation
(35,36).
The fidelity of the splice site sequence,
particularly the first two nucleotides in the donor site, is
essential for accurate splicing. The presence of a guanine base at
the +1 position at the intron-exon boundary of FGFR2 gene is
essential for splice site recognition. In patient #2, the splicing
site mutation (c.1084+1 G>T) can cause alternative splicing,
disrupt the third immunoglobulin-like domain of FGF2, and generate
pathogenic protein isoforms. However, compared to the cysteine
mutation in Patient #1, mutations affecting FGFR2 pre-mRNA splicing
usually cause relatively mild clinical manifestations (37–39).
Interestingly, a similar mutation in the FGFR2 gene at the
same position (c.1084+1 G>A) can cause mild bicoronal synostosis
(38).
Craniosynostosis may be complicated with other
ophthalmic anomalies. For example, some craniosynostosis patients
with FGFR2 mutation can also present with Peters anomaly (a
rare form of anterior segment dysgenesis), optic nerve hypoplasia,
scleralization of the cornea, and corectopia (13). In this study, patient #1 also had
strabismus, which expands the list of clinical manifestations
associated with Crouzon syndrome.
In summary, we identified two distinct mutations in
the FGFR2 gene in two Chinese patients with Crouzon syndrome
from unrelated families. These findings expand the mutational
spectrum of FGFR2, and provide valuable information for
genetic counseling and prenatal diagnosis in families with Crouzon
syndrome. Although our understanding of the function of FGFR is
still limited, the discovery of these mutant variants provides an
opportunity and rationale for in-depth mechanistic studies, and may
help to reveal critical pathophysiology underlying related skull
development disorders in general.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant nos. 81500709, 81570862,
81670872), the State Scholarship Fund from the China Scholarship
Council, the Medical Scientific Research Foundation of Guangdong
Province (grant no. A2016460) and the National Institute of Dental
and Craniofacial Research (grant no. DE020823).
References
|
1
|
Wilkie AO: Craniosynostosis: Genes and
mechanisms. Hum Mol Genet. 6:1647–1656. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Mitulla B, Hinkel GK and Lorenz P: Crouzon
syndrome (Mc K 12350). Kinderarztl Prax. 59:278–280. 1991.(In
German). PubMed/NCBI
|
|
3
|
Kaur H, Waraich H Singh and Sharma CM:
Crouzon syndrome: A case report and review of literature. Indian J
Otolaryngol Head Neck Surg. 58:381–382. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Khandelwal R, Agrawal P and Majumdar MR:
Crouzon syndrome. BMJ Case Rep 2012. 2012. View Article : Google Scholar
|
|
5
|
Reardon W, Winter RM, Rutland P, Pulleyn
LJ, Jones BM and Malcolm S: Mutations in the fibroblast growth
factor receptor 2 gene cause Crouzon syndrome. Nat Genet. 8:98–103.
1994. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu J, Kwon TG, Nam HK and Hatch NE:
Craniosynostosis-associated Fgfr2(C342Y) mutant bone marrow stromal
cells exhibit cell autonomous abnormalities in osteoblast
differentiation and bone formation. Biomed Res Int.
2013:2925062013.PubMed/NCBI
|
|
7
|
Bowling EL and Burstein FD: Crouzon
syndrome. Optometry. 77:217–222. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Giordano BP, Tuli SS, Ryan SF, Stern M and
Tuli SY: Crouzon syndrome: Visual diagnosis. J Pediatr Health Care.
30:270–273. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Barik M, Bajpai M, Malhotra A, Samantaray
JC, Dwivedi S and Das S: Novel mutation detection of fibroblast
growth factor receptor 1 (FGFR1) gene, FGFR2IIIa, FGFR2IIIb,
FGFR2IIIc, FGFR3, FGFR4 gene for craniosynostosis: A prospective
study in Asian Indian patient. J Pediatr Neurosci. 10:207–213.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kreiborg S: Crouzon syndrome: A clinical
and roentgencephalometric study. Scand J Plast Reconstr Surg Suppl.
18:1–198. 1981.PubMed/NCBI
|
|
11
|
Shotelersuk V, Mahatumarat C, Ittiwut C,
Rojvachiranonda N, Srivuthana S, Wacharasindhu S and Tongkobpetch
S: FGFR2 mutations among Thai children with Crouzon and Apert
syndromes. J Craniofac Surg. 14:101–107. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Sastre-Ibáñez M, Garcia-Asorey A,
Santos-Bueso E, Lerma-Gallardo JL, Garcia-Sáenz S and Garcia-Feijoo
J: Crouzon syndrome: Ophthalmologic complications in an untreated
adult patient. J Fr Ophtalmol. 38:e177–e178. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Okajima K, Robinson LK, Hart MA, Abuelo
DN, Cowan LS, Hasegawa T, Maumenee IH and Jabs EW: Ocular anterior
chamber dysgenesis in craniosynostosis syndromes with a fibroblast
growth factor receptor 2 mutation. Am J Med Genet. 85:160–170.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Guo L, Lai YN and Li LX: FGFR2 gene
mutation in a family with Crouzon syndrome and a sporadic Crouzon
syndrome patient. Zhonghua Yi Xue Yi Chuan Xue Za Zhi. 25:218–220.
2008.(In Chinese). PubMed/NCBI
|
|
15
|
Cunningham ML, Seto ML, Ratisoontorn C,
Heike CL and Hing AV: Syndromic craniosynostosis: From history to
hydrogen bonds. Orthod Craniofac Res. 10:67–81. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Gorry MC, Preston RA, White GJ, Zhang Y,
Singhal VK, Losken HW, Parker MG, Nwokoro NA, Post JC and Ehrlich
GD: Crouzon syndrome: Mutations in two spliceoforms of FGFR2 and a
common point mutation shared with Jackson-Weiss syndrome. Hum Mol
Genet. 4:1387–1390. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kitisin K, Saha T, Blake T, Golestaneh N,
Deng M, Kim C, Tang Y, Shetty K, Mishra B and Mishra L: Tgf-beta
signaling in development. Sci STKE: cm1. 2007. View Article : Google Scholar
|
|
18
|
Tan X, Zhu Y, Chen C, Chen X, Qin Y, Qu B,
Luo L, Lin H, Wu M, Chen W and Liu Y: Sprouty2 suppresses
epithelial-mesenchymal transition of human lens epithelial cells
through blockade of Smad2 and ERK1/2 pathways. PLoS One.
11:e01592752016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Turner N and Grose R: Fibroblast growth
factor signalling: From development to cancer. Nat Rev Cancer.
10:116–129. 2010. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Coutts JC and Gallagher JT: Receptors for
fibroblast growth factors. Immunol Cell Biol. 73:584–589. 1995.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zhang Y, Morgan R, Chen C, Cai Y, Clark E,
Khan WN, Shin SU, Cho HM, Al Bayati A, Pimentel A and Rosenblatt
JD: Mammary-tumor-educated B cells acquire LAP/TGF-β and PD-L1
expression and suppress anti-tumor immune responses. Int Immunol.
28:423–433. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chen C, Zhu Y, Lin Y, Liu Z, Wu M, Li D
and Cheng B: Suppression of retinal pigment epithelial cell
proliferation, migration and epithelial-mesenchymal transition by
proteasome inhibition, a potential defense against proliferative
vitreoretinopathy. Investigative Ophthalmol Visual Sci.
54:62542013.
|
|
23
|
Zhu Y, Chen C, Lin Y, Liu Y and Wu M;
Cataract Lab, : Downregulation of syndecan-4 by RNA interference
inhibits adhesion and bFGF-induced proliferation of lens epithelial
cells. Invest Ophthalmol Vis Sci. 54:4792013.
|
|
24
|
Lin Y, Ai S, Chen C, Liu X, Luo L, Ye S,
Liang X, Zhu Y, Yang H and Liu Y: Ala344Pro mutation in the FGFR2
gene and related clinical findings in one Chinese family with
Crouzon syndrome. Mol Vis. 18:1278–1282. 2012.PubMed/NCBI
|
|
25
|
Lin Y, Liang X, Ai S, Chen C, Liu X, Luo
L, Ye S, Li B, Liu Y and Yang H: FGFR2 molecular analysis and
related clinical findings in one Chinese family with Crouzon
syndrome. Mol Vis. 18:449–454. 2012.PubMed/NCBI
|
|
26
|
Matsumoto K, Urano Y, Kubo Y, Nakanishi H
and Arase S: Mutation of the fibroblast growth factor receptor 2
gene in Japanese patients with Apert syndrome. Plast Reconstr Surg.
101:307–311. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lin Y, Liu X, Yu S, Luo L, Liang X, Wang
Z, Chen C, Zhu Y, Ye S, Yan H and Liu Y: PAX6 analysis of two
sporadic patients from southern China with classic aniridia. Mol
Vis. 18:2190–2194. 2012.PubMed/NCBI
|
|
28
|
Li T, Lin Y, Gao H, Chen C, Zhu Y, Liu B,
Lian Y, Li Y, Zhou W, Jiang H and Li H: Two heterozygous mutations
identified in one Chinese patient with bilateral macular coloboma.
Mol Med Rep. Jun 29–2017.(Epub ahead of print).
|
|
29
|
Bagheri-Fam S, Ono M, Li L, Zhao L, Ryan
J, Lai R, Katsura Y, Rossello FJ, Koopman P, Scherer G, et al:
FGFR2 mutation in 46,XY sex reversal with craniosynostosis. Hum Mol
Genet. 24:6699–6710. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ke R, Yang X, Tianyi C, Ge M, Lei J and Mu
X: The C342R mutation in FGFR2 causes Crouzon syndrome with elbow
deformity. J Craniofac Surg. 26:584–586. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sun LL, Li M, Suo F, Liu XM, Shen EZ, Yang
B, Dong MQ, He WZ and Du LL: Global analysis of fission yeast
mating genes reveals new autophagy factors. PLoS Genet.
9:e10037152013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mangasarian K, Li Y, Mansukhani A and
Basilico C: Mutation associated with Crouzon syndrome causes
ligand-independent dimerization and activation of FGF receptor-2. J
Cell Physiol. 172:117–125. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pandey RK, Bajpai M, Ali A, Gayan S and
Singh A: Mutational identification of fibroblast growth factor
receptor 1 and fibroblast growth factor receptor 2 genes in
craniosynostosis in Indian population. Indian J Hum Genet.
19:449–453. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Glaser RL, Jiang W, Boyadjiev SA, Tran AK,
Zachary AA, Van Maldergem L, Johnson D, Walsh S, Oldridge M, Wall
SA, et al: Paternal origin of FGFR2 mutations in sporadic cases of
Crouzon syndrome and Pfeiffer syndrome. Am J Hum Genet. 66:768–777.
2000. View
Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kress W, Collmann H, Büsse M,
Halliger-Keller B and Mueller CR: Clustering of FGFR2 gene
mutations inpatients with Pfeiffer and Crouzon syndromes
(FGFR2-associated craniosynostoses). Cytogenet Cell Genet.
91:134–137. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Robertson SC, Meyer AN, Hart KC, Galvin
BD, Webster MK and Donoghue DJ: Activating mutations in the
extracellular domain of the fibroblast growth factor receptor 2
function by disruption of the disulfide bond in the third
immunoglobulin-like domain. Proc Natl Acad Sci USA. 95:4567–4572.
1998; View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Cohen MM and MacLean RE: Craniosynostosis:
Diagnosis, Evaluation and Management. 2nd edition. Oxford
University Press; New York, NY: pp. 4542000
|
|
38
|
Traynis I, Bernstein JA, Gardner P and
Schrijver I: Analysis of the alternative splicing of an FGFR2
transcript due to a novel 5′splice site mutation (1084+1G>A):
Case report. Cleft Palate Craniofac J. 49:104–108. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Burgess K and Price J: Whey to Go Whey
Protein Concentrate: A New Zealand Success Story. MacGibbon J:
Ngaio Press; Martinborough, New Zealand: pp. 2642014
|