Introduction
Sodium azide (NaN3), has a wide range of
applications. It is used in the military setting as a substrate in
explosive materials, a propulsion agent in jet aircraft and in
airplane escape chutes, and in the industrial setting as an
ingredient to inflate automobile airbag gas. It is also used as an
insecticide, herbicide, nematocide, fungicide and bactericide in
agriculture and as a potent preservative in clinical laboratories
and hospitals (1–3). Sodium azide is highly toxic,
similarly to cyanide poisoning, and poisoning by sodium azide poses
a serious risk of death. In recent years, cases involving sodium
azide poisonings were still reported in the literature, despite
limited access to chemicals of this type (4–8).
Various reports have demonstrated that sodium azide poisoning
causes severe hypoxemia (9–11),
and extensive damage in the nervous (10,12)
and cardiac systems (11,13,14).
A previous study demonstrated that sodium azide also causes acute
kidney injury (15). It is known
that aerobic organs, such as the heart, are highly sensitive to
hypoxia and susceptible to injury. Previous reports have confirmed
that sodium azide, a mitochondrial respiratory chain complex IV
inhibitor, could induce cell death when added to cultured neonatal
rat cardiac myocytes, and simulate chemical hypoxia, which was
associated with the proteolysis of biochemical indicators, such as
myocardial troponin I (11). This
process was demonstrated to be significantly inhibited by calcium
antagonists, such as nifedipine and benidipine (11,16).
Inhibition of Ca2+ influx, and preservation of
mitochondrial membrane potential (ΔΨm) and cellular ATP contents by
benidipine were important in the protection against sodium
azide-induced cardiac cell death (16). However, the exact mechanism of
sodium azide-induced cardiotoxicity remains not fully
understood.
Mdivi-1, a derivative of quinazolinone, is a novel
mitochondrial division inhibitor (17). It is a highly efficacious small
molecule serving as a selective inhibitor to suppress
dynamin-related protein 1 (Drp1) self-assembly and mitochondrial
fission (18–20). Drp1, a member of the dynamin family
of large GTPases, which is primarily found in the cytosol, is
recruited by mitochondrial fission 1 protein to translocate to the
outer mitochondrial membrane and is then localized to discrete
regions on the mitochondrial surface to initiate fission (21–24).
Previous studies have demonstrated that the small molecule
inhibitor Mdivi-1 attenuated both tubular cell apoptosis and acute
kidney injury (15), and has been
demonstrated to have protective effects by attenuating cell
apoptosis in both myocardial (18,25)
and cerebral ischemia/reperfusion (I/R) injury (19). In addition, Mdivi-1 has also been
reported to have cardioprotective effects by ameliorating pressure
overload during heart failure (26). However, the effects of Mdivi-1 on
sodium azide-induced cell death in H9c2 cardiac muscle cells remain
unclear.
Based on the above research literature, it was
hypothesized that Mdivi-1, a selective inhibitor of Drp1, may
prevent sodium azide-induced H9c2 cells death by improving
mitochondrial function and increasing reactive oxygen species (ROS)
production. Therefore, the present study assessed the effect of
Mdivi-1 in sodium azide-induced H9c2 cells and its mechanism. The
results revealed that inhibition of Drp1 by Mdivi-1 pretreatment
prevented sodium azide-induced H9c2 cell death, suggesting that it
may serve as a potential drug in the treatment of azide
poisonings.
Materials and methods
Materials
Mdivi-1 was purchased from Tocris Bioscience
(Bristol, UK) and dissolved in dimethyl sulfoxide (DMSO), assuring
that the final concentration of DMSO was <0.01% in all
experiments. Sodium azide (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany) was dissolved in medium as a 1M stock solution, and then
diluted to the indicated concentrations prior to use in
experiments.
Cell culture
The rat embryonic ventricular myocardial H9c2 cell
line (American Type Culture Collection, Manassas, VA, USA) was used
in the present study. H9c2 cells were cultured in Dulbecco's
modified Eagle's medium (DMEM; Hyclone; GE Healthcare Life
Sciences, Logan, UT, USA) supplemented with 10% fetal bovine serum
(FBS; Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Cells were incubated at 37°C in a humidified incubator containing
95% air and 5% CO2 and used at 70–80% of confluence. All
experimental procedures and protocols were approved by Ethics
Committee of Soochow University.
Cell viability assay
To assess cell viability, the Cell Counting kit
(CCK-8; Dojindo Molecular Technologies, Inc., Shanghai, China)
assay was used. Cells (5×104/well) were seeded in
96-well plates. Following culture for 24 h, sodium azide (0.1–70
mM) was added to the cells and incubated for 24 h to make the cell
injury model. For Mdivi-1 pretreatment, cells were cultured in the
presence of different doses of Mdivi-1 for 3 h, prior to the sodium
azide treatment and Mdivi-1 was kept in the same culture media
during the 24 h sodium azide treatment. Cells without any treatment
were used as control. Then, a total of 10 µl of CCK-8 solution
(Dojindo Molecular Technologies, Inc.) was added to each well and
incubated for another 3 h under standard cell-culture conditions
(37°C, 5% CO2). The absorbance was determined at 450 nm
wavelength (A450 nm) with a ELx808 microplate reader (BioTek
Instruments, Inc., Winooski, VT, USA). Cell viability was
calculated according to the mean optical density (OD) of 8 wells.
The experiments were repeated at least three times.
Nuclear morphology of DAPI-stained
H9c2 cells
H9c2 cells, with or without 1 µM Mdivi-1
pretreatment for 3 h, were incubated with 30 mM sodium azide for 24
h. In order to distinguish between programmed or non-apoptotic cell
death, nuclei were stained with DAPI. Briefly, cells were washed
twice with PBS and then fixed with 4% paraformaldehyde for 30 min
at room temperature. Following three washes, fixed cells were
stained with DAPI (1:5,000; dilution with PBS) for 5–10 min. Cells
were then washed with PBS and fluorescence images were captured
with a Leica DMI fluorescent microscope (Leica Microsystems GmbH,
Wetzlar, Germany). The experiments were repeated at least three
times.
Mitochondrial membrane potential (ΔΨm)
measurement
ΔΨm is a significant parameter of mitochondrial
function. ΔΨm was assessed by staining with the fluorescent probe
5,5′,6,6′-tetrachloro-1,1′,3,3′-tetraethylbenzimidazole-carbocyanide
iodide (JC-1; Beyotime Institute of Biotechnology, Haimen, China).
H9c2 cells were cultured in 24-well plates and either not treated
(control), or exposed to sodium azide for 24 h, with or without 1
µM Mdivi-1 pretreatment for 3 h. Subsequently, cells were stained
with JC-1, according to the manufacturer's protocol. Following
incubation at 37°C for 20 min, the cells were washed thrice and
fresh medium without serum was added. Images were observed and
captured with a fluorescent microscope: JC-1 monomer green
fluorescence (excitation 490 nm, emission 525 nm) denotes the
presence of low membrane potential and red J-aggregate fluorescence
(excitation 525 nm, emission 590 nm) denotes the presence of high
membrane potential. The positive control was treated with carbonyl
cyanide m-chlorophenylhydrazone (CCCP), as provided by the kit. In
order to quantify the changes of relative mitochondrial membrane
potential, ratios of red/green fluorescent densities were
calculated and analyzed with ImageJ v1.32 J software (National
Institutes of Health, Bethesda, MD, USA).
Measurement of cellular ATP
contents
H9c2 cells were cultured in 6-well plates,
pretreated with 1 µM Mdivi-1 for 3 h, then treated with 30 mM
sodium azide for 24 h. The measurement of cellular ATP contents was
performed with a firefly luciferase ATP assay kit (Beyotime
Institute of Biotechnology), according to the manufacturer's
instructions. In brief, cells were washed with pre-cooled PBS and
lysed with ATP lysis buffer on ice. Then samples were centrifuged
at 12,000 × g for 5 min in 4°C to collect the supernatant in 1.5 ml
tubes and stored at −80°C until measurement. ATP contents were
measured in 20 µl of each sample (including standard) and mixed
with 50 µl of ATP detection working dilution, which was placed in
advance at room temperature for 3–5 min. Luminescence (relative
light units, RLU) activity was measured immediately using a
luminometer (GloMax 20/20; Promega Corporation, Madison, WI, USA).
In each assay, a 7-point standard curve (range, 0.1–10 µM) for the
quantification was generated. Finally, the intracellular ATP
contents were expressed as nmol per mg of total protein.
Measurement of ROS production in H9c2
cells
Intracellular ROS production was assessed by using
the specific probe 2′,7′-dichlorofluorescein diacetate (DCFH-DA;
Beyotime Institute of Biotechnology), according to the
manufacturer's instructions. Intracellular ROS oxidize DCFH-DA,
yielding the fluorescent compound 2′,7′-dichlorofluorescein (DCF)
and measurement of the DCF fluorescence intensity is representative
of the amount of ROS in the cells. H9c2 cells were cultured in
6-well plates, then exposed to 30 mM sodium azide for 24 h, with or
without pretreatment with 1 µM Mdivi-1 for 3 h. Subsequently, cells
were treated with DCFH-DA (10 µM) dissolved in serum-free DMEM
(1:1,000) for 20 min at 37°C and then washed three times with
serum-free DMEM. The cells were then observed for green
fluorescence (excitation 488 nm, emission 525 nm) with a Leica DMI
fluorescent microscope (Leica Microsystems GmbH) and analyzed with
ImageJ v1.32 J software (National Institutes of Health).
Fluorescence intensities of ROS were presented in arbitrary units
(a.u).
Western blotting
H9c2 cells were exposed to 30 mM sodium azide for 24
h, with or without 1 µM Mdivi-1 pretreatment for 3 h. The cultured
cells were exposed to liquid nitrogen, lysed with
radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology) with protease inhibitors (1 mM PMSF; 1:100),
harvested in 1.5 ml tubes by scraping, and centrifuged at 4°C at
13,362 × g for 10 min in order to collect the supernatants.
Subsequently, the proteins concentrations were determined by
bicinchoninic acid assay (Pierce; Thermo Fisher Scientific, Inc.).
Equal amounts of proteins (50–60 µg) were separated by 10 and 15%
SDS-PAGE and transferred to polyvinylidene fluoride membrane by a
semidry electrotransferring unit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Following blocking with TBS containing 0.1%
Tween-20 (TBST) and 5% non-fat dry milk for 2 h at room
temperature, the membranes were incubated with primary antibodies
against Drp1 (cat. no. ABT155; dilution 1:1,000; EMD Millipore,
Billerica, MA, USA), BCL2 associated X (Bax; cat. no. sc-7480;
dilution, 1:200; Santa Cruz Biotechnology, Inc., Dallas, TX, USA)
and BCL2 apoptosis regulator (Bcl-2; cat. no. sc-7382; dilution,
1:200; Santa Cruz Biotechnology, Inc.) overnight at 4°C. The
following day, membranes were washed and incubated with horseradish
peroxidase-conjugated second antibodies (rabbit; cat. no. A0208;
1:1,000; Beyotime Institute of Biotechnology) or (mouse; cat. no.
A0216; 1:1,000; Beyotime Institute of Biotechnology) at room
temperature for 1 h. Finally, immunoreactivity was visualized by
the enhanced chemiluminescence system (ChemiScope 5200; Clinx
Science Instruments Co., Ltd., Shanghai, China) and quantitatively
analyzed with ImageJ v1.32 J software (National Institutes of
Health). GAPDH served as the loading control. Three independent
experiments were performed.
Statistical analysis
Statistical analysis was performed using SPSS 13.0
software (SPSS, Inc., Chicago, IL, USA). Data are presented as the
mean ± standard error of the mean and difference between groups was
evaluated by one-way analysis of variance followed by Bonferroni
post hoc tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
Mdivi-1 pretreatment inhibits sodium
azide-induced H9c2 cell death
In order to examine the viability of H9c2 cells
following treatment with sodium azide, the CCK-8 assay was used.
Cells were treated with different concentrations (0–70 mM) of
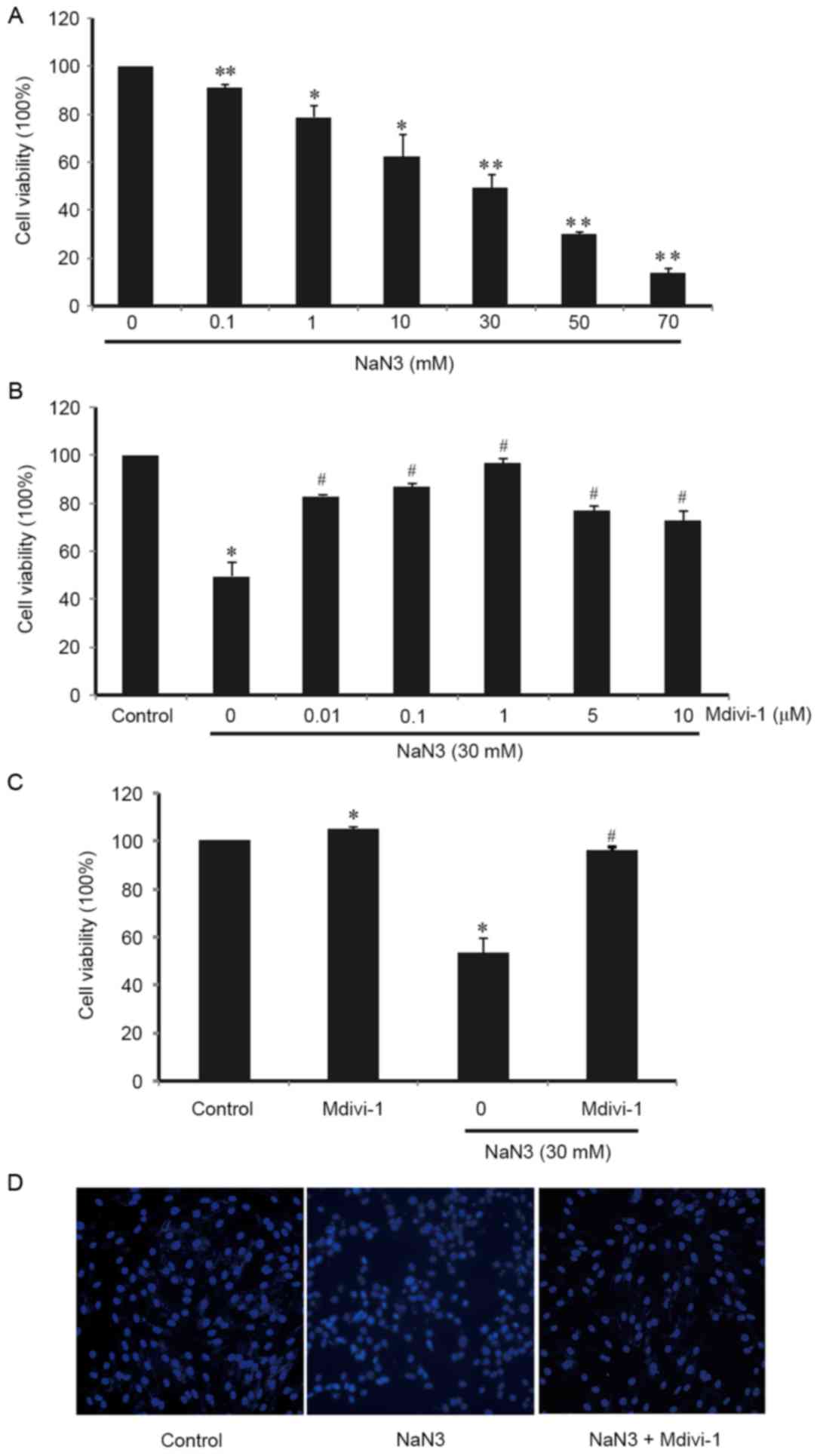
sodium azide for 24 h. As illustrated in Fig. 1A, cell viability was reduced
following sodium-azide treatment in a dose-dependent manner.
Treatment with 70 mM sodium azide resulted in ~90% of cells dying
in 24 h (Fig. 1A). These results
indicated that sodium azide induced cell death and cytotoxicity in
a dose-dependent manner. Exposure to sodium azide at a
concentration of30 mM caused prominent cell death in H9c2 cells (by
~50%; Fig. 1A).
To determine the role of Mdivi-1 in
sodium-azide-treated H9c2 cells, cells were pretreated with various
concentrations (0.01–10 µM) of Mdivi-1 for 3 h and then exposed to
30 mM sodium azide for 24 h. Using the CCK-8 assay as mentioned
above, the viability of cells was evaluated (Fig. 1B). The results indicated that there
was a dose-dependent response when treating with different dose
(0.01 to 10 µM) of Mdivi-1. Pretreatment with Mdivi-1 resulted in
an increase of cell viability compared with sodium azide-treated
cells alone (Fig. 1B). The optimal
concentration of Mdivi-1 to prevent sodium azide-induced H9c2 cells
death was 1 µM (Fig. 1B).
Furthermore, it was also confirmed that cell viability was
unaffected by Mdivi-1 treatment alone (Fig. 1C).
In addition, H9c2 cells were stained with DAPI and
observed by fluorescence microscopy. In sodium azide-treated H9c2
cells, atypical morphology of apoptosis was detected and Mdivi-1
pretreatment exhibited an obvious ameliorative effect (Fig. 1D), supporting the above results
from the CCK-8 assays. These findings suggest that Mdivi-1
protected against sodium azide-induced H9c2 cell death.
Mdivi-1 pretreatment moderates the
sodium azide-induced dissipation of ΔΨm in H9c2 cells
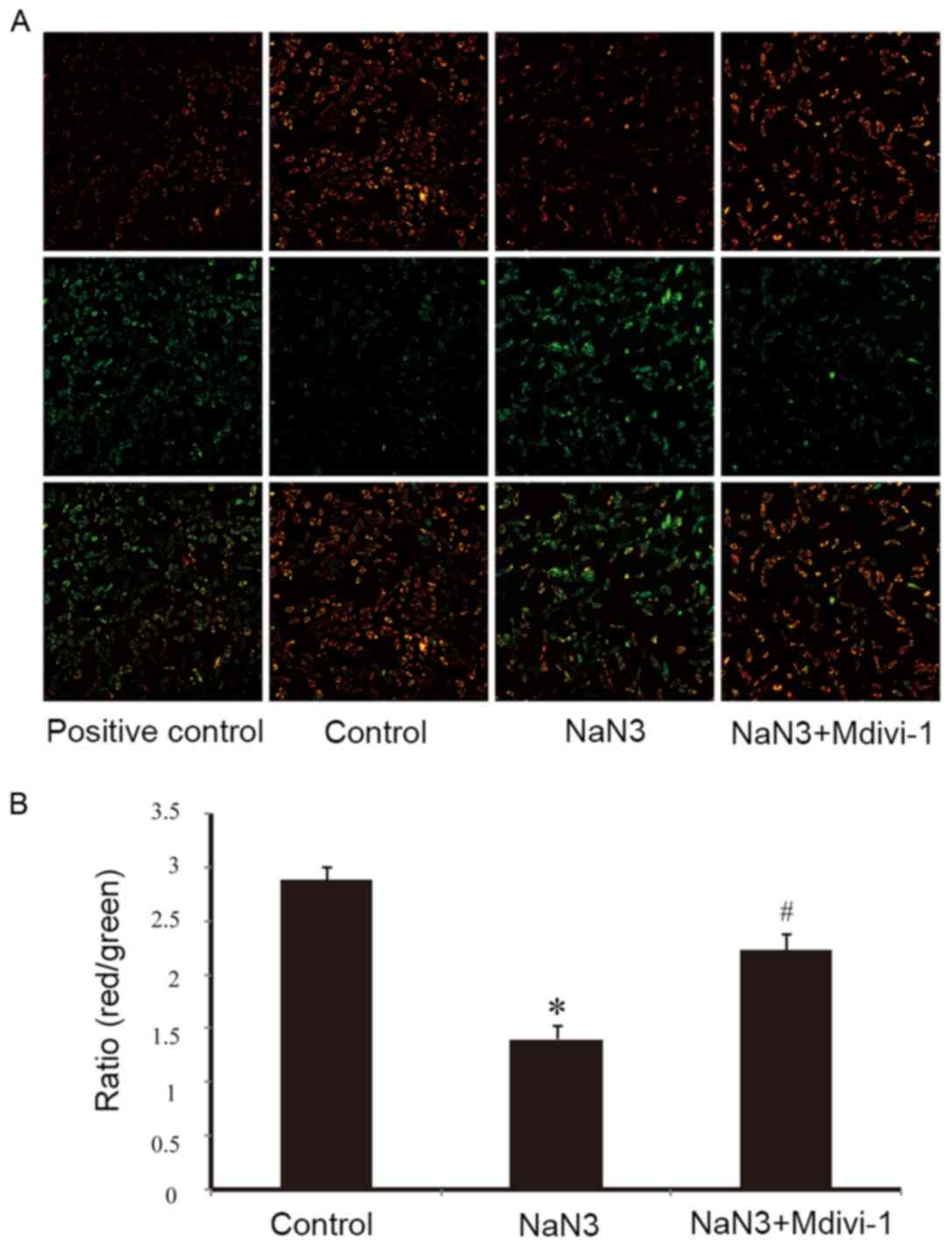
As illustrated in Fig.
2A by JC-1 staining, exposure of H9c2 cells to sodium azide (30
mM) for 24 h resulted in dissipation of ΔΨm, as observed by an
increase in green fluorescence compared to normal mitochondria that
exhibit red fluorescence. Pretreatment with Mdivi-1 (1 µM) was able
to moderate the decline of ΔΨm indicating the protective effects of
Mdivi-1 (Fig. 2A). CCCP, which can
induce mitochondrial membrane potential dissipation, was used as a
positive control (Fig. 2A). The
ratio of red/green fluorescence was used to quantify the effects of
sodium azide and Mdivi-1 on ΔΨm. As presented in Fig. 2B, the ratio of red to green
fluorescence was significantly decreased following sodium azide
treatment, while pretreatment with Mdivi-1 partially reversed this
effect and resulted in an increase in the ratio in H9c2 cells.
These results indicated that Mdivi-1 improved sodium azide-induced
mitochondrial dysfunction.
Mdivi-1 pretreatment reverses the
downregulation of sodium azide-induced mitochondrial ATP energy
production
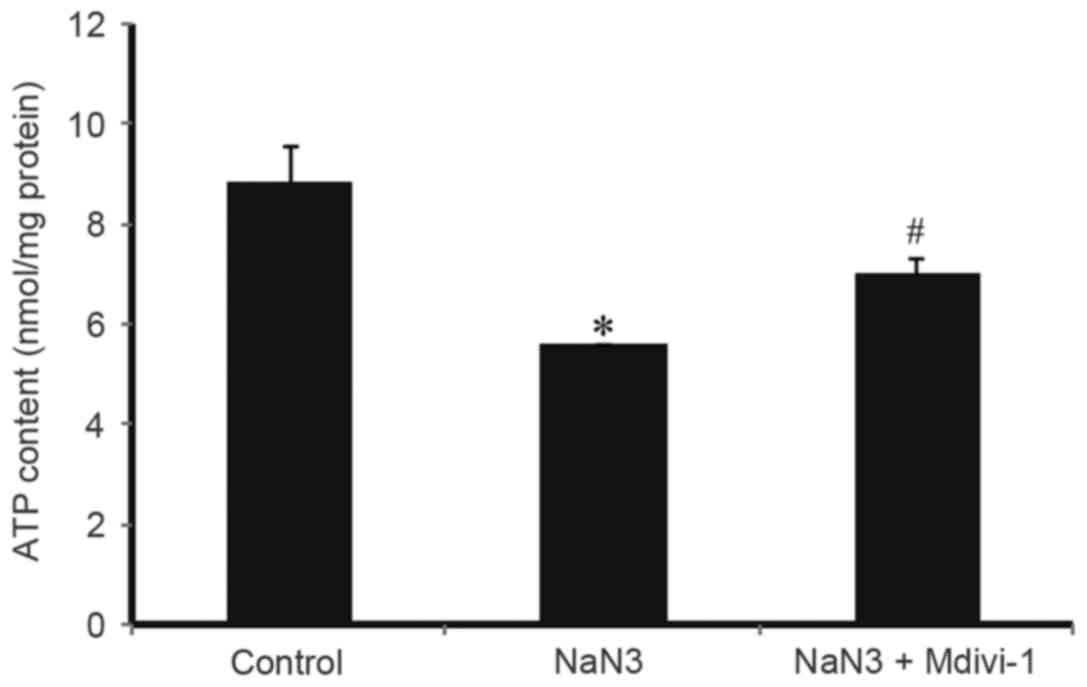
To evaluate whether Mdivi-1 could affect the ATP
contents in H9c2 cells exposed to sodium azide, ATP contents in
treated cells were quantitatively determined by a specific ATP
assay kit. As illustrated in Fig.
3, treatment with 30 mM sodium azide for 24 h resulted in a
significant decrease in the cellular ATP contents compared with
control, while pretreatment with1 µM Mdivi-1 reversed this effect.
These results suggested that Mdivi-1could reverse the sodium
azide-induced downregulation of mitochondrial ATP energy
production.
Mdivi-1 pretreatment inhibits sodium
azide-induced accumulation of mitochondrial ROS
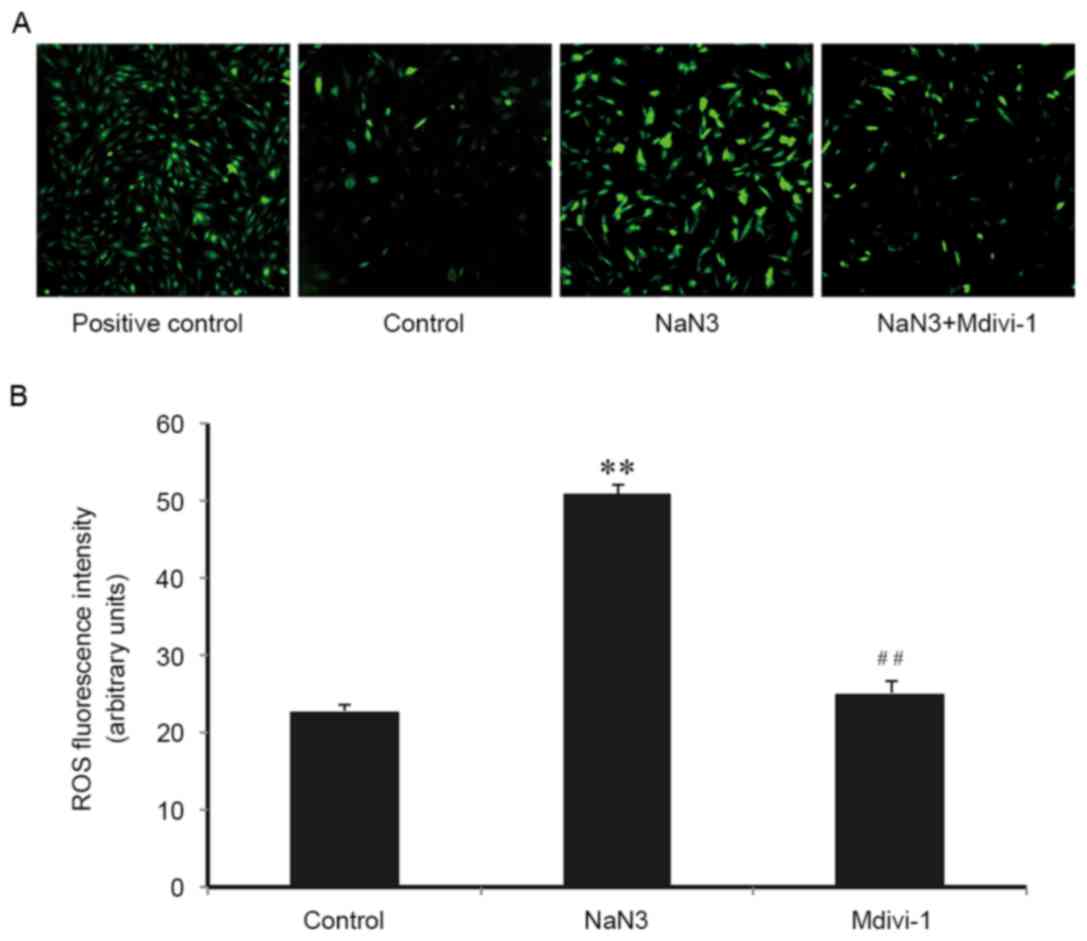
It is well-established that mitochondria are the
main source of cellular ROS, and ROS have an important role in
apoptosis activation. Therefore, the role of ROS in sodium
azide-induced H9c2 cell death and the implication of Mdivi-1 on
this process was examined. As presented in Fig. 4, compared with control cells, the
DCF fluorescence intensity increased significantly in cells
following sodium azide (30 mM) treatment for 24 h, indicating that
sodium azide resulted in mitochondrial ROS production. Mdivi-1 (1
µM) pretreatment markedly inhibited the accumulation of ROS
(Fig. 4A and B). These results
suggested that sodium azide-induced apoptosis was associated with
oxidative stress and indicated that Mdivi-1 may inhibit apoptosis
by alleviating ROS accumulation and protecting against oxidative
stress-induced cell injury.
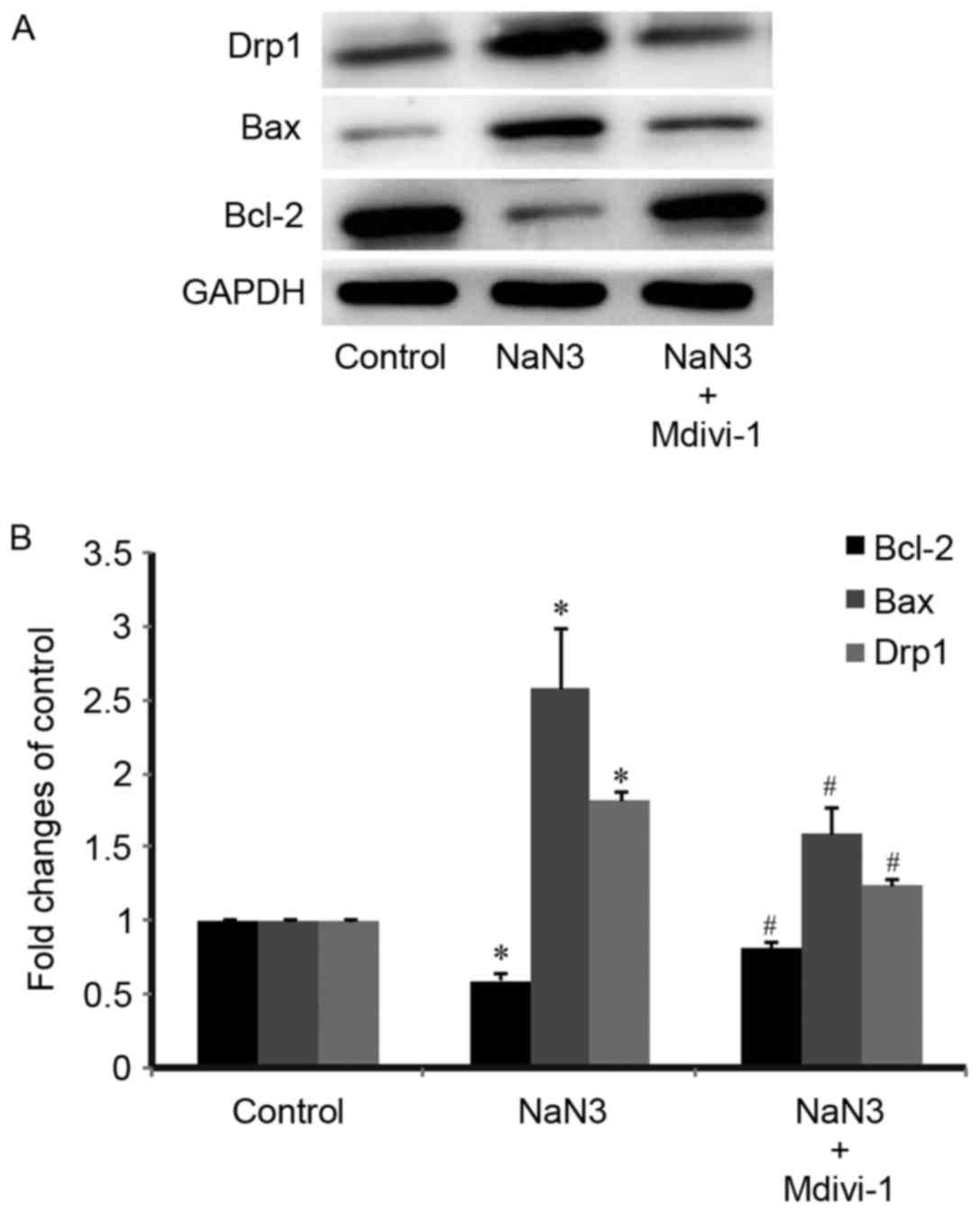
Mdivi-1 pretreatment inhibits
expression of Drp1 and apoptosis-related proteins
The present study further explored the protein
expression levels of Drp1 and apoptosis-related proteins in the
treated H9c2 cells. As presented in Fig. 5A, Drp1 and the proapoptotic protein
Bax were expressed at relatively low levels in the control
untreated cells. Protein expression levels of Drp1 and Bax were
significantly increased following treatment with sodium azide
(Fig. 5). Pretreatment with
Mdivi-1 (1 µM) for 3 h resulted in a significant decrease in both
Drp1 and Bax expression compared with sodium-azide treated cells
alone (Fig. 5). By contrast,
expression of the antiapoptotic protein Bcl-2 was decreased in the
sodium azide group compared with the untreated control group, while
Mdivi-1 pretreatment prevented the Bcl-2 expression decrease
(Fig. 5). These results indicated
that Mdivi-1 exhibited an antiapoptotic effect.
Discussion
The present study explored the mechanism underlying
the mitochondria-dependent apoptosis in sodium azide-induced
cardiotoxicity with an in vitro model of the H9c2 myocardial
cell line. Notably, the findings provided the first experimental
evidence that Mdivi-1, a mitochondrial division inhibitor, had
protective effects against sodium azide-induced cell death by
apoptosis. Pretreatment with Mdivi-1 inhibited the sodium
azide-induced upregulation of Drp1 expression, and attenuated H9c2
cell death. In addition, Mdivi-1 pretreatment inhibited the
apoptosis of H9c2 cells by modulating Bax and Bcl-2 expression. In
addition, Mdivi-1 pretreatment improved the sodium azide-induced
mitochondrial dysfunction by inhibiting mitochondrial membrane
potential dissipation, improving mitochondrial ATP energy
production, alleviating the overproduction of ROS and protecting
against oxidative stress-induced cell injury.
Previous studies suggest that mitochondria are
highly dynamic organelles that continually undergo fusion and
fission, which have been implicated in a variety of biological
processes, including cell apoptosis, autophagy, division, embryonic
development and metabolism (27,28).
Changes in mitochondrial dynamics, which can affect
cardioprotection, vascular smooth cell proliferation, myocardial
I/R and heart failure, have an important role in maintaining their
function in cardiovascular health and disease (29–31).
Mdivi-1, a novel mitochondrial division inhibitor, reduces
apoptotic cell death and has cadioprotective capacity to block
apoptotic cell death against I/R injury (25). In addition, inhibition of Drp1 by
Mdivi-1 attenuates cerebral ischemic injury via inhibition of the
mitochondria-dependent apoptotic pathway following cardiac arrest
(32). Therefore, in the present
study, Mdivi-1 was used in order to explore the mechanism in sodium
azide-induced apoptosis in terms of mitochondria function and
oxidative stress.
As the main regulators of energy production and
apoptosis in the cells, mitochondria have key roles in cell
function, whose structural, biochemical, or functional abnormality
can lead to cell injury (33,34).
It is known that this organelle is not only the major site of ATP
production, but also serve an important role in apoptosis (35). To explore the impact of sodium
azide on mitochondria in the present study, the changes of
mitochondrial membrane potential (ΔΨm) were first explored in H9c2
cells treated with sodium azide, with the hypothesis that these
changes likely also affect the energy production. JC-1 staining was
used to evaluate changes in ΔΨm. The results demonstrated a decline
in mitochondrial membrane potential following sodium azide
treatment, but this decline was reversed by Mdivi-1 treatment.
These data indicated that the sodium azide-induced dissipation of
ΔΨm in mitochondria was moderated by Mdivi-1. Then, the ATP
contents were also quantitatively determined. The results
demonstrated that the cellular ATP contents in the sodium
azide-treated cells were decreased compared with the control cells,
suggesting that mitochondrial function was hindered by sodium azide
treatment. The present results demonstrated that Mdivi-1
pretreatment had a protective effect in this sodium azide-induced
mitochondrial dysfunction.
Furthermore, mitochondria are a major source of ROS
in myocytes. Increasing evidence has suggested that ROS overload is
associated with the pathogenesis of cardiovascular diseases, such
as myocardial infarction and heart failure (36). A previous study has demonstrated
that ROS is important in apoptosis of myocytes (37). However, whether ROS has a role in
apoptosis of sodium azide-treated H9c2 cells remained unclear. In
the present study, sodium azide treatment was demonstrated to
result in an increase of mitochondrial ROS production in H9c2
cardiomyocytes. Notably, Mdivi-1 pretreatment significantly
inhibited the accumulation of ROS.
Previous studies have suggested that sodium azide
could induce cell apoptosis in neonatal rat cardiac myocytes
(38,39). To identify the molecular mechanism
of apoptosis in the sodium azide-treated H9c2 cells, the expression
levels of the Bcl-2 family proteins were examined in the present
study. This family of proteins, consisting of both proapoptotic and
antiapoptotic members, includes Bax, Bcl-2 and BCL2 extra-large.
Bcl-2 is an important cellular protein, which prevents the release
of proapoptotic factors, such as cytochrome c, from the
mitochondria into the cytosol, and thus prevents apoptotic cell
death (40). By contrast, Bax, as
a proapoptotic factor, causes the collapse of the mitochondrial
membrane potential and subsequent increase in mitochondrial
permeability, triggers the caspase cascade and finally leads to
apoptosis (41,42). In the present study, the results
demonstrated that sodium azide-induced H9c2 cell apoptosis was
associated with decrease of Bcl-2 and increase of Bax expression.
Of note, pretreatment with Mdivi-1 attenuated the sodium
azide-induced upregulation of Bax and downregulation of Bcl-2.
In conclusion, the present study indicates that the
mechanism of sodium azide-induced cardiotoxicity may involve the
mitochondria-dependent apoptotic pathway. Mdivi-1 was demonstrated
to have a protective effect on the sodium azide-induced H9c2 cell
damage, suggesting that it may serve as a therapeutic agent in the
treatment of sodium azide-induced cardiotoxicity. However, the
present study was limited as Mdivi-1 was only used as a
pretreatment, and therefore, only a preventative, and not a
therapeutic, effect has been demonstrated. Thus, further studies
are required to confirm whether Mdivi-1 may also act as a
therapeutic agent.
Acknowledgements
This study was supported by the National Natural
Science Foundation of China (grant no. 81571848), and a Project
Funded by the Priority Academic Program Development of Jiangsu
Higher Education Institutions.
References
|
1
|
Chang S and Lamm SH: Human health effects
of sodium azide exposure: A literature review and analysis. Int J
Toxicol. 22:175–186. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Qamirani E, Razavi HM, Wu X, Davis MJ, Kuo
L and Hein TW: Sodium azide dilates coronary arterioles via
activation of inward rectifier K+ channels and
Na+-K+-ATPase. Am J Physiol Heart Circ
Physiol. 290:H1617–1623. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
El-Shenawy NS, Al-Harbi MS and Hamza RZ:
Effect of vitamin E and selenium separately and in combination on
biochemical, immunological and histological changes induced by
sodium azide in male mice. Exp Toxicol Pathol. 67:65–76. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Meatherall R and Oleschuk C: Suicidal
fatality from Azide ingestion. J Forensic Sci. 60:1666–1667. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Wiergowski M, Galer-Tatarowicz K,
Krzyzanowski M, Jankowski Z and Sein AJ: Suicidal intoxication with
sodium azide-a case report. Przegl Lek. 69:568–571. 2012.PubMed/NCBI
|
|
6
|
Mutz S, Meatherall R and Palatnick W:
Fatal intentional sodium azide poisoning. Clin Tox. 47:7132009.
|
|
7
|
Meatherall R and Palatnick W: Convenient
headspace gas chromatographic determination of azide in blood and
plasma. J Anal Toxicol. 33:525–531. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kikuchi M, Sato M, Ito T and Honda M:
Application of a new analytical method using gas chromatography and
gas chromatography-mass spectrometry for the azide ion to human
blood and urine samples of an actual case. J Chromatogr B Biomed
Sci Appl. 752:149–157. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Downes MA, Taliana KE, Muscat TM and Whyte
IM: Sodium azide ingestion and secondary contamination risk in
healthcare workers. Eur J Emerg Med. 23:68–70. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Marino S, Marani L, Nazzaro C, Beani L and
Siniscalchi A: Mechanisms of sodium azide-induced changes in
intracellular calcium concentration in rat primary cortical
neurons. Neurotoxicology. 28:622–629. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Chen SJ, Bradley ME and Lee TC: Chemical
hypoxia triggers apoptosis of cultured neonatal rat cardiac
myocytes: Modulation by calcium-regulated proteases and protein
kinases. Mol Cell Biochem. 178:141–149. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Szabados T, Dul C, Majtényi K, Hargitai J,
Pénzes Z and Urbanics R: A chronic alzheimer's model evoked by
mitochondrial poison sodium azide for pharmacological
investigations. Behav Brain Res. 154:31–40. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hessel MH, Michielsen EC, Atsma DE,
Schalij MJ, Van Der Valk EJ, Bax WH, Hermens WT, van Dieijen-Visser
MP and van der Laarse A: Release kinetics of intact and degraded
troponin I and T after irreversible cell damage. Exp Mol Pathol.
85:90–95. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Swafford AJ Jr, Bratz IN, Knudson JD,
Rogers PA, Timmerman JM, Tune JD and Dick GM: C-reactive protein
does not relax vascular smooth muscle: Effects mediated by sodium
azide in commercially available preparations. Am J Physiol Heart
Circ Physiol. 288:H1786–H1795. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Brooks C, Wei Q, Cho SG and Dong Z:
Regulation of mitochondrial dynamics in acute kidney injury in cell
culture and rodent models. J Clin Invest. 119:1275–1285. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Inomata K and Tanaka H: Protective effect
of benidipine against sodium azide-induced cell death in cultured
neonatal rat cardiac myocytes. J Pharmacol Sci. 93:163–170. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Cassidy-Stone A, Chipuk JE, Ingerman E,
Song C, Yoo C, Kuwana T, Kurth MJ, Shaw JT, Hinshaw JE, Green DR
and Nunnari J: Chemical inhibition of the mitochondrial division
dynamin reveals its role in Bax/Bak dependent mitochondrial outer
membrane permeabilization. Dev Cell. 14:193–204. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Sharp WW, Fang YH, Han M, Zhang HJ, Hong
Z, Banathy A, Morrow E, Ryan JJ and Archer SL: Dynamin-related
protein1 (Drp1)-mediated diastolic dysfunction in myocardial
ischemia-reperfusion injury: Therapeutic benefits of Drp1
inhibition to reduce mitochondrial fission. FASEB J. 28:316–326.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang N, Wang S, Li Y, Che L and Zhao Q: A
selective inhibitor of Drp1, Mdivi-1, acts against cerebral
ischemia/reperfusion injury via an anti-apoptotic pathway in rats.
Neurosci Lett. 535:104–109. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Wu Q, Xia SX, Li QQ, Gao Y, Shen X, Ma L,
Zhang MY, Wang T, Li YS, Wang ZF, et al: Mitochondrial division
inhibitor 1 (Mdivi-1) offers neuroprotection through diminishing
cell death and improving functional outcome in a mouse model of
traatic brain injury. Brain Res. 1630:134–143. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Labrousse AM, Zappaterra MD, Rube DA and
van der Bliek AM: C. Elegans dynamin-related protein DRP-1 controls
severing of the mitochondrial outer membrane. Mol Cell. 4:815–826.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Chan DC: Mitochondria: Dynamic organelles
in disease, aging, and development. Cell. 125:1241–1252. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Smirnova E, Griparic L, Shurland DL and
Van der Bliek AM: Dynamin-related protein drp1 is required for
mitochondrial division in mammalian cells. Mol Biol Cell.
12:2245–2256. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Boland K, Flanagan L and Prehn JH:
Paracrine control of tissue regeneration and cell proliferation by
caspase-3. Cell Death Dis. 4:e7252013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ong SB, Subrayan S, Lim SY, Yellon DM,
Davidson SM and Hausenloy DJ: Inhibiting mitochondrial fission
protects the heart against ischemia/reperfusion injury.
Circulation. 121:2012–2022. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Givvimani S, Munjal C, Tyagi N, Sen U,
Metreveli N and Tyagi SC: Mitochondrial division/mitophagy
inhibitor (Mdivi) ameliorates pressure overload induced heart
failure. PLoS One. 7:e323882012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Cipolat S, de Brito O Martins, Dal Zilio B
and Scorrano L: OPA1 requires mitofusin 1 to promote mitochondrial
fusion. Proc Natl Acad Sci USA. 101:15927–15932. 2004; View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Claycomb WC, Lanson NA Jr, Stallworth BS,
Egeland DB, Delcarpio JB, Bahinski A and Izzo NJ Jr: HL-1 cells: A
cardiac muscle cell line that contracts and retains phenotypic
characteristics of the adult cardiomyocyte. Proc Natl Acad Sci USA.
95:2979–2984. 1998; View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Hom J and Sheu SS: Morphological dynamics
of mitochondria - A special emphasis on cardiac muscle cells. J Mol
Cell Cardiol. 46:811–820. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ong SB and Hausenloy DJ: Mitochondrial
morphology and cardiovascular disease. Cardiovasc Res. 88:16–29.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ong SB, Hall AR and Hausenloy DJ:
Mitochondrial dynamics in cardiovascular health and disease.
Antioxid Redox Signal. 19:400–414. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Li Y, Wang P, Wei J, Fan R, Zuo Y, Shi M,
Wu H, Zhou M, Lin J, Wu M, et al: Inhibition of DRP1 by Mdivi-1
attenuates cerebral ischemic injury via inhibition
ofthemitochondria-dependent apoptotic pathway after cardiac arrest.
Neuroscience. 311:67–74. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Crompton M: The mitochondrial permeability
transition pore and its role in cell death. Biochem J. 341:233–249.
1999. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Di Lisa F and Bernardi P: Mitochondrial
function as a determinant of recovery or death in cell response to
injury. Mol Cell Biochem. 184:379–391. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Green DR and Reed JC: Mitochondria and
apoptosis. Science. 281:1309–1312. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Zhang ZW, Xu XC, Liu T and Yuan S:
Mitochondrion-permeable antioxidants to treat ROS-burst-mediated
acute diseases. Oxid Med Cell Longev. 2016:68595232016.PubMed/NCBI
|
|
37
|
Giordano FJ: Oxygen, oxidative stress,
hypoxia, and heart failure. J Clin Invest. 115:500–508. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Li Z, Cheng XR, Juan-Juan HU, Lan S and Du
GH: Neuroprotective effects of hyperoside on sodium azide-induced
apoptosis in pc12 cells. Chin J Nat Med. 9:450–455. 2011.
|
|
39
|
Wang J, Wei Q, Wang CY, Hill WD, Hess DC
and Dong Z: Minocycline up-regulates bcl-2 and protects against
cell death in mitochondria. J Biol Chem. 279:19948–19949. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chao DT and Korsmeyer SJ: BCL-2 family:
Regulators of cell death. Annu Rev Immunol. 16:395–419. 1998.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Schulze K, Dorner A and Schultheiß HP:
Mitochondrial function in heart failure. Heart Fail Rev. 4:229–244.
1999. View Article : Google Scholar
|
|
42
|
Moorjani N, Catarino P, Trabzuni D, Saleh
S, Moorji A, Dzimiri N, Al-Mohanna F, Westaby S and Ahmad M:
Upregulation of bcl-2 proteins during the transition to pressure
overload-induced heart failure. Int J Cardiol. 116:27–33. 2007.
View Article : Google Scholar : PubMed/NCBI
|