Introduction
Hepatocellular carcinoma (HCC) is a common type of
the primary malignancy of liver cancer, which is the third leading
cause of cancer-related deaths worldwide (1). The incidence of HCC is increasing in
both developing countries and economically developed regions
(2,3). A variety of risk factors are
contributed to the development of HCC, including chronic liver
inflammation caused by hepatitis B and C infection, obesity
(4), diabetes-induced liver
fibrosis and the environmental factors (5–7).
Currently, surgical resection and liver transplant have been the
two major therapeutic options in the treatment of liver cancer
(6). Nevertheless, since patients
are most often diagnosed at the advantaged stage with tumor
metastasis, surgery is only favorable for about 20% of liver cancer
cases. For patients harboring cancer metastasis, chemotherapy has
been one of the most important methods used in clinical practice,
however, with the more concern on the side-effect and resistance,
the amplification of chemotherapy in cancer is limited (8). Therefore, developing new strategies
for liver cancer treatment requires further investigation.
Increasing evidence has suggested that epigenetic
changes are involved in the development and progression of
malignant cancers (9–12). The acetylation modification of
histone is generally considered as the most extensively studied
epigenetic event (13), which is
regulated by histone acetyltransferases (HATs) and histone
deacetylases (HDACs) (14). HDACs
are overexpressed in a variety of cancers and correlated with the
poor prognosis of cancer patients (15–18).
Currently, histone deacetylase inhibitors (HDACi) are a major focus
of accumulating interests as anticancer agents, which function
through blocking histone deacetylation and modifying chromatin
structure and gene expression (17,19,20).
In addition to histone, a wide range of non-histone proteins are
also modified by acetylation and regulated by HDACi. Quisinostat
(JNJ-26481585), a novel second-generation HDACi, has high
specificity toward class I and II HDACs (21,22).
Quisinostat has shown anti-proliferative activity against non-small
cell lung cancer (NSCLC) (22),
however, the therapeutic effect of quisinostat on HCC remains
largely unknown.
In this study, we investigated the effect of
quisinostat on the cell growth of HCC. Our results showed that HCC
cells exposed to quisinostat treatment exhibited decreased cell
viability. Quisinostat treatment increased the acetylation of p53
and induced cell cycle arrest and cell apoptosis. Our results
provide insights into the potential use for quisinostat as a novel
chemotherapeutic agent in HCC treatment.
Materials and methods
Cell culture
HCC cell line HepG2 (harboring wild-type p53
(23,24)) was purchased from the Cell Bank of
the Chinese Academy of Sciences (Shanghai, China). Cells were
cultured in RPMI-1640 medium (GIBCO, Grand Island, NY, USA)
containing 10% fetal bovine serum at 37°C in a 5% CO2
incubator and passaged once every 2–3 days.
Cell viability assay
The cell viability assay was performed using the
Cell Counting Kit-8 (CCK-8) (Beyotime Institute of Biotechnology,
Shanghai, China) according to the manufacturer's instructions.
Briefly, HepG2 cells were cultured in the 96-plates with the cell
density at 1×103 per well. Cells were exposed to the
indicated concentration of quisinostat (dissolved in DMSO as stock
solutions and kept at room temperature) at different time point.
And then medium was removed, 10 µl of CCK-8 and 100 µl of serum
free medium were added. Cells were incubated at 37°C for 1 h. The
absorbance of each well was measured at 450 nm using the microplate
reader (Thermo Fisher Scientific, Waltham, MA, USA). The experiment
was performed in triplicate.
Cell apoptosis analysis
Cell apoptosis assay was performed with the Annexin
V-FITC Apoptosis Detection kit (Invitrogen, Carlsbad, CA, USA)
according to the protocol of manufacturer. HepG2 cells pretreated
with quisinostat were trypsinized and washed with pre-cold PBS. The
samples were centrifuged for 5 min at 400 × g. Discard the
supernatant and suspend the cells with 1× Annexin-binding buffer
with a final density of ~1×106 cells/ml. Cells were then
stained with Annexin V-fluorescein isothiocyanate (FITC) and PI
working solution for 15 min in the darkness at room temperature.
The cell apoptosis rate was analyzed using flow cytometry.
In vitro colony formation assay
Five hundred HepG2 cells were seeded into the 6-well
plate and cultured for 7 days. And then the culture medium was
replaced with fresh medium containing quisinostat with the
indicated concentration and cultured for another 7 days. The colony
formation of HepG2 cells was stained with 0.05% crystal violet for
5 min at room temperature.
Western blot analysis
Cells with the indicated treatment were harvested
and lyzed with NP-40 lysis buffer. The protein concentration was
determined by the Bradford assay (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Equal quantities of proteins were separated by
12% SDS-PAGE and then transferred into the nitrocellulose membrane
(Millipore Corp., Billerica, MA, USA). The membrane was blocked
with 5% milk at room temperature for 1 h and then incubated with
the primary antibodies for 2 h. After this step, the membrane was
incubated with horseradish peroxidase-conjugated secondary
antibodies (cat. no. 7072; 1:1,000; Cell Signaling Technology,
Inc., Danvers, MA, USA) for 1 h at room temperature. The bands were
visualized with KeyGEN Enhanced ECL detection kit (Keygen, Nanjing,
China). The following antibodies used: anti-p53 (#9282, 1:1,000;
Cell Signaling Technology, Inc.), anti-p21 (#2947, 1:1,000; Cell
Signaling Technology, Inc.), anti-cleaved caspase 3 (#9661,
1:1,000; Cell Signaling Technology, Danvers, MA, USA), anti-cleaved
caspase 8 (NB100-56116, 1:2,000; Novus Biologicals, Littleton, CO,
USA), anti-cleaved caspase 9 (no. 9501, 1:2,000; Cell Signaling
Technology, Inc.), anti-GAPDH mAb (3H12, 1:3,000; MBL, Nagoya,
Japan).
Co-immunoprecipitation (Co-IP)
The Co-IP assay was performed according to the
previous publications (25).
Briefly, HepG2 cells transfected with the indicated plasmids were
treated with quisinostat. Cells were harvested and lysed with the
NP-40 lysis buffer for 2 h at 4°C. The cell lysates were pretreated
with protein G beads for 1 h and then primary antibody was added to
the supernatant and incubated overnight at 4°C. Protein G beads
were added to the lysates to pull down the immunocomplexs. The
interacting proteins were detected by western blot.
Xenograft animal model
HepG2 cells were suspended with sterile PBS to make
a final density of 5×106 cells/ml. 200 µl of cell
suspension was injected subcutaneously into the flank of the nude
mice (BALB/c, 5–6 weeks of age, female) and left to grow for 2
weeks. The tumor development was checked daily. Mice were divided
randomly into control group and quisinostat group. Quisinostat was
given to the mice with the dosage of 2 mg/kg once daily for 15
consecutive days as previously described with minor modification
(22). The control group received
equal volume of 0.9% saline. The tumor volume was measured with the
digital calipers and calculated with the formula (length ×
width2)/2 (26,27). After 15 days, the mice were
sacrificed by cervical dislocation. The tumor weight and mice body
were also measured, respectively. This assay was approved by the
Animal Research Ethics Committee of Cangzhou Central Hospital. All
animals were handled following the ‘Guide for the Care and Use of
Laboratory Animals’ and the ‘Principles for the Utilization and
Care of Vertebrate Animals’.
Cell cycle
HepG2 cells treated with quisinostat were washed
with pre-cold PBS and then fixed in 70% ethanol. Cells were then
incubated with PBS containing 40 µg/ml RNase A for 30 min at 37°C.
Followed this, cells were resuspended in PBS containing 50 µg/ml
propidium iodide. The cell cycle distribution was detected by a BD
FACScan Cytometer (BD Biosciences, Franklin Lakes, NJ, USA).
Generation of the p53 null HepG2
cells
The p53-null HepG2 cells was generated with the
TP53-human gene knockout kit via CRISPR (KN200003; OriGene
Technologies, Inc., Rockville, MD, USA) according to the
manufacturer's instructions. Briefly, two targeting sequences (TCG
ACG CTA GGA TCT GACTG; CTG TGA GTG GAT CCA TTG GA) were selected.
To facilitate the cloning of target sequences into the pCas-Guide
vector, extra bases of ‘gatcg’ was added to the 5′-end of the
forward sequence and ‘g’ was added to the 3′ end. Meanwhile, add
‘aaaac’ to the 5′ end of the reverse complementary sequence and ‘c’
to its 3′ end. The two oligos were annealed to form the double
strand duplexes. The double-strand oligo DNA were ligated into the
pre-cut pCAS-Guide vector according to the manufacturer's
recommendation. The ligation product was transformed into the
competent cells. Sequence the purified DNA to identify correct
clones for proper insertion. The vector was transfected into the
HepG2 cells. Positive cells harboring the vector were selected. The
knockout efficiency of p53 was validated by PCR and western blot
analysis.
Statistical analysis
Differences between groups were determined by
Student's t-test or one way-ANOVA using the GraphPad Prism 5
(GraphPad Software, Inc., La Jolla, CA, USA). P<0.05 was
considered to indicate a statistically significant difference. Data
are presented as mean ± SD from three independent experiments.
Results
Quisinostat suppressed the viability
of HepG2 cells
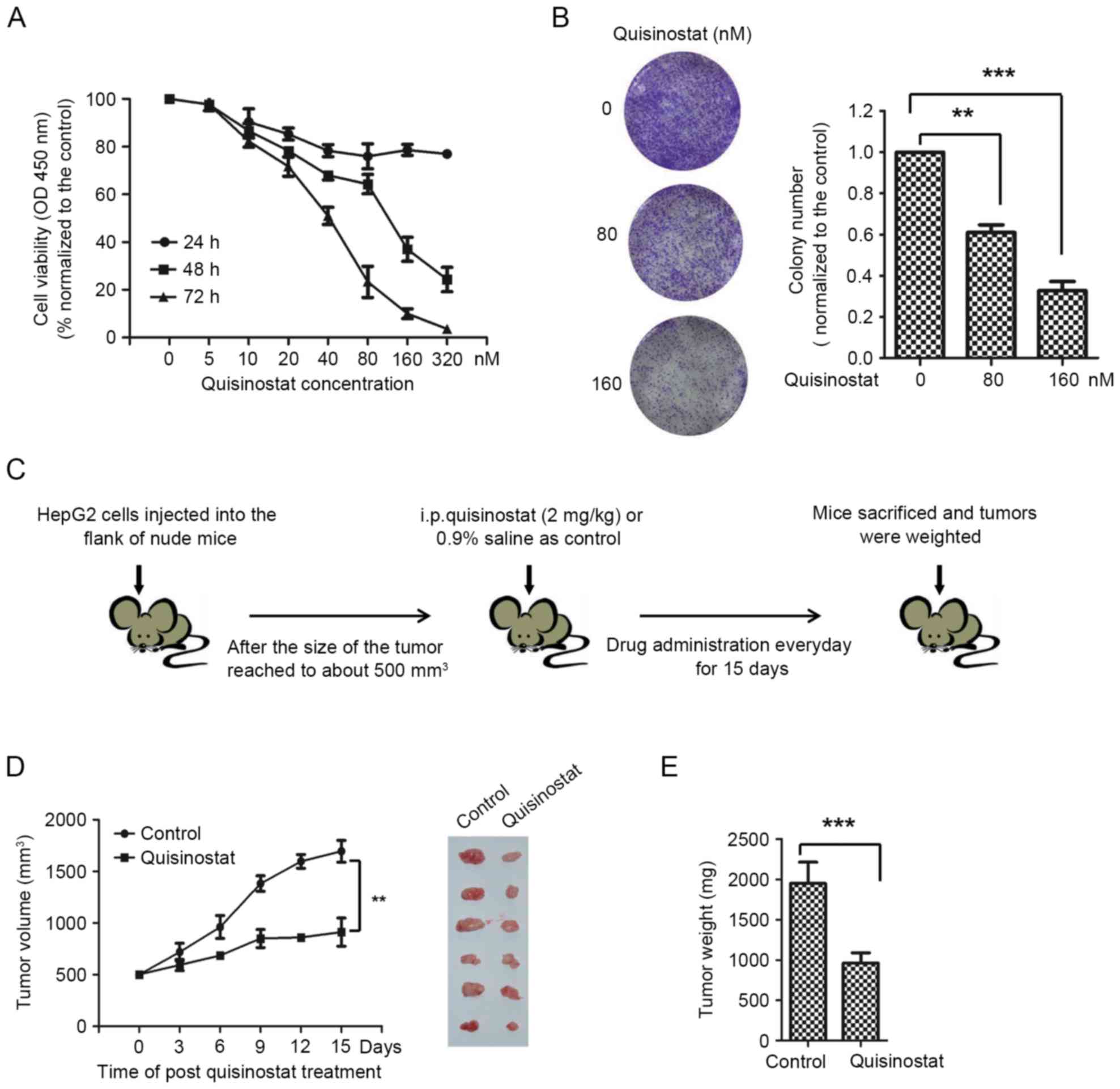
To determine the effect of quisinostat on HepG2
cell, CCK-8 assay was performed to detect the cell viability of
HepG2 cell exposed to quisinostat. Cells were treated for 24, 48
and 72 h with quisinostat diluted to concentrations of 5, 10, 20,
40, 80, 160 and 320 nM. Quisinostat inhibited the viability of
HepG2 cells in dose- and time-dependent manners (Fig. 1A). The IC50 values of
cells for quisinostat treatment at 48 and 72 h were 81.2 and 30.8
nM, respectively. The growth inhibitory effect of quisinostat was
also evaluated by in vitro colony formation assay. HepG2
cells in control group generated a number of visible colonies in 15
days, however, the number of colony formed by HepG2 cells cultured
with quisinostat was significantly less than that of the control
group (Fig. 1B).
To further detect the in vivo
anti-proliferative effect of quisinostat on HCC, HepG2 cells were
subcutaneously injected into the flanks of nude mice. When tumors
were grown for 2 weeks, mice were treated with quisinostat for a
consecutive 15 days (Fig. 1C).
Significantly decreased tumor volume and tumor weight were observed
with exposure of quisinostat in comparison with that of control
group only receiving saline (Fig. 1D
and E). In addition, no significant body weight loss was
observed among mice treated with quisinostat (data not shown),
suggesting no apparent toxicity of quisinostat on mice.
Collectively, these results demonstrated that quisinostat has
anti-proliferative effect on HepG2 cell growth.
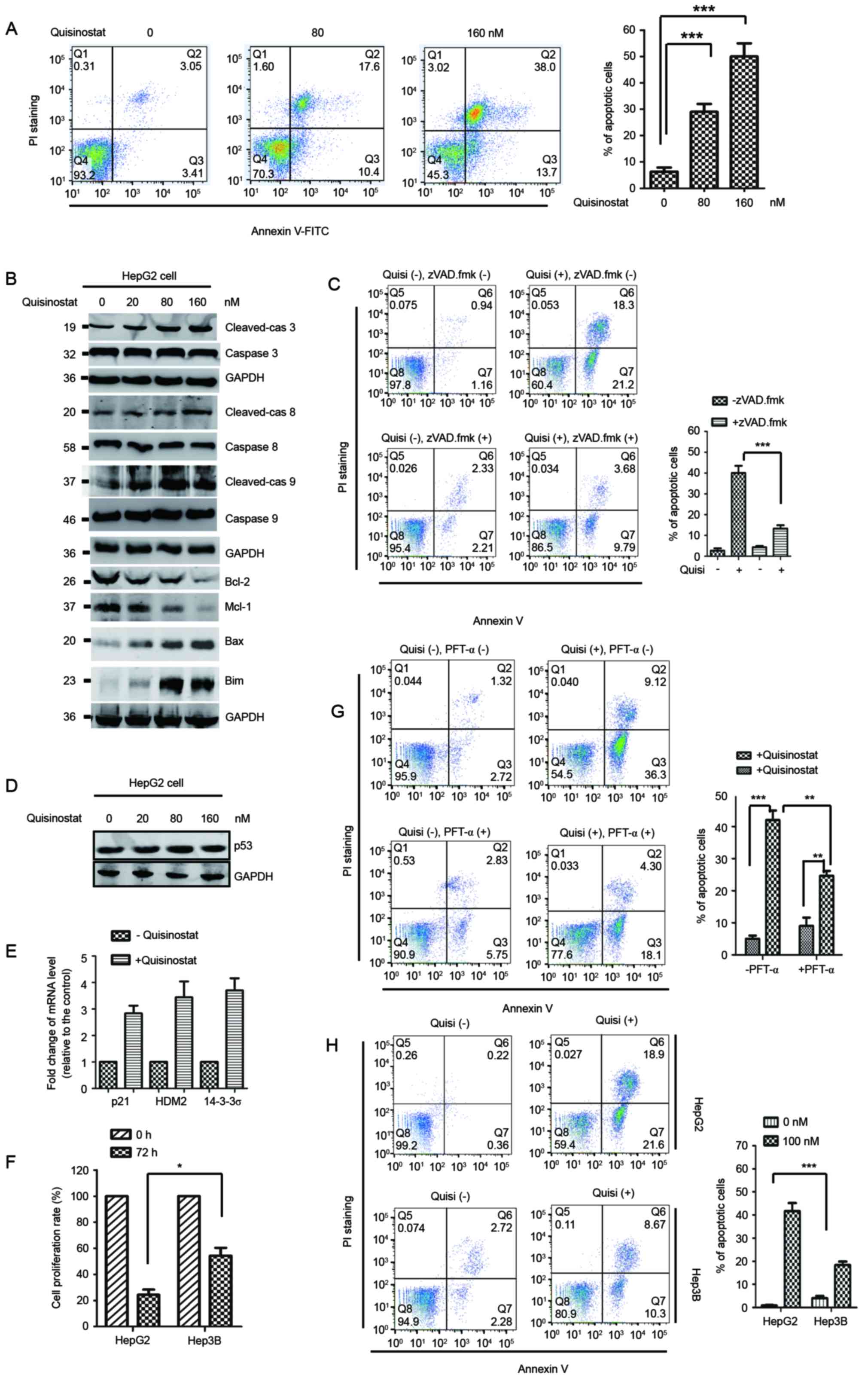
Quisinostat promotes cell apoptosis
via activation of caspase and p53 signaling
Apoptosis has been considered as a major approach to
eliminate cancer cells. To detect whether the decreased cell growth
induced by quisinostat was associated with the activation of cell
apoptosis, HepG2 cells treated with quisinostat were subjected to
FACS analysis to evaluate the cell apoptosis rate. The result
showed that quisinostat treatment obviously increased the apoptosis
of HepG2 cells compared with that of control cells (Fig. 2A). It has been reported that
mitochondria-mediated apoptotic pathway plays important roles in
inducing cell apoptosis, which releases Cyto c from the
mitochondrial inner space to cytosol, and activates caspase 9 and
caspase 3 via cleavage (28). To
define the molecular mechanism of quisinostat-induced cell
apoptosis, HepG2 cells were treated with quisinostat and the
caspase activation was investigated. Western blot analysis data
showed that quisinostat apparently resulted in higher expression of
the cleaved caspase family proteins including caspase 8, caspase 9
and caspase 3 (Fig. 2B).
Additionally, we found that HepG2 cells exposed to quisinostat
exhibited a noticeably decreased expression of anti-apoptotic
proteins Bcl-2 and Mcl-1 (Fig.
2B), while the abundance of pro-apoptotic protein Bax and Bim
were significantly increased (Fig.
2B). To precisely determine the cell death induced by
quisinostat treatment was via apoptosis, HepG2 cells were treated
with quisinostat in the presence of the apoptosis inhibitor
N-benzyloxycarbonyl-Val-Ala-Asp-fluoromethyl ketone (zVAD.fmk). The
results showed that zVAD.fmk treatment significantly inhibited the
cell apoptosis induced by quisinostat (Fig. 2C).
P53 activation is considered to be involved in
apoptotic response. To detect whether quisinostat-induced cell
apoptosis was also associated with p53, the expression level of p53
was detected in HepG2 cells treated with quisinostat. The protein
abundance of p53 was not significantly changed with the addition of
quisinostat (Fig. 2D). To
determine whether quisinostat activates p53 without increasing the
level of p53, the mRNA expression of p53 down-stream targets
including p21, HDM2 and 14-3-3σ were evaluated by RT-PCR. The
result showed that upon the treatment of quisinostat, the
expression of p21, HDM2, 14-3-3σ was significantly increased
(Fig. 2E), which suggested that
quisinostat activates p53. To determine the contribution of p53 in
quisinostat-induced HCC cell growth inhibition, HepG2 cells with
wild-type p53 and Hep3B cells harboring mutated p53 were treated
with quisinostat and the cell proliferation was monitored by CCK-8
assay. The result showed that upon treatment of quisinostat,
significantly decreased cell proliferation was observed in HepG2
cells in comparison with that of Hep3B cells with mutated p53
(Fig. 2F). To detect the
involvement of p53 in quisinostat-induced cell apoptosis, p53
inhibitor PFT-α was used to treat the cells and the cell apoptosis
was measured. As shown in Fig. 2G,
quisinostat treatment induced cell apoptosis, while PFT-α
significantly inhibited the cell apoptosis caused by quisinostat
treatment. To further determine the involvement of p53 in
quisinostat induced cell apoptosis, we detected the cell apoptosis
rate with Hep3B cells harboring mutated p53. Compared with that of
HepG2 cells, quisinostat treatment induced a significant decreased
cell apoptosis rate in Hep3B cells (Fig. 2H). These results indicated that the
cell growth inhibition trigged by quisinostat is tightly associated
with the activation of p53 signaling pathway.
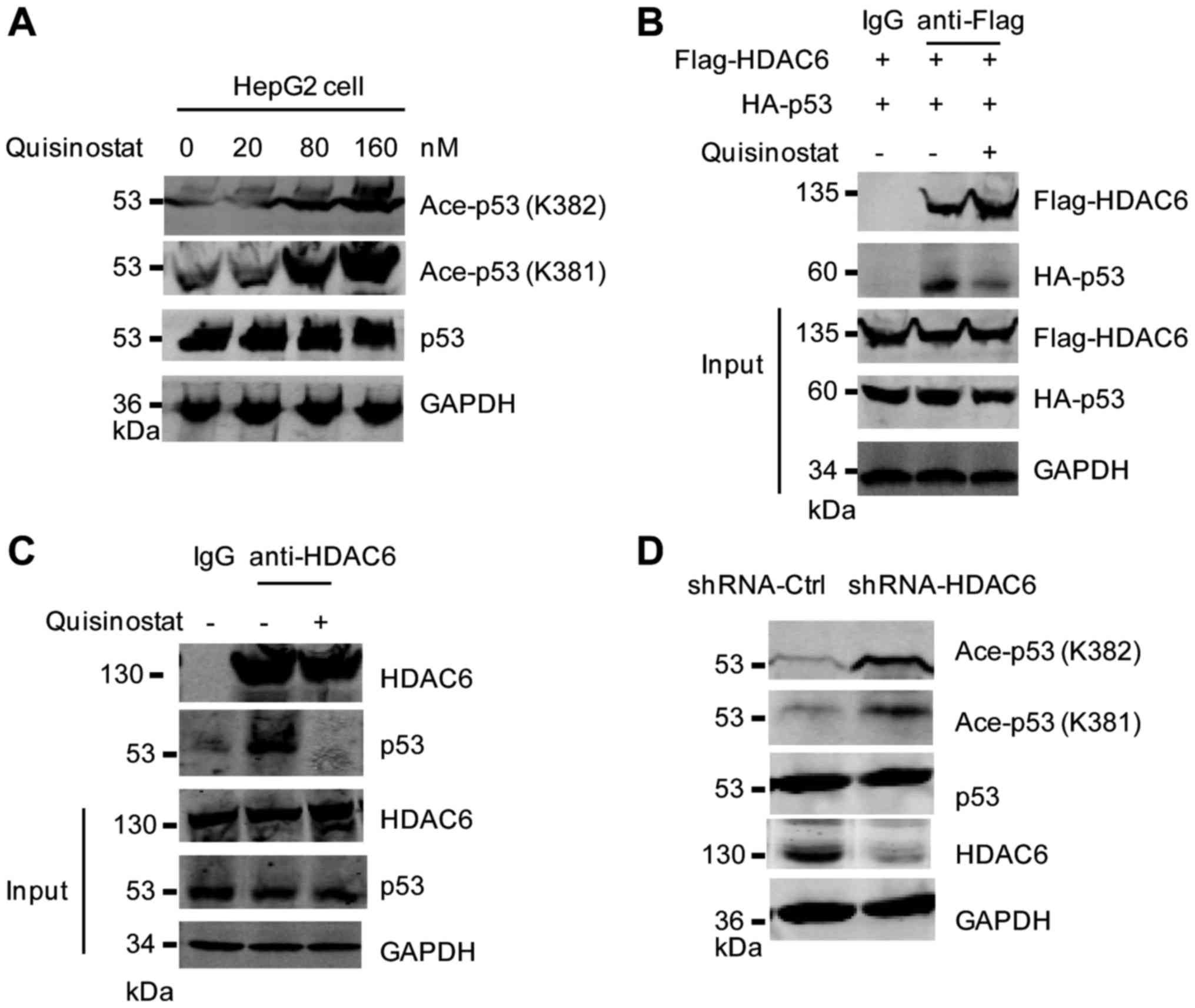
Quisinostat increased the acetylation
of p53
Acetylation is essential for the activation of p53
(29). To detect whether the
activation of p53 by quisinostat treatment is associated with the
acetylation status, western blot analysis was performed with HepG2
cells treated with quisinostat. The result showed that the
acetylation of p53 at K381/K382 was significantly increased with
quisinostat treatment (Fig. 3A).
Previous studies demonstrated that the histone deacetylase 6
(HDAC6) deacetylase p53 at lysine 381/382 (30,31).
To further understand the molecular mechanism of the upregulated
p53 acetylation induced by quisinostat, we hypothesized that
addition of quisinostat may block the deacetylation of p53 by
HDAC6. To determine this, we first detected the interaction between
HDAC6 and p53 in the presence of quisinostat. HepG2 cells were
transfected with Flag-HDAC6 and HA-p53. The Co-immunoprecipitation
(Co-IP) experiment showed that the interaction between HDAC6 and
p53 was weakened with the addition of quisinostat (Fig. 3B). Consistently, the endogenous
binding between HDAC6 and p53 was also impaired with quisinostat
treatment (Fig. 3C). To mimic the
inhibitory effect of quisinostat on the binding of HDAC6 and p53,
HepG2 cells were transfected with shRNA-HDAC6 to deplete the
endogenous expression of HDAC6, and then the acetylation status of
p53 was detected. The result showed that downregulation of HDAC6
significantly increased the acetylation of p53 at lysine 381/382
(Fig. 3D). Our data supported the
conclusion that quisinostat inhibited the binding between HDAC6 and
p53 and increased the acetylation of p53.
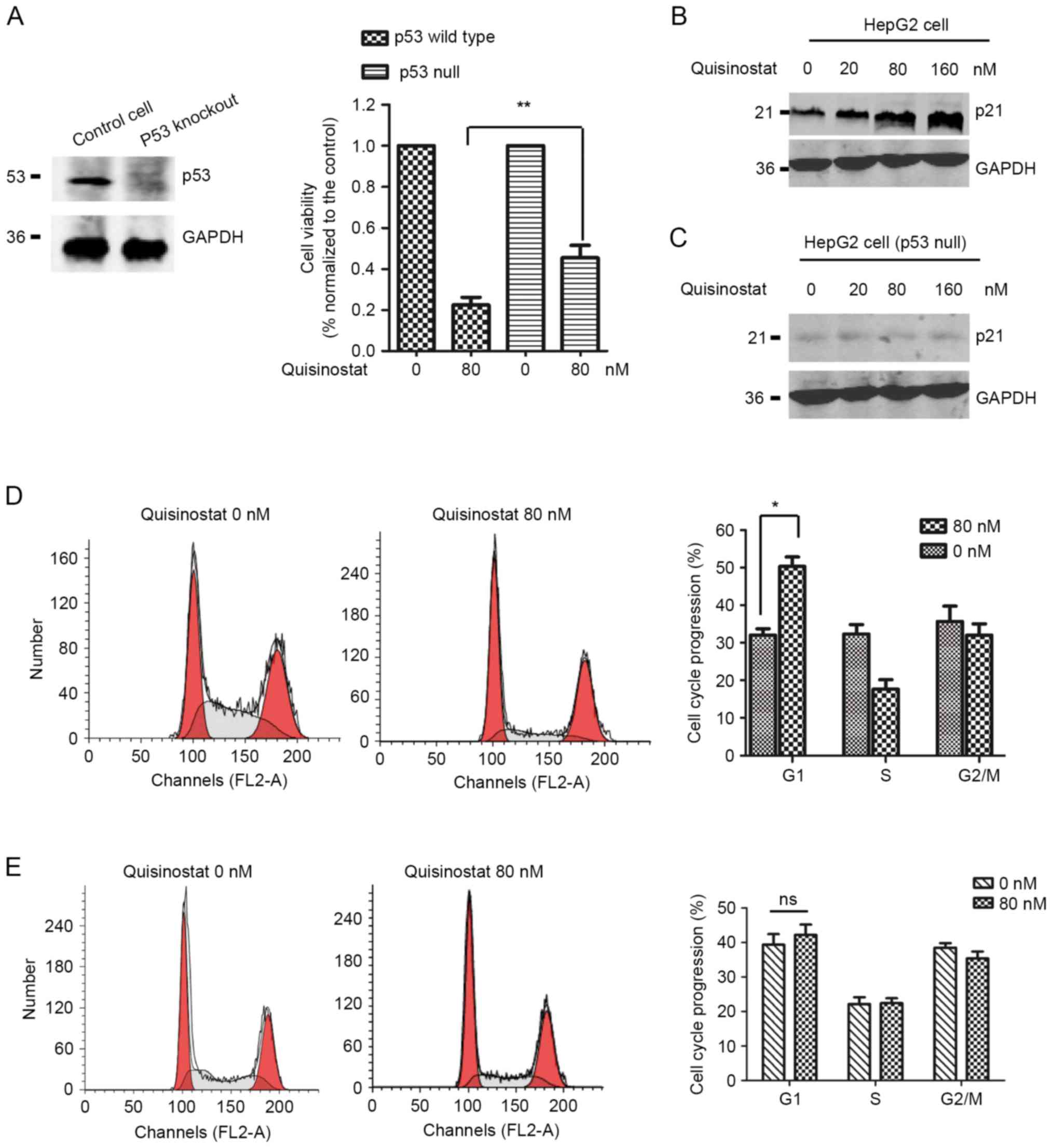
HepG2 cells with wild-type p53 are
more sensitive to quisinostat treatment
Given that the activation of p53 induced by
quisinostat, we determined the function of p53 in mediating the
inhibitory role of quisinostat in HepG2 cells. To this end, HepG2
cells with wild-type p53 or p53-null were selected for the
treatment of quisinostat, respectively. The knockout efficiency of
p53 was detected by western blot analysis (Fig. 4A, left panel). Cell viability assay
suggested that quisinostat inhibited the cell viability of HepG2
cells with or without p53 (Fig.
4A, right panel). However, the inhibitory effect was much more
significant in HepG2 cells with wild-type p53. This data
illustrated that p53 plays important role in mediating
quisinostat-induced cell growth inhibition.
To support this conclusion, we also monitored the
down-stream targets of p53. The result suggested that p21, an
important cell cycle regulator governing the cell progression from
G1 to S phase, is significantly highly expressed in HepG2 cells
with quisinostat treatment (Fig.
4B). In p53-depleted HepG2 cells, the expression level of p21
has not obviously changed (Fig.
4C). As p21 is tightly associated with cell cycle progression,
we performed FACS analysis with HepG2 cells pre-treated with
quisinostat. As shown in Fig. 4D,
the cell cycle distribution in HepG2 cells with quisinostat
treatment exhibited a significant accumulation in the G1 phase,
which suggested cell cycle arrest from G1 to S phase, however, no
significant cell cycle arrest was observed in p53 null HepG2 cells
(Fig. 4E). There results suggested
that HepG2 cells with wild-type p53 are more sensitive to
quisinostat treatment.
Discussion
HCC cells display a marked resistance to currently
available chemotherapeutic treatment strategies resulting in
disappointing clinical outcomes in the cancer patients.
It has been recognized that epigenetic changes play
important roles in the pathogenesis of a variety of human cancers
(9). Therefore, alternation of
epigenetic modification is identified as a novel therapeutic
approach. In recent years, HDAC inhibitors have improved the
treatment outcome of conventional standard chemotherapy (32–34).
Among these, vorinostat (SAHA) and rimidepsin (depsipetide, FK288)
have been approved by the US Food and Drug Administration (FDA) for
the treatment of patients with cutaneous T cell lymphoma (35–37).
Quisinostat has been reported to exhibit improved
anti-proliferative activity over HDAC inhibitory compounds. Recent
studies demonstrated that quisinostat inhibits the tumorigenesis of
NSCLC (10,22). Notably, Heinicke U and collages
demonstrated that quisinostat induced apoptosis and inhibited the
growth of rhabdomyosarcoma through activating the mitochondrial
pathway of apoptosis (21). This
is a very interesting study, which provides novel insights into the
underlying molecular mechanism of the anticancer activity of
quisinostat, and more importantly, suggests the promising
application of quisinostat for the treatment of rhabdomyosarcoma.
This paper provides critical evidence for exploring the function of
quisinostat in other types of cancers. In the present study, we
showed that quisinostat suppressed the cell viability of HepG2
cells. Activation of caspase and p53 were observed in
quisinostat-treated HepG2 cells. Specifically, for the activation
of p53, we found that quisinostat disrupted the interaction between
HDAC6 and p53, which consequently increases the acetylation of p53
and induces p53 mediated cell apoptosis.
Induction of apoptosis upon HDAC inhibitors has
previously been shown in various cancers. Cell apoptosis, inducing
cell death, has been a major mechanism in anti-cancer drug
development. A recent study documented that quisinostat treatment
in NSCLC cells increased reactive oxygen species (ROS) production
and destroyed mitochondrial membrane potential, which resulting in
mitochondrial-mediated cell apoptosis (10). Our results also demonstrated the
activation of mitochondrial-mediated apoptotic pathway in
quisinostat treated HepG2 cells. In addition, the present study
demonstrated the involvement of p53 signaling in quisinostat
induced cell apoptosis and growth inhibitory in HepG2 cells.
Aberrantly loss-of function of p53 is critical in malignant
progression of cancer cells. Increasing the expression level of
wild-type p53 or activating p53 has been one of the main strategies
to trigger p53-mediate physiological function, including cell
apoptosis and cell cycle arrest.
Acetylation of p53 is essential for its activation
(29). Upregulated p53 acetylation
is involved in HDACi treatment on NSCLC cells (10). The present study found that in
HepG2 cells, quisinostat increased the acetylation of p53. Recent
published data demonstrated that HDAC6 catalyzed the deacetylation
of p53 at lysine 381/382 (30).
For the underlying mechanism, we found that addition of quisinostat
disrupted the physical interaction between HDAC6 and p53, which
impaired the deacelytation of p53 and increased the p53 activity. A
recent study by Bao et al (10) showed that quisinostat treatment in
NSCLC cells also resulted in the accumulation of p53 acetylation at
lysine 372. Further investigation may be required to detect whether
the lysine 372 acetylation of p53 is also regulated by quisinostat
in HCC cells.
In summary, the present study demonstrated that
quisinostat inhibited the proliferation of HepG2 cells, causing
cell cycle arrest and cell apoptosis partially via upregulating the
acetylation of p53 and activating caspase cleavage.
Quisinostat-mediated cell growth inhibition may be helpful for HCC
treatment as a potential candidate agent in clinical practice.
References
|
1
|
El-Serag HB and Rudolph KL: Hepatocellular
carcinoma: Epidemiology and molecular carcinogenesis.
Gastroenterology. 132:2557–2576. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Erichsen R, Jepsen P, Jacobsen J, Norgaard
M, Vilstrup H and Sørensen HT: Time trends in incidence and
prognosis of primary liver cancer and liver metastases of unknown
origin in a Danish region, 1985–2004. Eur J Gastroenterol Hepatol.
20:104–110. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Bosch FX, Ribes J, Díaz M and Cléries R:
Primary liver cancer: Worldwide incidence and trends.
Gastroenterology. 127 5 Suppl 1:S5–S16. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Ray K: Gut microbiota: Obesity-induced
microbial metabolite promotes HCC. Nat Rev Gastroenterol Hepatol.
10:4422013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Starzl TE, Marchioro TL, Vonkaulla KN,
Hermann G, Brittain RS and Waddell WR: Homotransplantation of the
liver in humans. Surg Gynecol Obstet. 117:659–676. 1963.PubMed/NCBI
|
|
6
|
Liao PH, Hsu HH, Chen TS, Chen MC, Day CH,
Tu CC, Lin YM, Tsai FJ, Kuo WW and Huang CY: Phosphorylation of
cofilin-1 by ERK confers HDAC inhibitor resistance in
hepatocellular carcinoma cells via decreased ROS-mediated
mitochondria injury. Oncogene. 36:1978–1990. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Ande SR, Nguyen KH, Grégoire Nyomba BL and
Mishra S: Prohibitin-induced, obesity-associated insulin resistance
and accompanying low-grade inflammation causes NASH and HCC. Sci
Rep. 6:236082016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Baylin SB: Resistance, epigenetics and the
cancer ecosystem. Nat Med. 17:288–289. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lund AH and van Lohuizen M: Epigenetics
and cancer. Genes Dev. 18:2315–2335. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bao L, Diao H, Dong N, Su X, Wang B, Mo Q,
Yu H, Wang X and Chen C: Histone deacetylase inhibitor induces cell
apoptosis and cycle arrest in lung cancer cells via mitochondrial
injury and p53 up-acetylation. Cell Biol Toxicol. 32:469–482. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Cui Y, Gao D, Linghu E, Zhan Q, Chen R,
Brock MV, Herman JG and Guo M: Epigenetic changes and functional
study of HOXA11 in human gastric cancer. Epigenomics. 7:201–213.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Downs B and Wang SM: Epigenetic changes in
BRCA1-mutated familial breast cancer. Cancer Genet. 208:237–240.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Allfrey VG, Faulkner R and Mirsky AE:
Acetylation and methylation of histones and their possible role in
the regulation of Rna synthesis. Proc Natl Acad Sci USA.
51:786–794. 1964; View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Kurdistani SK and Grunstein M: Histone
acetylation and deacetylation in yeast. Nat Rev Mol Cell Biol.
4:276–284. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
15
|
Yoon S and Eom GH: HDAC and HDAC
inhibitor: From cancer to cardiovascular diseases. Chonnam Med J.
52:1–11. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li Y and Seto E: HDACs and HDAC inhibitors
in cancer development and therapy. Cold Spring Harb Perspect Med.
6(pii): a0268312016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang XJ and Seto E: HATs and HDACs: From
structure, function and regulation to novel strategies for therapy
and prevention. Oncogene. 26:5310–5318. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Osada H, Tatematsu Y, Saito H, Yatabe Y,
Mitsudomi T and Takahashi T: Reduced expression of class II histone
deacetylase genes is associated with poor prognosis in lung cancer
patients. Int J Cancer. 112:26–32. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
West AC and Johnstone RW: New and emerging
HDAC inhibitors for cancer treatment. J Clin Invest. 124:30–39.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Kim MS, Son MW, Kim WB, In Park Y and Moon
A: Apicidin, an inhibitor of histone deacetylase, prevents
H-ras-induced invasive phenotype. Cancer Lett. 157:23–30. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Heinicke U, Kupka J, Fichter I and Fulda
S: Critical role of mitochondria-mediated apoptosis for
JNJ-26481585-induced antitumor activity in rhabdomyosarcoma.
Oncogene. 35:3729–3741. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Arts J, King P, Mariën A, Floren W, Beliën
A, Janssen L, Pilatte I, Roux B, Decrane L, Gilissen R, et al:
JNJ-26481585, a novel ‘second-generation’ oral histone deacetylase
inhibitor, shows broad-spectrum preclinical antitumoral activity.
Clin Cancer Res. 15:6841–6851. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cheung ST, Wong SY, Lee YT and Fan ST: GEP
associates with wild-type p53 in hepatocellular carcinoma. Oncol
Rep. 15:1507–1511. 2006.PubMed/NCBI
|
|
24
|
Dai Q, Yin Y, Liu W, Wei L, Zhou Y, Li Z,
You Q, Lu N and Guo Q: Two p53-related metabolic regulators, TIGAR
and SCO2, contribute to oroxylin A-mediated glucose metabolism in
human hepatoma HepG2 cells. Int J Biochem Cell Biol. 45:1468–1478.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang CZ, Chen GG, Merchant JL and Lai PB:
Interaction between ZBP-89 and p53 mutants and its contribution to
effects of HDACi on hepatocellular carcinoma. Cell Cycle.
11:322–334. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Laporte AN, Barrott JJ, Yao RJ, Poulin NM,
Brodin BA, Jones KB, Underhill TM and Nielsen TO: HDAC and
proteasome inhibitors synergize to activate pro-apoptotic factors
in synovial sarcoma. PLoS One. 12:e01694072017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Faustino-Rocha A, Oliveira PA,
Pinho-Oliveira J, Teixeira-Guedes C, Soares-Maia R, da Costa RG,
Colaço B, Pires MJ, Colaço J, Ferreira R and Ginja M: Estimation of
rat mammary tumor volume using caliper and ultrasonography
measurements. Lab Anim (NY). 42:217–224. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Green DR: Apoptotic pathways: Ten minutes
to dead. Cell. 121:671–674. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Tang Y, Zhao W, Chen Y, Zhao Y and Gu W:
Acetylation is indispensable for p53 activation. Cell. 133:612–626.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ryu HW, Shin DH, Lee DH, Choi J, Han G,
Lee KY and Kwon SH: HDAC6 deacetylates p53 at lysines 381/382 and
differentially coordinates p53-induced apoptosis. Cancer Lett.
391:162–171. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Ding G, Liu HD, Huang Q, Liang HX, Ding
ZH, Liao ZJ and Huang G: HDAC6 promotes hepatocellular carcinoma
progression by inhibiting P53 transcriptional activity. FEBS Lett.
587:880–886. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mariadason JM: HDACs and HDAC inhibitors
in colon cancer. Epigenetics. 3:28–37. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pelicci PG: A new class of anti-cancer
drugs: HDAC-inhibitors. Suppl Tumori. 1:S662002.PubMed/NCBI
|
|
34
|
Secrist JP, Zhou X and Richon VM: HDAC
inhibitors for the treatment of cancer. Curr Opin Investig Drugs.
4:1422–1427. 2003.PubMed/NCBI
|
|
35
|
Duvic M and Vu J: Vorinostat in cutaneous
T-cell lymphoma. Drugs Today (Barc). 43:585–599. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Duvic M and Vu J: Vorinostat: A new oral
histone deacetylase inhibitor approved for cutaneous T-cell
lymphoma. Expert Opin Investig Drugs. 16:1111–1120. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Murata M, Towatari M, Kosugi H, Tanimoto
M, Ueda R, Saito H and Naoe T: Apoptotic cytotoxic effects of a
histone deacetylase inhibitor, FK228, on malignant lymphoid cells.
Jpn J Cancer Res. 91:1154–1160. 2000. View Article : Google Scholar : PubMed/NCBI
|