Introduction
It is well established that smoking is a serious
health concern and is associated with cancer and various other
diseases (1–3). Cardiovascular disease is a
frequently-occurring and common disease, with a higher incidence
and mortality among smokers (4).
Nicotine is the key ingredient in smoking products and there are
two distinctive arguments regarding nicotine and apoptosis. Certain
reports have indicated that nicotine promotes proliferation and
inhibits apoptosis by acting on neuronal nicotinic acetylcholine
receptors (nAChRs) (5,6). However, another report indicated that
nicotine induces human cell apoptosis by influencing Hsp90 α
expression (7). Therefore, the
effect of nicotine on apoptosis requires further investigation.
Previously, it was demonstrated that nicotine has a harmful effect
on cardiomyocytes via the promotion of apoptosis (8), which is associated with various
cardiology diseases and high cardiovascular mortality, however, the
underlying mechanism is not well established.
Akt, a serine/threonine kinase, has an important
role in cell survival, proliferation, migration and apoptosis. Akt
is phosphorylated and activated by phosphoinositide 3-kinase
(PI3K), which is mediated by the insulin pathway, and subsequently
regulates fatty acid β-oxidation and promotes survival (9,10).
An et al (11) reported
that melatonin attenuates sepsis-induced cardiac dysfunction via
activation of Akt. Due to the critical role of Akt, the regulatory
mechanism of Akt is important. Various methods of Akt regulation
exist, one of which is Akt protein degradation (12). A previous study demonstrated that
ubiquitin-mediated Akt protein degradation leads to normal human
lung fibroblast apoptosis (13).
Tetratricopeptide repeat domain 3 (TTC3) and mitochondrial E3
ubiquitin protein ligase 1 (MUL1), which are important regulating
factors of E3 ligases for Akt, have been reported to cause Akt
ubiquitination and proteasomal degradation (14,15).
The present study, to the best of our knowledge,
demonstrated for the first time that nicotine induced H9C2 cell
apoptosis via Akt protein degradation. In addition, the results
indicated that nicotine upregulated the level of TTC3 mRNA, which
may be responsible for Akt protein degradation.
Materials and methods
Cell culture
H9C2 embryonic rat myocardium-derived cells were
obtained from Shanghai Institutes for Biological Sciences, Chinese
Academy of Sciences (Shanghai, China). The cells were cultured in
Dulbecco's modified Eagle's medium (Hyclone; GE Healthcare Life
Sciences, Logan, UT, USA) containing 10% heat-inactivated fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) and 100 U/ml penicillin and 100 µg/ml streptomycin at 37°C in
a humidified incubator with 5% CO2 until 70–80%
confluence, and sub-cultured (1:3 split ratio) using trypsin
(0.25%) containing EDTA (0.02%).
Cell treatments
Nicotine (Sigma Aldrich, Merck KGaA, Darmstadt,
Germany) was dissolved in DMEM containing 10% FBS to concentration
of 10 and 100 µM, then 80% confluent cells were incubated with
different concentrations of nicotine (10 or 100 µM) or standard
medium for 48 h at 37°C, or together with a PYR-41 pretreatment for
30 min. PYR-41 (Selleckchem, TX, USA) was dissolved in DMEM
containing 10% FBS to concentration of 5 and 10 µM.
Cell viability assay
Cell Counting Kit-8 (CCK-8) assay was purchased from
Dojindo Molecular Technologies, Inc. (Kumamoto, Japan) and was used
to measure the cell viability in different nicotine treatment
groups, according to the manufacturer's protocol. Briefly, cells
were seeded in a 96-well plate at a density of 5×103
cells/well with four replicates for each group and cultured
overnight at 37°C, cells were subsequently exposed to various
concentrations of nicotine (10 or 100 µM) or standard medium as a
control for 48 h at 37°C. Following treatment, 100 µl fresh medium
containing 10 µl CCK-8 solution was added to each well in an
incubator for 30 min at 37°C. The optical density of each well was
determined at 450 nm by a microplate reader to calculate the cell
viability.
Quantification of apoptotic cells
According to the manufacturer's protocol, the cell
apoptosis at single-cell level was detected and quantified by TUNEL
assay, using the in situ Cell Death Detection Fluorescein
kit (Roche Molecular Systems, Inc., Pleasanton, CA, USA). Briefly,
cells cultured on glass coverslips at 37°C at 70–80% confluence,
were treated with various concentrations of nicotine (0, 10 or 100
µM) for 48 h at 37°C, washed with PBS, fixed with 4%
paraformaldehyde in PBS for 1 h at 37°C and permeabilized with 0.5%
Triton X-100 in 0.1% sodium citrate for 2 min on ice. Following
incubation with the enzyme solution mixture and label solution for
1 h at 37°C in a dark, humid chamber, nuclei were counterstained
with 100 ng/ml DAPI for 3–5 min at 37°C. The percentage of cells
undergoing apoptosis was determined with a fluorescence microscope
and 25 random fields were quantified by an investigator who was
blind to the treatment.
mRNA expression analysis
Total RNA was prepared from 95% confluent cultured
cells using TRIzol reagent (Invitrogen; Thermo Fisher Scientific,
Inc.), according to the manufacturer's protocol. RNase-free DNase I
(cat. no. RR047A; Takara Biotechnology, Co., Ltd., Dalian, China)
was used to remove DNA contamination and cDNA was generated from 2
µg total RNA using the PrimeScript™ RT reagent kit with gDNA Eraser
(cat. no. RR047A; Takara Biotechnology, Co., Ltd.), according to
the manufacturer's protocol. The 20 µl reaction mixture was
incubated as 50°C of 15 min followed with enzyme inactivation by
incubation at 85°C for 5 sec. Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) was performed using a CFX96
Real-Time PCR System (Bio-Rad Laboratories, Inc., Hercules, CA,
USA) and the QuantiTect SYBR-Green PCR kit (Takara Biotechnology,
Co., Ltd.), with the following An initial predenaturation step at
95°C for 30 sec, followed by 40 cycles of denaturation at 95°C for
5 sec and annealing at 60°C for 30 sec. Data processing and
normalization was undertaken as previously described (16). All RT-qPCR experiments were
performed in triplicate. Primers used are listed in Table I.
 | Table I.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction. |
Table I.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction.
|
|
| Primer sequence |
|---|
|
|
|
|
|---|
| Gene | Accession no. | Forward | Reverse |
|---|
| GAPDH | NM_017008.4 |
5′-GGCACAGTCAAGGCTGAGAATG-3′ |
5′-ATGGTGGTGAAGACGCCAGTA-3′ |
| Total-Akt | NM_033230.2 |
5′-ATGGACTTCCGGTCAGGTTCA-3′ |
5′-GCCCTTGCCCAGTAGCTTCA-3′ |
|
| NM_017093.1 |
|
|
|
| NM_031575.1 |
|
|
| MUL1 | NM_001106695.1 |
5′-GCAAGTTACAGCCACCACCTGA-3′ |
5′-CCAGACTTTGTGTTGCTGCTGA-3′ |
| TTC3 | NM_001108315 |
5′-GTGGGCACAAGTTTCACAAAGG-3′ |
5′-AAGTGCATGGTGCATTAGTGAGG-3′ |
Gene silencing with small interfering
RNA (siRNA) and gene overexpression with plasmid vector
The siRNA against TTC3 mRNA
(5′-UUGCAACUUGCUAGAAGAAUU-3′) and scrambled siRNA
(5′-UUAACGUUGAACGAUCUUCUU-3′) were designed and purchased from
Shanghai GenePharma Co., Ltd. (Shanghai, China). The Akt1
overexpression pCMV6-XL5 plasmid vector and control pCMV6-XL5
plasmid vector were designed by GeneCopoeia, Inc. (Rockville, MD,
USA). H9C2 cells were plated onto 6-well plates at a density of
1.5×104 cells/well and transfected with TTC3 siRNA (75
pmol) or pCMV6-XL5-Akt1 (2 µg) at 80% confluence at 37°C using
Lipofectamine 2000 according to the manufacturer's protocol
(Invitrogen; Thermo Fisher Scientific, Inc.). At 48 h
post-transfection, RT-qPCR was used to determine the expression
levels of TTC3 and western blotting was performed to determine the
expression levels of Akt1. At 36 h post-transfection, the cells
were subjected to treatment with nicotine (10 µM) or standard
medium for 48 h at 37°C. Subsequently, the cells were harvested for
further analysis.
Western blot analysis
Cells were cultured in a 6-well plates until 95%
confluence and were lysed in ice-cold radioimmunoprecipitation
assay lysis buffer (Beyotime Institute of Biotechnology, Haimen,
China) supplemented with 1% phenylmethylsulfonyl fluoride (Beyotime
Institute of Biotechnology). Protein concentrations were detected
with a bicinchoninic acid assay. Lysates (40 µg/lane) were
separated on 12% SDS-PAGE gels, transferred to nitrocellulose
filter membranes. Subsequently, the membrane was incubated with 10%
skim milk for 1 h at 37°C to block nonspecific binding, and
incubated overnight at 4°C with antibodies directed against Akt
(cat. no. 9272; 1:1,000), phosphorylated (p)-Akt (ser473; cat. no.
4060; 1:1,000), caspase-3 (cat. no. 14220; 1:1,000),
cleaved-caspase-3 (cat. no. 9664; 1:1,000; all from Cell Signaling
Technology, Inc., Danvers, MA, USA), β-actin (cat. no. sc-130300;
1:500, Santa Cruz Biotechnology, Inc., Dallas, TX, USA). The blots
were subsequently incubated with horseradish peroxidase-conjugated
anti-rabbit IgG secondary antibody (cat. no. 7074; 1:2,000) or an
anti-mouse IgG (cat. no. 7076; 1:2,000; Cell Signaling Technology,
Inc.) for 1 h at 37°C. Bands were visualized with an enhanced
chemiluminescence kit (EMD Millipore, Billerica, MA, USA) in a
ChemiDoc XRS system (Bio-Rad Laboratories, Inc.). The protein level
was quantified with Quantity One software version 4.6.2 (Bio-Rad
Laboratories, Inc.).
Statistical analysis
Data are presented as the mean ± standard error of
the mean. SPSS 15.0 software (SPSS, Inc., Chicago, IL, USA) was
used for all statistical analyses. Statistical comparisons between
two groups were performed with the unpaired Student's t-test. The
differences between more than two groups were analyzed with one-way
analysis of variance followed by Student-Newman-Keuls post hoc
test. P≤0.05 was considered to indicate a statistically significant
difference.
Results
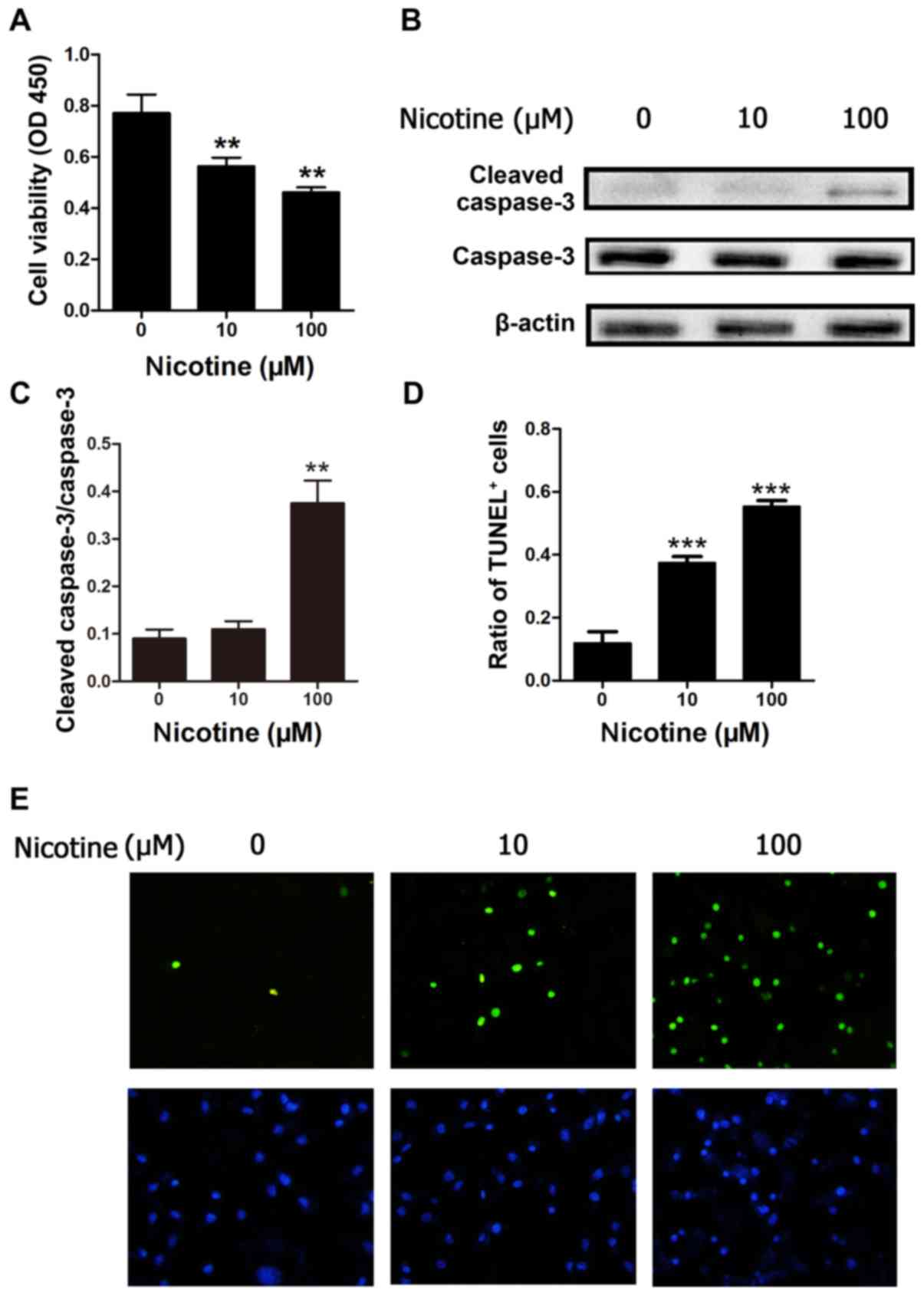
Nicotine promotes H9C2 cell apoptosis
in vitro
To investigate whether nicotine may promote cardiac
myoblast cell apoptosis, the present study employed H9C2 cells to
determine the role of nicotine on apoptosis in cultured cells. Cell
viability was determined by CCK-8 assay. The viability of H9C2
cells exposed to nicotine at 10 and 100 µM was significantly
reduced in a concentration-dependent manner after 48 h, compared
with control cells (P<0.01; Fig.
1A). To further investigate the effect of the nicotine on H9C2
cells, the protein expression level of caspase-3, which is involved
in apoptosis, was also investigated by western blotting. The
results demonstrated that nicotine led to activation of caspase-3
(cleaved-caspase-3) in the 100 µM nicotine treatment, but not the
10 µM treatment group compared with the control (Fig. 1B and C). In addition, apoptosis was
evaluated by TUNEL analysis. Compared with control cells, nicotine
at 10 and 100 µM induced a significant increase in the number of
TUNEL-positive cells after 48 h of treatment (Fig. 1D and E). These results indicate
that nicotine may inhibit H9C2 cell viability and promote H9C2 cell
apoptosis.
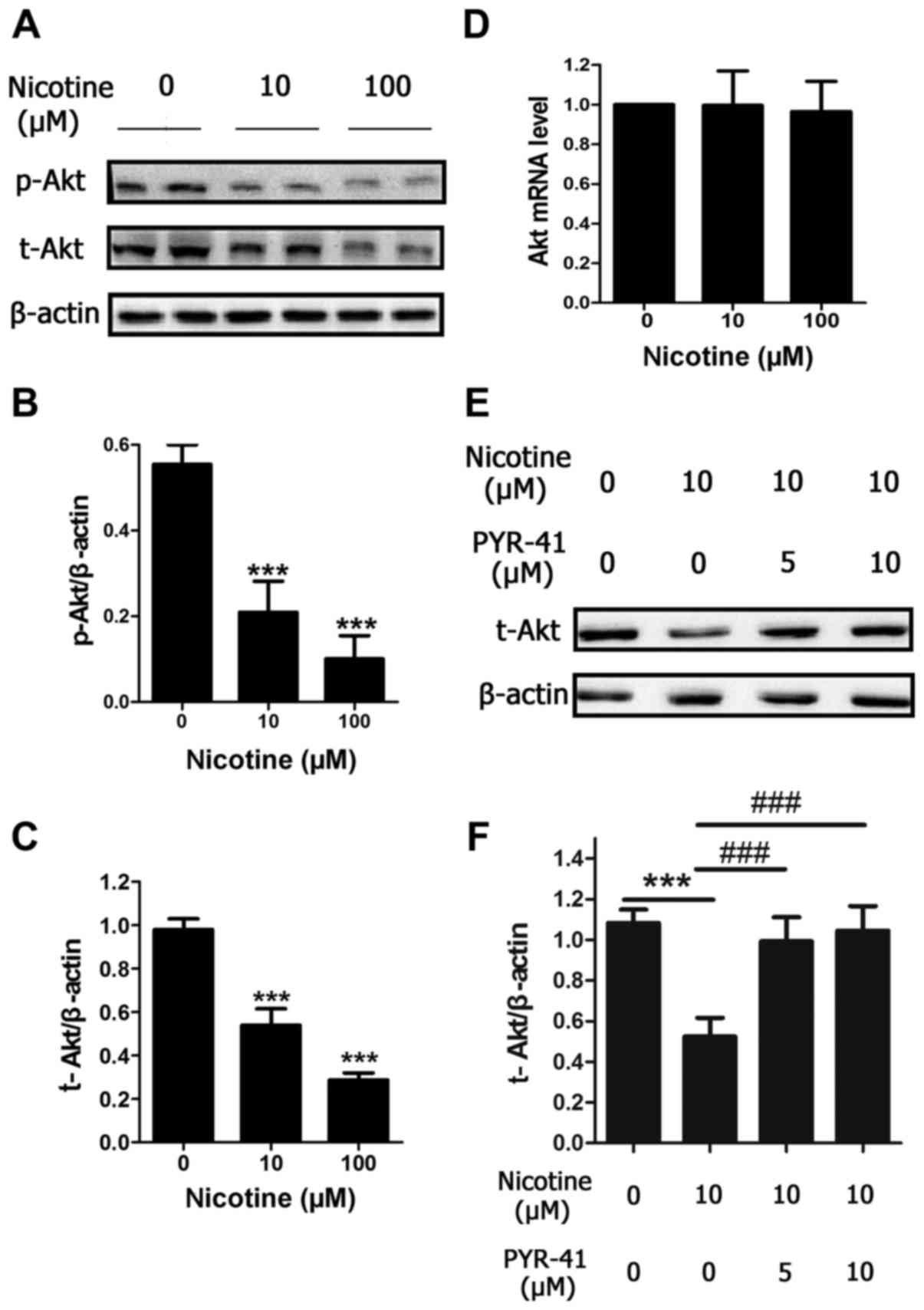
Nicotine induces Akt protein
degradation through the ubiquitin-proteasome system in a
concentration-dependent manner
To investigate the mechanism of nicotine-induced
cells apoptosis, the present study investigated whether nicotine
altered Akt and p-Akt protein levels. Similar to the reduction in
cell viability, protein levels of p-Akt and total-Akt were reduced
by a 48 h nicotine exposure in a concentration-dependent manner
(Fig. 2A-C), compared with control
cells. In order to further investigate the mechanism, the level of
Akt mRNA expression was determined, and it was observed that Akt
mRNA expression was unchanged following 48 h exposure to 10 and 100
µM nicotine (Fig. 2D). To
investigate the involvement of the ubiquitin-proteasome system in
the reduced protein expression of Akt following nicotine treatment,
H9C2 cells were treated with different concentrations of PYR-41 (5
or 10 µM), which is a ubiquitin E1 inhibitor and also a valuable
tool for investigating ubiquitination (17). After a 48 h period of nicotine
exposure at 10 µM, which is the concentration that is closest to
the levels observed in the blood of smokers, the reduction in the
protein expression of Akt induced by 10 µM nicotine was
significantly blocked by PYR-41 (Fig.
2E and F). Taken together, the results indicate that nicotine
may cause Akt protein degradation via the ubiquitin-proteasome
system in H9C2 cells.
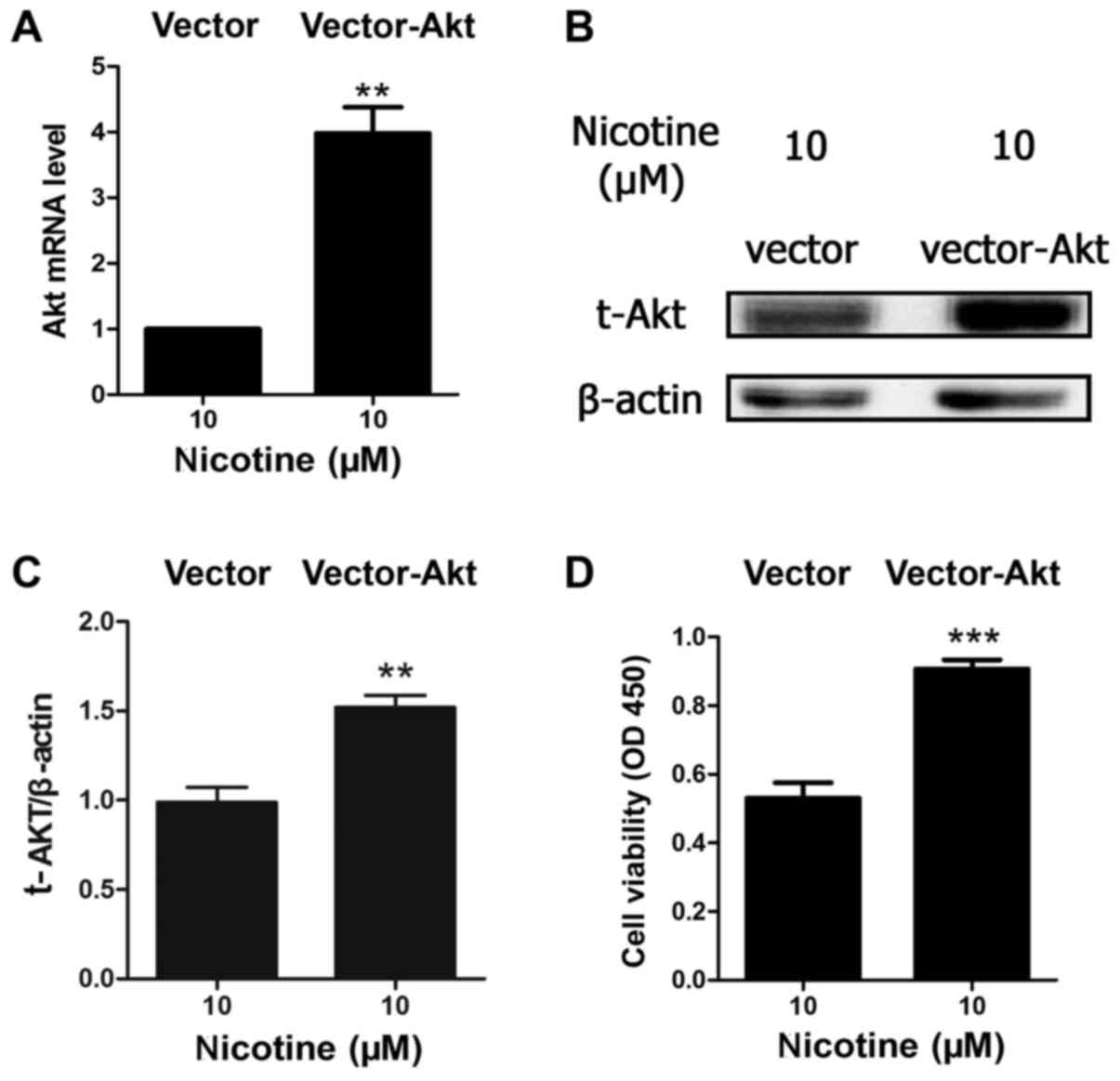
Akt overexpression improves cell
viability when exposed to nicotine
Due to the important role of Akt in cell survival
and apoptosis, and as the results of the present study indicated
that nicotine induced Akt protein degradation in a
concentration-dependent manner, the present study further
investigated the association between apoptosis and Akt. H9C2 cells
were transfected with an Akt1 overexpression plasmid or
vector-only. The results indicated that the level of Akt mRNA and
protein were significantly upregulated in the vector-Akt group
compared with the vector-only group when treated with 10 µM
nicotine (Fig. 3A and B).
Furthermore, the results demonstrated that Akt overexpression
significantly inhibited nicotine-induced reductions in cell
viability (Fig. 3C).
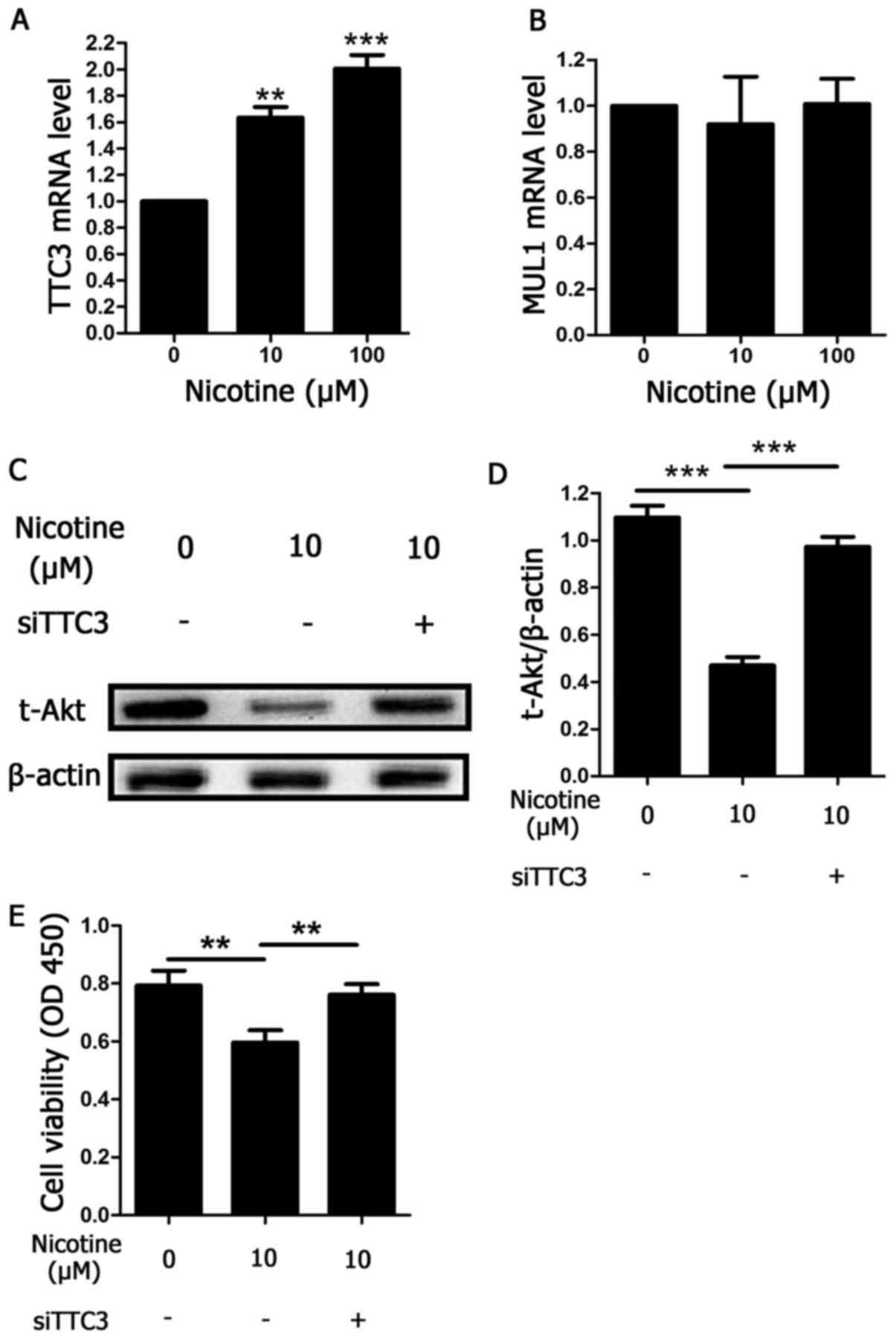
Nicotine induces Akt ubiquitination by
TTC3
The results discussed in Fig. 2, where an inhibitor of ubiquitin E1
(PYR-41) was used, indicated that Akt protein degradation following
exposure to nicotine occurred via ubiquitination and the
ubiquitin-proteasome system. TTC3 and MUL1 are both E3 ligases that
facilitate Akt ubiquitination. Therefore, to determine the type of
E3 ligase against Akt that was involved in nicotine-induced Akt
ubiquitination, the mRNA levels of TTC3 and MUL1 were determined
using RT-qPCR in nicotine-exposed H9C2 cells. Nicotine augmented
TTC3 mRNA expression in a concentration-dependent manner after 48 h
of treatment (Fig. 4A). By
contrast, MUL1 mRNA was unchanged under the same conditions
(Fig. 4B). To further demonstrate
the role of TTC3 in nicotine-induced Akt ubiquitination and
degradation, TTC3 siRNA was used to inhibit TTC3 mRNA expression.
Knockdown of TTC3 expression by TTC3 siRNAs led to a significant
reduction in Akt ubiquitination and degradation induced by
nicotine, compared with nicotine-treated cells transfected with
scrambled siRNA (Fig. 4C and D).
Furthermore, the current study determined whether knockdown of TTC3
expression may affect nicotine-induced reductions in cell
viability. Compared with scrambled siRNA, cell viability was
significantly increased by TTC3 siRNA (Fig. 4E), indicating that apoptosis levels
may have been reduced.
Discussion
The present study demonstrated that nicotine reduced
the viability and induced H9C2 cells apoptosis, Akt protein
expression was downregulated when exposed to nicotine at various
concentrations for 48 h, PYR-41, a ubiquitin E1 inhibitor, restored
the protein expression of Akt, Akt overexpression inhibited H9C2
cell apoptosis induced by nicotine, nicotine upregulated the
expression of TTC3 mRNA, nicotine-induced reductions in the protein
expression of Akt protein level were reversed when TTC3 was
silenced by siRNA and nicotine-induced reductions in cell viability
were inhibited when TTC3 was silenced by siRNA.
It is clear that smoking is associated with numerous
diseases, particularly cardiovascular diseases. Recent study
reported by Baber et al (18) reported that smoking was an
independent predictor of major bleeding following percutaneous
coronary intervention with drug-eluting stents. In addition, Stone
et al (19) discovered that
smoking was associated with hospitalization for heart failure.
Furthermore, Das et al (20), using pigs as experimental animals,
revealed that smoking induced myocardial injury by release of
cardiac troponin-T and -I in the serum, oxidative stress,
inflammation, apoptosis, thrombosis and collagen deposition in the
myocardium. Sumanasekera et al (21) also reported that smoking induced
reduced cardiac stem cell viability, migration reduction and led to
exacerbation of the damage.
Nicotine, which is a key ingredient in smoking
products, may be responsible for various risks associated with
smoking (22). Marrs and Maynard
(23) reported that nicotine is
the classic nAChR agonist and it has also been used as an
insecticide. Li et al (24)
discovered that nicotine induced cardiomyocyte hypertrophy through
the transient receptor potential cation channel subfamily C member
3-mediated Ca2+/nuclear factor of activated T-cells
signaling pathway. Furthermore, Zhou et al (8) reported that nicotine promoted
cardiomyocyte apoptosis via oxidative stress and altered expression
of apoptosis-associated genes. In the present study, the reduction
of cell viability, the increased percentage of TUNEL-positive cells
and the activation of caspase-3 indicated that nicotine also
promoted H9C2 cell apoptosis.
Akt, a serine/threonine kinase, is a crucial
molecule that is involved in various biological actions, including
apoptosis, survival, proliferation and cell migration. Activated
Akt, which is phosphorylated by PI3K, suppresses apoptosis by
inactivating proapoptotic factors, maintaining mitochondrial
integrity and stabilizing anti-apoptotic factors (25). In addition, Akt prevents apoptosis
by phosphorylating apoptosis signal-regulating kinase 1 (26), which causes the activation of c-Jun
N-terminal kinase and p38 mitogen-activated protein kinase
(27). Furthermore, Akt leads to
p53 ubiquitination and degradation by phosphorylating the ubiquitin
ligase mouse double minute homolog 2, which leads to apoptosis
inhibition (28). Stulpinas et
al (29) previously
demonstrated that inhibition of Akt kinase signaling pathways leads
to muscle-derived stem cell death. Regarding the cardiovascular
system, Kerr et al (30)
discovered that endothelial Akt deletion induced retinal vascular
smooth muscle cell loss and basement membrane deterioration, and
resulted in vascular regression and retinal atrophy. In addition,
Fujio et al (31) reported
that Akt protected cardiomyocytes against ischemia-reperfusion
injury. The results of the present study indicated that the protein
level of Akt was downregulated when exposed to nicotine in a
concentration-dependent manner, while the cell viability in H9C2
cells was increased following nicotine treatment when Akt
expression was upregulated.
There are various methods of Akt regulation, and one
mechanism is Akt degradation. Kim et al (32) reported that non-thermal plasma
induced Akt degradation via the ubiquitin-proteasome system,
subsequently leading to head and neck cancer cell death. They also
demonstrated that cigarette smoke induced normal human lung
fibroblast cell apoptosis by Akt protein degradation via the
ubiquitin-proteasome system (13).
The present study demonstrated that PYR-41, a ubiquitin E1
inhibitor, restored the protein expression of Akt following
nicotine treatment. Therefore, it was hypothesized that nicotine
induced Akt protein degradation via the ubiquitin-proteasome
system. The results of the current study also indicated that the
cell viability was restored when Akt was overexpressed.
TTC3 is an Akt-specific E3 ligase that binds to Akt
and facilitates its ubiquitination and degradation within the
nucleus (14,33,34).
The present study demonstrated that nicotine upregulated the level
of TTC3 mRNA, whereas the level of Akt mRNA was not changed.
Furthermore, when TTC3 was silenced by siRNA, the nicotine-induced
reduction in the protein expression of Akt was significantly
reversed, and the cell viability was also improved. Taken together,
these results indicate that nicotine may induce Akt protein
degradation by the ubiquitin-proteasome system via TTC3
upregulation.
In conclusion, the current study demonstrated that
nicotine induced H9C2 cell apoptosis by facilitating Akt protein
degradation, it was also demonstrated that inhibition of the
degradation of Akt protein protected H9C2 cells from reduction of
cell viability induced by nicotine. These results may contribute to
the investigation of the mechanism of diseases caused by smoking
and provide a novel therapeutic target for smoking-associated
diseases. Due to the effect of nicotine on the apoptosis of H9C2
cells, and the role of nicotine on Akt protein degradation, we
hypothesize that nicotine may aggravate apoptosis following
ischemia/reperfusion, which requires further investigation.
Acknowledgements
The present study was supported by the Program for
National Science Fund for Distinguished Young Scholars of China
(grant no. 81225001), the National Key Basic Research Program of
China (973 Program; grant no. 2013CB531204), the Key Science and
Technology Innovation Team in Shaanxi Province (grant no.
2014KCT-19), the Program for Changjiang Scholars and Innovative
Research Team in University (grant no. PCSIRT-14R08), the National
Science Funds of China (grant nos. 81170186, 81470478 and
81400201), the Major Science and Technology Project of China
‘Significant New Drug Development’ (grant no. 2012ZX09J12108-06B)
and the Fourth Military Medical University's Young Talent Project
(the first level).
References
|
1
|
Glickman MS and Schluger N: Adding insult
to injury: Exacer-bating TB risk with smoking. Cell Host Microbe.
19:432–433. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Purdue MP and Silverman DT: Clearing the
air: Summarizing the smoking-related relative risks of bladder and
kidney cancer. Eur Urol. 70:467–468. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sharma R, Harlev A, Agarwal A and Esteves
SC: Cigarette smoking and semen quality: A new meta-analysis
examining the effect of the 2010 World Health Organization
laboratory methods for the examination of human semen. Eur Urol.
70:635–645. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Office of the Surgeon General (US); Office
on Smoking and Health (US), . The Health Consequences of Smoking: A
Report of the Surgeon General. Atlanta (GA): Centers for Disease
Control and Prevention (US); 2004
|
|
5
|
Singh S, Pillai S and Chellappan S:
Nicotinic acetylcholine receptor signaling in tumor growth and
metastasis. J Oncol. 2011:4567432011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Lau JK, Brown KC, Thornhill BA, Crabtree
CM, Dom AM, Witte TR, Hardman WE, McNees CA, Stover CA, Carpenter
AB, et al: Inhibition of cholinergic signaling causes apoptosis in
human bronchioalveolar carcinoma. Cancer Res. 73:1328–1339. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wu YP, Kita K and Suzuki N: Involvement of
human heat shock protein 90 alpha in nicotine-induced apoptosis.
Int J Cancer. 100:37–42. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou X, Sheng Y, Yang R and Kong X:
Nicotine promotes cardiomyocyte apoptosis via oxidative stress and
altered apoptosis-related gene expression. Cardiology. 115:243–250.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Franke TF: PI3K/Akt: Getting it right
matters. Oncogene. 27:6473–6488. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Manning BD and Cantley LC: AKT/PKB
signaling: Navigating downstream. Cell. 129:1261–1274. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
An R, Zhao L, Xi C, Li H, Shen G, Liu H,
Zhang S and Sun L: Melatonin attenuates sepsis-induced cardiac
dysfunction via a PI3K/Akt-dependent mechanism. Basic Res Cardiol.
111:82016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Liao Y and Hung MC: Physiological
regulation of Akt activity and stability. Am J Transl Res. 2:19–42.
2010.PubMed/NCBI
|
|
13
|
Kim SY, Lee JH, Huh JW, Ro JY, Oh YM, Lee
SD, An S and Lee YS: Cigarette smoke induces Akt protein
degradation by the ubiquitin-proteasome system. J Biol Chem.
286:31932–31943. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Suizu F, Hiramuki Y, Okumura F, Matsuda M,
Okumura AJ, Hirata N, Narita M, Kohno T, Yokota J, Bohgaki M, et
al: The E3 ligase TTC3 facilitates ubiquitination and degradation
of phosphorylated Akt. Dev Cell. 17:800–810. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bae S, Kim SY, Jung JH, Yoon Y, Cha HJ,
Lee H, Kim K, Kim J, An IS, Kim J, et al: Akt is negatively
regulated by the MULAN E3 ligase. Cell Res. 22:873–885. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Yang Y, Kitagaki J, Dai RM, Tsai YC,
Lorick KL, Ludwig RL, Pierre SA, Jensen JP, Davydov IV, Oberoi P,
et al: Inhibitors of ubiquitin-activating enzyme (E1), a new class
of potential cancer therapeutics. Cancer Res. 67:9472–9481. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Baber U, Mehran R, Giustino G, Cohen DJ,
Henry TD, Sartori S, Ariti C, Litherland C, Dangas G, Gibson CM, et
al: Coronary thrombosis and major bleeding after PCI with
drug-eluting stents: Risk scores from PARIS. J Am Coll Cardiol.
67:2224–2234. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Stone GW, Selker HP, Thiele H, Patel MR,
Udelson JE, Ohman EM, Maehara A, Eitel I, Granger CB, Jenkins PL,
et al: Relationship between infarct size and outcomes following
primary PCI: Patient-level analysis from 10 randomized trials. J Am
Coll Cardiol. 67:1674–1683. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Das A, Dey N, Ghosh A, Das S,
Chattopadhyay DJ and Chatterjee IB: Molecular and cellular
mechanisms of cigarette smoke-induced myocardial injury: Prevention
by vitamin C. PLoS One. 7:e441512012. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sumanasekera WK, Tran DM, Sumanasekera TU,
Le N, Dao HT and Rokosh GD: Cigarette smoke adversely affects
functions and cell membrane integrity in c-kit+ cardiac
stem cells. Cell Biol Toxicol. 30:113–125. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Lee J and Cooke JP: Nicotine and
pathological angiogenesis. Life Sci. 91:1058–1064. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Marrs TC and Maynard RL: Neurotranmission
systems as targets for toxicants: A review. Cell Biol Toxicol.
29:381–396. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li N, Si B, Ju JF, Zhu M, You F, Wang D,
Ren J, Ning YS, Zhang FQ, Dong K, et al: Nicotine induces
cardiomyocyte hypertrophy through TRPC3-mediated
Ca2+/NFAT signalling pathway. Can J Cardiol.
32:1260.e1–1260.e10. 2016. View Article : Google Scholar
|
|
25
|
Zhou BP, Liao Y, Xia W, Spohn B, Lee MH
and Hung MC: Cytoplasmic localization of p21Cip1/WAF1 by
Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat
Cell Biol. 3:245–252. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Kim AH, Khursigara G, Sun X, Franke TF and
Chao MV: Akt phosphorylates and negatively regulates apoptosis
signal-regulating kinase 1. Mol Cell Biol. 21:893–901. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tobiume K, Matsuzawa A, Takahashi T,
Nishitoh H, Morita K, Takeda K, Minowa O, Miyazono K, Noda T and
Ichijo H: ASK1 is required for sustained activations of JNK/p38 MAP
kinases and apoptosis. EMBO Rep. 2:222–228. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Zhou BP, Liao Y, Xia W, Zou Y, Spohn B and
Hung MC: HER-2/neu induces p53 ubiquitination via Akt-mediated MDM2
phosphorylation. Nat Cell Biol. 3:973–982. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Stulpinas A, Imbrasaitė A and Kalvelytė
AV: Daunorubicin induces cell death via activation of apoptotic
signalling pathway and inactivation of survival pathway in
muscle-derived stem cells. Cell Biol Toxicol. 28:103–114. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kerr BA, West XZ, Kim YW, Zhao Y,
Tischenko M, Cull RM, Phares TW, Peng XD, Bernier-Latmani J,
Petrova TV, et al: Stability and function of adult vasculature is
sustained by Akt/Jagged1 signalling axis in endothelium. Nat
Commun. 7:109602016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Fujio Y, Nguyen T, Wencker D, Kitsis RN
and Walsh K: Akt promotes survival of cardiomyocytes in vitro and
protects against ischemia-reperfusion injury in mouse heart.
Circulation. 101:660–667. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim SY, Kim HJ, Kang SU, Kim YE, Park JK,
Shin YS, Kim YS, Lee K and Kim CH: Non-thermal plasma induces AKT
degradation through turn-on the MUL1 E3 ligase in head and neck
cancer. Oncotarget. 6:33382–33396. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Dey-Guha I, Alves CP, Yeh AC, Salony, Sole
X, Darp R and Ramaswamy S: A mechanism for asymmetric cell division
resulting in proliferative asynchronicity. Mol Cancer Res.
13:223–230. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Toker A: TTC3 ubiquitination terminates
Akt-ivation. Dev Cell. 17:752–754. 2009. View Article : Google Scholar : PubMed/NCBI
|