Introduction
The distal region of chromosome 8p is prone to
various genomic rearrangements, mainly due to the presence of low
copy repeats (LCRs). The LCRs flank an approximately 5 Mb region on
8p23.1 and contain two olfactory receptors gene clusters or
defensin repeats, repeat-distal (REPD) in the distal 8p23.1 region
and repeat-proximal (REPP) in the proximal 8p23.1 region (1,2).
These LCRs are believed to mediate a U-type exchange, commonly
resulting in a recurrent interstitial 8p23 deletion, a terminal
8p23 deletion and a variably sized interstitial inverted
duplication in 8p with an 8p terminal deletion (1,3–6). The
distal deletions of 8p23, including either the terminal or the
sub-terminal 8p region, appear as an isolated finding or as a part
of a complex chromosomal rearrangement. Both interstitial or
terminal 8p23 deletions are relatively frequent among the
structural variations associated with 8p, and patients with a
deletion in this region are generally characterized by
developmental delay (DD)/intellectual disability (ID), mildly
dysmorphic features, microcephaly, congenital heart defects (CHD),
congenital diaphragmatic hernia (CDH), seizure, and
neuropsychiatric disorders (4,5,7,8). The
deleted region often spans a large genomic segment containing
numerous genes. Therefore, the genotype-phenotype correlations
associated with the deletion are largely unknown. However, an
increasing number of studies have contributed to narrowing the
critical region (CR) to search for the candidate genes associated
with the clinical features. A deletion at 8p23.1 was proposed as a
CR for CHD, CDH and other common clinical features (4,5,8–10).
Although genes in the deleted region may interact or participate in
the same pathways, specific candidate genes are expected to play
important roles in certain phenotypes. Notably, haploinsufficiency
of GATA4, NEIL2 and/or SOX7 located at 8p23.1
were shown to be associated with CHD and CDH in several studies
(8,11–13).
Another gene in the region, MCPH1, has also been implicated
as a candidate gene for primary microcephaly (14–16).
In addition to the common 8p23.1 deletion, a
terminal deletion more distal to the REPD deletion involving 8p23.2
to 8pter has increasingly been reported as a CR associated with
DD/ID, seizure and neurobehavioral disorders, such as autism
spectrum disorder (ASD) and attention-deficit/hyperactivity
disorders (ADHD) (7,17–19).
Isolated 8p23.2-pter deletions are a rare chromosomal aberration.
The recurrence of an isolated distal deletion involving the region
from 8p23.2 to 8pter in patients with similar phenotypes is
consistent with a distinctive microdeletion syndrome. To the best
of our knowledge, cytogenetic and molecular characteristics have
only been reported for four patients (7,17,19).
The locations and sizes of the deletions detected in this region
vary, similar to most of the sub-telomeric microdeletion syndromes.
The majority of the examined patients with an 8p23 deletion were
characterized using traditional techniques, including G-banded
karyotyping, fluorescence in situ hybridization (FISH),
microsatellite marker genotyping and multiplex ligation-dependent
probe amplification (MLPA), with limited resolution and low
efficiency. Therefore, an accurate definition of the locations,
sizes and genes contained in the deletions has been difficult to
obtain (5,9,20–23).
Nevertheless, few microarray-based molecular studies have been
performed to investigate these deletions. Current high-resolution
chromosomal microarray analyses (CMA), including array comparative
genome hybridization and single-nucleotide polymorphism (SNP)
arrays, can now accurately determine the locations, sizes and genes
contained in 8p23 deletions. This technique allows researchers to
obtain more precise genotype-phenotype correlations and aids in
identifying CRs and candidate genes. Here, we present a new case
study of a patient with an isolated 8p23.2-pter deletion that was
determined using a SNP array and compare the clinical features with
patients with isolated deletions involving the region from 8p23.2
to 8pter that were previously identified using CMA or recorded in
the Database of Chromosomal Imbalance and Phenotype in Humans using
Ensembl Resources (DECIPHER; https://decipher.sanger.ac.uk/). The accumulation of
patients with isolated 8p23.2-pter deletions at various locations
and of different sizes will help in further defining the CRs and
candidate genes for certain phenotypes.
Materials and methods
Case presentation
The patient was a 5-year-old Chinese male, who was
the first child of healthy, unrelated parents that had no personal
or family history of DD/ID, congenital malformations or psychiatric
disorders. The patient was born at term via a natural delivery. His
weight, length and head circumference at birth were 2.5 kg (<3rd
percentile), 49 cm (<25th percentile) and 29 cm [≤3 standard
deviations (SD)], respectively. The child progressed
physiologically during the neonatal period. He sat alone at 7
months, crawled at 10 months, walked at 13 months and spoke his
first words at 16 months. The parents noted that the child appeared
to experience a growth delay, psychomotor delay and poor
language/motor skills during preschool. He understood simple and
blended verbal orders, but he had more difficulty in understanding
complex verbal orders. He would sometimes talk to himself. He had
deficiencies in his interactions with others, would not hold a
direct gaze and often performed certain stereotyped or repetitive
behaviours. He also had poor balance and coordination. At the age
of 3 years and 11 months old, the child was diagnosed with ASD by
qualified child psychiatrists based on both the childhood autism
rating scale (CARS) and the autistic behaviour checklist (ABC).
Additionally, attention deficits, impulsivity, unstable emotion and
hyperactivity were also observed. The patient's intelligence
quotient (IQ) was measured with the Wechsler Young Children Scale
of Intelligence (C-WYCSI) at the age of 4 years and 4 months old.
Psychomotor development was slightly delayed (IQ 50, verbal IQ 49,
performance IQ 60). At the age of 5 years and 5 months, he weighed
15 kg (<3rd percentile) and had a height of 104 cm (<3rd
percentile). He showed mild microcephaly, with a head circumference
of 47.6 cm (≤2 SD). Mildly dysmorphic craniofacial abnormalities
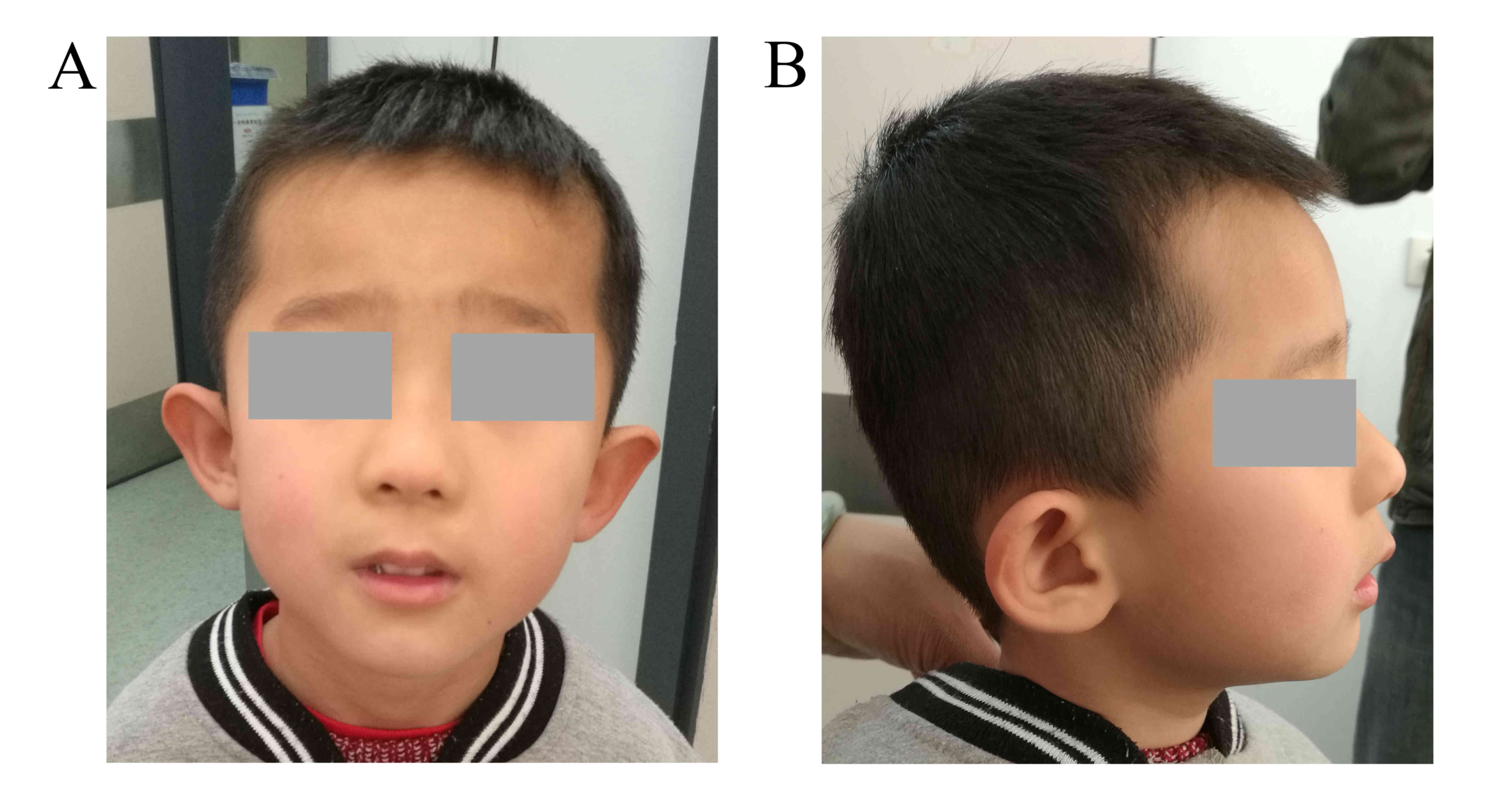
were noted, including low-set ears with a bilateral prominence of
the antitragus, epicanthal folds and a long philtrum (Fig. 1). Electroencephalograms and
cerebral magnetic resonance imaging (MRI) did not reveal any
anomalies. The parents provided informed consent for clinical
information and photographs of the child to be published.
Cytogenetic analysis
Using standard procedures, routine G-banded
karyotyping was performed on cultured peripheral blood lymphocytes
from the patient and his parents. A chromosome analysis was
performed at the 400-band level in both the patient and his parents
according to the International System for Human Cytogenomic
Nomenclature (ISCN) 2013 (50 metaphases each).
High-resolution SNP array
analysis
Genomic DNA was isolated from peripheral blood
lymphocytes with the QIAamp DNA Blood Mini kit (Qiagen, Inc.,
Valencia, CA, USA) according to the manufacturer's instructions.
The concentration of the extracted genomic DNA was determined using
a NanoDrop ND-1000 spectrophotometer (NanoDrop Technologies,
Berlin, Germany). The DNA sample (250 ng) was hybridized to
CytoScan HD arrays on an Affymetrix SNP array platform (Affymetrix;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) according to the
manufacturer's protocol. The CytoScan HD array contains more than
2.6 million markers for the copy number analysis. Of these markers,
1,950,000 are unique, non-polymorphic oligonucleotide probes, and
750,000 are SNP probes used for genotyping. The average marker
spacing is one probe per 1.1 kb, with a mean spacing of one probe
per 1.7 kb on non-gene backbones and one probe per 880 bp in
intragenic regions. Using ChAS 3.0 software (Affymetrix; Thermo
Fisher Scientific, Inc.), aberrations were filtered up to a minimum
size of 100 kb, with at least 50 probe calls for deletions and
duplications.
FISH analysis
A metaphase FISH analysis was performed on the
patient and his parents using standard methods. Fluorescent probes
specific for the sub-telomere region of chromosome 8p and the
centromere of chromosome 8 (Cytocell, Inc., Cambridge, UK) were
used for the FISH analysis and were labelled with SpectrumOrange
and SpectrumGreen, respectively. FISH signals were observed using
an Olympus BX-51 microscope (Olympus Corp., Tokyo, Japan).
Results
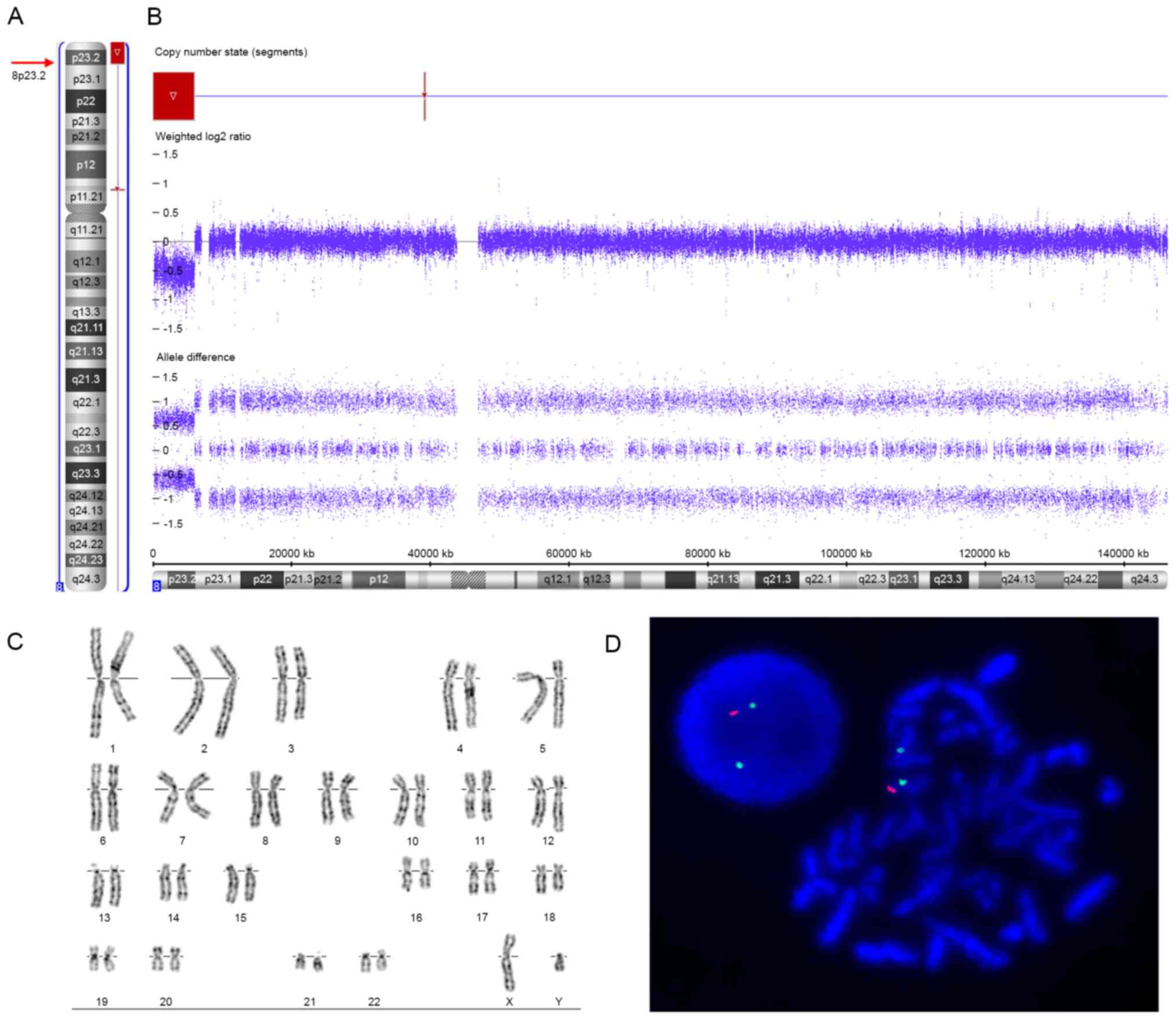
Routine G-banded karyotyping showed normal
karyotypes in the patient (46, XY, Fig. 2C) and his parents (46, XX and 46,
XY, respectively). The SNP array revealed an 8p23.2-pter terminal
deletion in the patient, represented as arr[hg19] 8p23.3p23.2(158,
048 - 6, 004, 205)x1 (Fig. 2A and
B). The deleted region was ~6.0 Mb in size and contained 23
RefSeq genes, including 11 protein-coding genes and 12 non-coding
genes; 6 of the protein-coding genes were annotated in OMIM
(FBXO25, DLGAP2, CLN8, ARHGEF10,
MYOM2, and CSMD1) (Fig.
3A). Other copy number variants related to pathogenicity or of
unknown significance were not identified. Furthermore, the
two-colour FISH analysis of the patient's metaphase chromosomes
using specific probes identified a terminal deletion of chromosome
8p (Fig. 2D), confirming the 8p
terminal deletion identified by SNP array. However, FISH studies of
the parents were normal, excluding cryptic rearrangements of the 8p
terminal region in the parents and indicating the de novo
deletion of 8p23.2-pter in the patient.
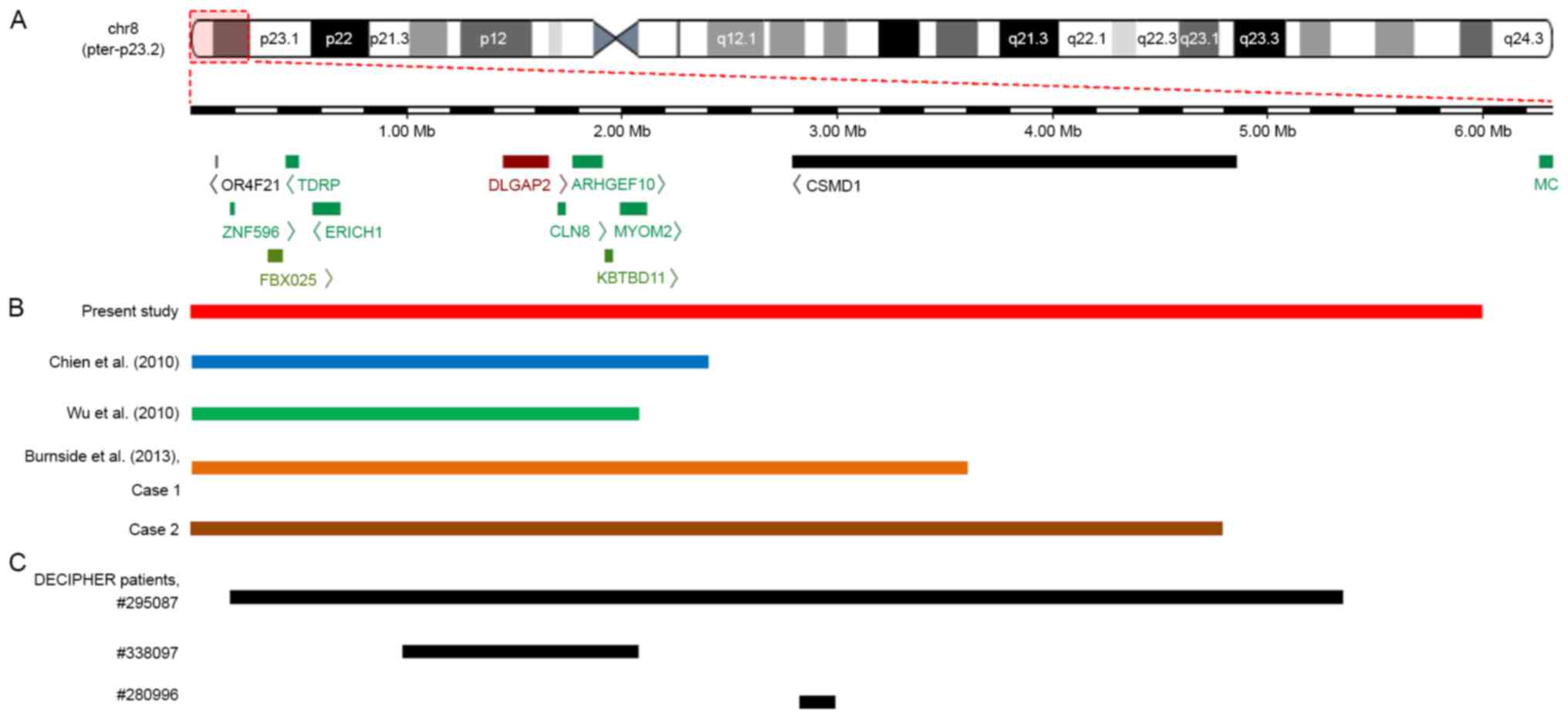 | Figure 3.An overlapping map of 8p23.2-pter
deletions in the CMA-identified patients reported in the present
study, previous studies and the DECIPHER database. (A) Schematic
presentation of the 8p23.2-pter region and the genes located in
this region, as determined by the University of California Santa
Cruz Genome Browser. (B) An overview of CMA-identified patients
with a deletion involving 8p23.2 to 8pter who were described in
various reports. Among five patients, the deleted segment in the
patient reported by Wu et al (19) was the smallest. Both deletions
reported by Chien et al (17) and the deletion reported by Wu et
al (19) encompassed
DLGAP2, CLN8 and ARHGEF10 genes, whereas both
deletions reported by Burnside et al (7) and the current study not only covered
the DLGAP2, CLN8 and ARHGEF10 genes but also
the CSMD1 gene. (C) An overview of patients from the
DECIPHER database with deletions involving the 8p23.2 to the 8pter
region that were regarded as definitely or likely pathogenic. A
total of three patients presented with ID, and patient no. 338097
also exhibited behavioural problems (not specifically described).
The deleted region in patient no. 338097 covered the DLGAP2,
CLN8 and ARHGEF10 genes, whereas the deletion in
patient no. 280996 only partially overlapped with the CSMD1
gene. CMA, chromosomal microarray analysis. |
To date, only four patients with isolated deletions
involving the region from 8p23.2 to 8pter that were identified
using CMA have been reported in previous studies; all of them
shared common clinical features. In addition, three patients were
included in DECIPHER harbored deletions from 8p23.2 to 8pter that
were recognized as definitely or likely pathogenic (Table I). The locations, sizes and genes
contained in the deleted region in these patients were compared
based on the genetic map (Fig. 3).
In addition, we also performed a phenotypic comparison between the
8p23.2-pter deletion and 8p23.1 deletion (Table II).
 | Table I.Summary of the clinical features of
patients characterized by a CMA in the present study, previous
studies and the DECIPHER database. |
Table I.
Summary of the clinical features of
patients characterized by a CMA in the present study, previous
studies and the DECIPHER database.
| Case study | Sex | Age, years | Deleted region | Genomic breakpoint
(hg19), bp | Size, Mb | Intellectual
disability | Developmental
delay | Language delay | Neurobehavioral
disorders | Craniofacial
abnormalities | Other
abnormalities | Other | Refs. |
|---|
| Chien et
al | M | 12 | 8p23.2-pter | ~2,410,000 | 2.4 | + | + | + | ASD and ADHD | − | Seizure | − | (17) |
| Wu et
al | F | 1 | 8p23.3-pter | ~2,067,000 | 2.1 | + | + | ND | ND | Microcephaly,
hypertelorism, long philtrum, malformed ears | − | − | (19) |
| Burnside et
al |
|
|
|
|
|
|
|
|
|
|
|
|
|
| Patient
1 | F | 2 | 8p23.2-pter | ~3,623,904 | 3.6 | − | + | + | − | Microcephaly,
brachycephaly and mild maxillary, flattening medial epicanthal
folds with upslanting palpebral a fissures, preauricular pit and
bilateral prominence of the antitragus |
Balance/coordination problems | − | (7) |
| Patient
2 | M | 4 | 8p23.2-pter |
~4,832,934 | 4.8 | − | + | − | − | Microcephaly, a
palpable metopic ridge, upslanting palpebral fissures, nystagmus,
aniridia, low-set and posteriorly rotated ears with overfolded
helices, small upturned nose, and downturned corners of the
mouth | Coarctation of the
aorta and nail hypoplasia | − |
| Present
study | M | 5 | 8p23.2-pter |
~6,004,205 | 6.0 | + | + | + | ASD and ADHD | Microcephaly,
low-set ears with bilateral prominence of the antitragus,
epicanthal folds, and a long philtrum |
Balance/coordination problems | − |
| DECIPHER
patients |
|
|
|
|
|
|
|
|
|
|
|
| No.
295087 | F | 10 | 8p23.2-pter |
191,560–5,353,580 | 5.2 | + | unk | unk | unk | unk | − | Definitely
pathogenica |
| No.
338097 | M | unk | 8p23.3 |
974,820–2,067,679 | 1.1 | + | unk | unk | Behavioural
abnormalities | unk | − | Likely
pathogenica |
| No.
280996 | M | 6 | 8p23.2 |
2,832,422–296,643 | 0.1 | + | unk | unk | unk | unk | unk | Likely
pathogenica |
| Table II.Comparison of phenotypes in patients
with the 8p23.2-pter deletion and 8p23.1 deletion. |
Table II.
Comparison of phenotypes in patients
with the 8p23.2-pter deletion and 8p23.1 deletion.
| Clinical
features | 8p23.2-pter
deletion | Current
patient | 8p23.1 deletion
(terminal or interstitial) |
|---|
| DD/ID | + | + | + |
| Speech delay | + | + | + |
| Growth
retardation | + | + | + |
| Neurobehavioral
disorders | + | + | + |
|
ASD | + | + | + |
|
ADHD | + | + | + |
|
Aggressiveness | + | − | + |
|
Impulsiveness | + | − | + |
| Craniofacial
features | + | + | + |
|
Microcephaly | + | + | + |
|
Brachycephaly | + | − | − |
|
Palpable metopic ridge | + | − | − |
| Low-set
ears | + | + | + |
|
Malformed ears | + | + | + |
|
Upslanting palpebral
fissures | + | − | + |
|
Epicanthal folds | + | + | + |
| Eye
abnormalities | + | − | + |
| Bulbous
nasal tip | − | − | + |
| Long
philtrum | + | + | − |
| Widely
spaced nipples | − | − | + |
| Thin
upper lip | − | − | + |
|
Micrognathia | − | − | + |
| Congenital heart
disease | − | − | + |
| Congenital
diaphragmatic hernia | − | − | + |
| Hypospadias | − | − | + |
| Cryptorchism | − | − | + |
| Short fingers | − | − | + |
| Seizure | + | − | + |
Discussion
Deletions of varying sizes at the distal region of
the 8p chromosome are frequently reported, particularly the 8p23
deletion, but most have not been investigated using molecular
cytogenetics. With the increasing use of high-resolution microarray
technologies, a better understanding of the genotype-phenotype
correlations of the distal 8p deletion have been obtained with the
identification of CRs or candidate genes. Among distal 8p
deletions, the 8p23.1 deletion is frequently observed, mainly due
to the presence of LCRs. The recurrence of CHD and CDH in patients
with various 8p23.1 deletions has led to the identification of a CR
at 8p23.1 for CHD and CDH (4,5,8–10).
Furthermore, the GATA4, NEIL2 and SOX7 genes in the
deleted region have been recognized as candidate genes for CHD and
CDH (8,11–13).
In addition, a more distal deletion to 8p23.1 is the 8p23.2-pter
deletion, a relatively rare aberration. Patients with an
8p23.2-pter deletion usually do not present CHD, CDH and urogenital
abnormalities, but instead appear to have milder phenotypes than
patients with an 8p23.1 deletion (Table II). The diversity in phenotypes
suggests that the 8p23.2-pter deletion does not encompass the CR or
candidate genes (such as the GATA4, NEIL2 and SOX7
genes) on 8p23.1 associated with these clinical features. Moreover,
based on these data, the 8p23.1 region plays an important role in
the manifestation of these clinical features.
However, common clinical features, including DD/ID,
mildly dysmorphic features, microcephaly and neurobehavioral
disorders, are usually shared by patients with either an
8p23.2-pter deletion or an 8p23.1 deletion (not only individuals
with a large terminal 8p23.1 deletion but also in individuals with
small interstitial 8p23.1 deletions) (Table II). Either an isolated 8p23.1
deletion or an isolated 8p23.2-pter deletion seems to contribute to
these common clinical features, suggesting that the isolated
8p23.2-pter region may harbour other CRs and candidate genes
associated with these features. The present study compared the
phenotypic features between the present patient and four previously
reported patients with isolated terminal deletions involving the
region from 8p23.2 to 8pter that were characterized by CMA
(7,17,19).
Of these five patients, the most common phenotypes included growth
retardation (5/5), microcephaly (4/5) and mildly dysmorphic
features (4/5), followed by ID (3/5), language delay (3/5) and
behavioural problem (3/5, including ASD and ADHD). In the DECIPHER
database, three patients with isolated deletions involving the
8p23.2 to 8pter region were classified as definitely or likely
pathogenic and all of them presented with ID. Taken together,
although the phenotypes listed in DECIPHER might be incomplete and
limited, ID was present in 6 out of the 8 patients with a deletion
involving the region from 8p23.2 to 8pter reported in the
literature and DECIPHER. A terminal 2.1 Mb deletion of 8p23.3
reported by Wu et al (19)
was the smallest segment observed in the five patients from the
present study and the literature. This deletion might be proposed
as a CR for DD/ID, dysmorphic features and microcephaly (Fig. 2). The CR contains 5 OMIM genes,
including FBXO25, DLGAP2, CLN8,
ARHGEF10 and MYOM2. Most of these genes are known to
be expressed in the brain.
The product encoded by FBXO25 is a member of
the F-box protein family and is involved in
phosphorylation-dependent ubiquitination. Fbxo25 may act as a
ubiquitin E3 ligase to degrade cardiac transcription factors and
control the transcriptional activity of the cardiac transcription
factors Tbx5 and Nkx2-5, which play key roles in human cardiac
development (24). The protein
encoded by DLGAP2 is a membrane-associated protein that is
biallelically expressed in the brain and plays roles in synapse
organization and signalling in neuronal cells. It has been
implicated as a candidate gene in neurobehavioral disorders, such
as ASD and schizophrenia (25). Li
et al (26) identified 5
rare, isolated variants and a DLGAP2 haplotype (CCACCAACT)
in a cohort of patients with schizophrenia and showed that
increased expression of the DLGAP2 gene is associated with
schizophrenia. As shown in a study by Chien et al (17), an individual with DD/ID, ASD and
seizures carried an 8p23.2-pter deletion. The DLGAP2 gene
located in this region was suggested to be a candidate gene for
ASD. Subsequently, the authors performed deep exon resequencing of
DLGAP2 and detected two SNPs and some rare missense
DLGAP2 mutations that might contribute to ASD (27). In a large autism case-control
study, a de novo 817 kb duplication that partially
overlapped with DLGAP2 was detected in a child with
non-syndromic autism, and the authors also proposed DLGAP2
as a candidate gene involved in the pathogenesis of ASD (28). Notably, two SNPs in DLGAP2
(rs6558484 and rs7014992) were also revealed to be associated with
alterations in brain volume (white matter volume of the prefrontal
cortex) (29). CLN8 is
recognized to have roles in lipid synthesis, transport, and
sensing, thus contributing to cell proliferation during neuronal
differentiation and defence against cell death stimuli (30). Mutations in this gene alter the
levels of sphingolipids and phospholipids in the brain (30,31).
A homozygous CLN8 mutation is one of the causes of autosomal
recessive neuronal ceroid lipofuscinoses, which are mainly
associated with progressive seizures and DD/ID (30). Allen et al (32) reported a patient with seizures,
microcephaly, DD and behavioural problems, which were thought to
result from the co-existence of a heterozygous CLN8 mutation
and a de novo 8p23.3 terminal deletion. Notably, the 8p23.3
deletion was only identified by a FISH analysis using a probe
(D8S504) located on the telomeric region of the chromosome
(8:1017556-1017754) that did not cover the CLN8 gene.
Therefore, the authors did not clearly determine whether this
patient truly had a CLN8 gene deletion. The actual size and
genes contained in the 8p23.3 deletion should be further
investigated to obtain a better understanding of the
genotype-phenotype correlation in this patient. ARHGEF10
encodes a Rho guanine nucleotide exchange factor and may play a
role in neural morphogenesis (33). Mutations in the ARHGEF10
gene may cause demyelination and re-myelination of the peripheral
nervous system and are associated with a slower nerve conduction
velocity through an autosomal dominant pattern (33). Some SNPs in ARHGEF10 might
confer a risk of developing schizophrenia (34). Notably, in DECIPHER, the
8p23.3-p23.2 and 8p23.3 deletions presented by two patients (nos.
295087 and 338097, respectively) with ID covered DLGAP2,
CLN8 and ARHGEF10. Among all the patients carrying
these genes (literature and DECIPHER), the deleted segment in
patient no. 338097 was the smallest. Further studies of patients
with similar phenotypes are required to determine whether the
deleted region in patient no. 338097 is a potential CR for ID (only
this phenotype was presented in DECIPHER). Although limited
clinical features are listed in DECIPHER, the two patients also
provided evidence for the potential role for either the deleted
region or these genes in the pathogenesis of ID.
Furthermore, among the five patients with an
8p23.2-pter deletion from the present study and the literature,
only the deletions reported by both Chien et al (17) and Wu et al (19) did not contain the CSMD1
gene. CSMD1 encodes a large multi-domain protein that mainly
consists of CUB and Sushi domains. This protein may function as an
important regulator of complement activation and inflammation in
the developing central nervous system and is involved in brain
mechanisms associated with memory and learning (35,36).
Based on the evidence from previous studies, a SNP (rs10503253)
located within the CSMD1 gene may be associated with the
risk of multiple neurodevelopmental phenotypes, such as
schizophrenia, seizure, speech delay, cognitive impairment and
learning difficulties (37–39).
Regarding cognitive ability, the ‘A’ allele of rs10503253 is
considered to have deleterious impacts on verbal or performance
intelligence, verbal episodic memory, and spatial working memory
(35). In DECIPHER, patient no.
280996 had a maternally inherited 134 kb deletion that partially
overlapped with CSMD1, and the deletion was recognized as a
likely pathogenic variant. Both the patient and his mother showed
moderate ID. Thus, CSMD1 might be a potential candidate gene
for ID.
Microcephaly is caused by a congenital insufficiency
during foetal brain (mainly cerebral cortex) development and is
frequently observed in patients with either an 8p23.2-pter deletion
or an 8p23.1 deletion (7,16,19).
A candidate gene for microcephaly, the MCPH1 gene (16,40),
is located on 8p23.1, but no candidate genes have been proposed for
the region from 8p23.2 to 8pter. However, the DLGAP2,
CLN8, ARHGEF10 and CSMD1 genes mentioned above are
all expressed in the brain and implicated as being associated with
multiple neurodevelopmental phenotypes. These genes should not be
excluded as possibly having a role in microcephaly, particularly
DLGAP2, which has been shown to be associated with
alterations in brain volume alterations (29).
However, haploinsufficiency of each gene alone has
not proven sufficient to explain the clinical phenotypes. These
genes might function together, forming an interaction network in
some pathway or may be modified by other genetic or environmental
factors (27). Thus, the deletion
of all these genes might conceivably have adverse effects on
neurodevelopment. Compound inheritance of a rare null mutation and
a common risk haplotype should also be considered in studies aiming
to understand genotype-phenotype correlations. According to the
study by Wu et al (41),
compound inheritance of a heterozygous deletion and a common risk
haplotype of the TBX6 allele are responsible for congenital
scoliosis. Therefore, some genes in the deleted region, including
DLGAP2, CLN8, ARHGEF10 and CSMD1, might
contribute to DD/ID, microcephaly and neurobehavioral disorders,
although the pathogenic mechanisms remain unclear.
In summary, the current study reported a new case
study of a patient with a rarely isolated 8p23.2-pter deletion, and
performed a review of patients with deletions in the region from
8p23.2 to 8pter that have been reported in the literature and the
DECIPHER database. Similar clinical characteristics were observed
among patients with the deletions, which were consistent with a
distinctive microdeletion syndrome. Although a refined CR for
DD/ID, microcephaly and neurobehavioral disorders was not
conclusively determined due to the currently limited number of
patients, the deleted region reported by Wu et al (19) as being responsible for these
features is regarded as the smallest deletion among the five
deletions reviewed in the present study. Furthermore, after a
detailed review of the potential correlations between the genes
located in 8p23.2 to 8pter region and their clinical significance,
we hypothesized that DLGAP2, CLN8, ARHGEF10 and
CSMD1 might be plausible candidate genes for DD/ID,
microcephaly and neurobehavioral disorders. However, further
high-resolution studies of patients with small isolated,
overlapping and interstitial deletions involving the region from
8p23.2 to 8pter may allow us to obtain genotype-phenotype
correlations for specific genes crucial to 8p23.2-pter.
Acknowledgements
No funding was applicable to this study. We are
grateful to the patient and his parents for participating in the
study.
References
|
1
|
Hollox EJ, Barber JC, Brookes AJ and
Armour JA: Defensins and the dynamic genome: What we can learn from
structural variation at human chromosome band 8p23.1. Genome Res.
18:1686–1697. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Yu S, Fiedler S, Stegner A and Graf WD:
Genomic profile of copy number variants on the short arm of human
chromosome 8. Eur J Hum Genet. 18:1114–1120. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rowe LR, Lee JY, Rector L, Kaminsky EB,
Brothman AR, Martin CL and South ST: U-type exchange is the most
frequent mechanism for inverted duplication with terminal deletion
rearrangements. J Med Genet. 46:694–702. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Páez MT, Yamamoto T, Hayashi K, Yasuda T,
Harada N, Matsumoto N, Kurosawa K, Furutani Y, Asakawa S, Shimizu N
and Matsuoka R: Two patients with atypical interstitial deletions
of 8p23.1: Mapping of phenotypical traits. Am J Med Genet A.
146A:1–1165. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ballarati L, Cereda A, Caselli R,
Selicorni A, Recalcati MP, Maitz S, Finelli P, Larizza L and
Giardino D: Genotype-phenotype correlations in a new case of 8p23.1
deletion and review of the literature. Eur J Med Genet. 54:55–59.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
García-Santiago FA, Martínez-Glez V,
Santos F, García-Minañr S, Mansilla E, Meneses AG, Rosell J,
Granero ÁP, Vallespín E, Fernández L, et al: Analysis of
invdupdel(8p) rearrangement: Clinical, cytogenetic and molecular
characterization. Am J Med Genet A. 167A:1–1025. 2015.PubMed/NCBI
|
|
7
|
Burnside RD, Pappas JG, Sacharow S,
Applegate C, Hamosh A, Gadi IK, Jaswaney V, Keitges E, Phillips KK,
Potluri VR, et al: Three cases of isolated terminal deletion of
chromosome 8p without heart defects presenting with a mild
phenotype. Am J Med Genet A. 161A:1–828. 2013.PubMed/NCBI
|
|
8
|
Wat MJ, Shchelochkov OA, Holder AM, Breman
AM, Dagli A, Bacino C, Scaglia F, Zori RT, Cheung SW, Scott DA and
Kang SH: Chromosome 8p23.1 deletions as a cause of complex
congenital heart defects and diaphragmatic hernia. Am J Med Genet
A. 149A:1–1677. 2009. View Article : Google Scholar
|
|
9
|
Devriendt K, Matthijs G, Van Dael R,
Gewillig M, Eyskens B, Hjalgrim H, Dolmer B, McGaughran J,
Bröndum-Nielsen K, Marynen P, et al: Delineation of the critical
deletion region for congenital heart defects, on chromosome 8p23.1.
Am J Hum Genet. 64:1119–1126. 1999. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Molck MC, Monteiro FP, Simioni M and
Gil-da-Silva-Lopes VL: 8p23.1 interstitial deletion in a patient
with congenital cardiopathy, neurobehavioral disorders and minor
signs suggesting 22q11.2 deletion syndrome. J Dev Behav Pediatr.
36:544–548. 2015.PubMed/NCBI
|
|
11
|
Longoni M, Lage K, Russell MK, Loscertales
M, Abdul-Rahman OA, Baynam G, Bleyl SB, Brady PD, Breckpot J, Chen
CP, et al: Congenital diaphragmatic hernia interval on chromosome
8p23.1 characterized by genetics and protein interaction networks.
Am J Med Genet A. 158A:1–3158. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Wat MJ, Beck TF, Hernández-García A, Yu Z,
Veenma D, Garcia M, Holder AM, Wat JJ, Chen Y, Mohila CA, et al:
Mouse model reveals the role of SOX7 in the development of
congenital diaphragmatic hernia associated with recurrent deletions
of 8p23.1. Hum Mol Genet. 21:4115–4125. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Keitges EA, Pasion R, Burnside RD, Mason
C, Gonzalez-Ruiz A, Dunn T, Masiello M, Gebbia JA, Fernandez CO and
Risheg H: Prenatal diagnosis of two fetuses with deletions of
8p23.1, critical region for congenital diaphragmatic hernia and
heart defects. Am J Med Genet A. 161A:1–1758. 2013.PubMed/NCBI
|
|
14
|
Garshasbi M, Motazacker MM, Kahrizi K,
Behjati F, Abedini SS, Nieh SE, Firouzabadi SG, Becker C,
Rüschendorf F, Nurnberg P, et al: SNP array-based homozygosity
mapping reveals MCPH1 deletion in family with autosomal recessive
mental retardation and mild microcephaly. Hum Genet. 118:708–715.
2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Jackson AP, Eastwood H, Bell SM, Adu J,
Toomes C, Carr IM, Roberts E, Hampshire DJ, Crow YJ, Mighell AJ, et
al: Identification of microcephalin, a protein implicated in
determining the size of the human brain. Am J Hum Genet.
71:136–142. 2002. View
Article : Google Scholar : PubMed/NCBI
|
|
16
|
Faheem M, Naseer MI, Rasool M, Chaudhary
AG, Kumosani TA, Ilyas AM, Pushparaj P, Ahmed F, Algahtani HA,
Al-Qahtani MH and Jamal H Saleh: Molecular genetics of human
primary microcephaly: An overview. BMC Med Genomics. 8 Suppl
1:S42015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chien WH, Gau SS, Wu YY, Huang YS, Fang
JS, Chen YJ, Soong WT, Chiu YN and Chen CH: Identification and
molecular characterization of two novel chromosomal deletions
associated with autism. Clin Genet. 78:449–456. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Nucaro A, Pisano T, Chillotti I, Montaldo
C and Pruna D: Chromosome 8p23. 2-pter: A critical region for
mental retardation, autism and epilepsy? Clin Genet. 79:394–396.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wu Y, Ji T, Wang J, Xiao J, Wang H, Li J,
Gao Z, Yang Y, Cai B, Wang L, et al: Submicroscopic subtelomeric
aberrations in Chinese patients with unexplained developmental
delay/mental retardation. BMC Med Genet. 11:722010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Reddy KS: A paternally inherited terminal
deletion, del(8)(p23.1)pat, detected prenatally in an amniotic
fluid sample: A review of deletion 8p23.1 cases. Prenat Diagn.
19:868–872. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Giglio S, Graw SL, Gimelli G, Pirola B,
Varone P, Voullaire L, Lerzo F, Rossi E, Dellavecchia C, Bonaglia
MC, et al: Deletion of a 5-cM region at chromosome 8p23 is
associated with a spectrum of congenital heart defects.
Circulation. 102:432–437. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Claeys I, Holvoet M, Eyskens B,
Adriaensens P, Gewillig M, Fryns JP and Devriendt K: A recognisable
behavioural phenotype associated with terminal deletions of the
short arm of chromosome 8. Am J Med Genet. 74:515–520. 1997.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Novo-Filho GM, Montenegro MM, Zanardo ÉA,
Dutra RL, Dias AT, Piazzon FB, Costa TV, Nascimento AM, Honjo RS,
Kim CA, et al: Subtelomeric copy number variations: The importance
of 4p/4q deletions in patients with congenital anomalies and
developmental disability. Cytogenet Genome Res. 149:241–246. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Jeong HS, Jung ES, Sim YJ, Kim SJ, Jang
JW, Hong KS, Lee WY, Chung HM, Park KT, Jung YS, et al: Fbxo25
controls Tbx5 and Nkx2-5 transcriptional activity to regulate
cardiomyocyte development. Biochim Biophys Acta. 1849:709–721.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Li JM, Lu CL, Cheng MC, Luu SU, Hsu SH and
Chen CH: Exonic resequencing of the DLGAP3 gene as a candidate gene
for schizophrenia. Psychiatry Res. 208:84–87. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li JM, Lu CL, Cheng MC, Luu SU, Hsu SH, Hu
TM, Tsai HY and Chen CH: Role of the DLGAP2 gene encoding the
SAP90/PSD-95-associated protein 2 in schizophrenia. PLoS One.
9:e853732014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Chien WH, Gau SS, Liao HM, Chiu YN, Wu YY,
Huang YS, Tsai WC, Tsai HM and Chen CH: Deep exon resequencing of
DLGAP2 as a candidate gene of autism spectrum disorders. Mol
Autism. 4:262013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Pinto D, Pagnamenta AT, Klei L, Anney R,
Merico D, Regan R, Conroy J, Magalhaes TR, Correia C, Abrahams BS,
et al: Functional impact of global rare copy number variation in
autism spectrum disorders. Nature. 466:368–372. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Wu K, Hanna GL, Easter P, Kennedy JL,
Rosenberg DR and Arnold PD: Glutamate system genes and brain volume
alterations in pediatric obsessive-compulsive disorder: A
preliminary study. Psychiatry Res. 211:214–220. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Vantaggiato C, Redaelli F, Falcone S,
Perrotta C, Tonelli A, Bondioni S, Morbin M, Riva D, Saletti V,
Bonaglia MC, et al: A novel CLN8 mutation in late-infantile-onset
neuronal ceroid lipofuscinosis (LINCL) reveals aspects of CLN8
neurobiological function. Hum Mutat. 30:1104–1116. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Katata Y, Uematsu M, Sato H, Suzuki S,
Nakayama T, Kubota Y, Kobayashi T, Hino-Fukuyo N, Saitsu H and Kure
S: Novel missense mutation in CLN8 in late infantile neuronal
ceroid lipofuscinosis: The first report of a CLN8 mutation in
Japan. Brain Dev. 38:341–345. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Allen NM, O'HIci B, Anderson G, Nestor T,
Lynch SA and King MD: Variant late-infantile neuronal ceroid
lipofuscinosis due to a novel heterozygous CLN8 mutation and de
novo 8p23.3 deletion. Clin Genet. 81:602–604. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Verhoeven K, De Jonghe P, Van de Putte T,
Nelis E, Zwijsen A, Verpoorten N, De Vriendt E, Jacobs A, Van
Gerwen V, Francis A, et al: Slowed conduction and thin myelination
of peripheral nerves associated with mutant rho Guanine-nucleotide
exchange factor 10. Am J Hum Genet. 73:926–932. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Jungerius BJ, Hoogendoorn ML, Bakker SC,
Van't Slot R, Bardoel AF, Ophoff RA, Wijmenga C, Kahn RS and Sinke
RJ: An association screen of myelin-related genes implicates the
chromosome 22q11 PIK4CA gene in schizophrenia. Mol Psychiatry.
13:1060–1068. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Donohoe G, Walters J, Hargreaves A, Rose
EJ, Morris DW, Fahey C, Bellini S, Cummins E, Giegling I, Hartmann
AM, et al: Neuropsychological effects of the CSMD1 genome-wide
associated schizophrenia risk variant rs10503253. Genes Brain
Behav. 12:203–209. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Kraus DM, Elliott GS, Chute H, Horan T,
Pfenninger KH, Sanford SD, Foster S, Scully S, Welcher AA and
Holers VM: CSMD1 is a novel multiple domain complement-regulatory
protein highly expressed in the central nervous system and
epithelial tissues. J Immunol. 176:4419–4430. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Glancy M, Barnicoat A, Vijeratnam R, de
Souza S, Gilmore J, Huang S, Maloney VK, Thomas NS, Bunyan DJ,
Jackson A and Barber JC: Transmitted duplication of 8p23.1–8p23.2
associated with speech delay, autism and learning difficulties. Eur
J Hum Genet. 17:37–43. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Havik B, Le Hellard S, Rietschel M, Lybaek
H, Djurovic S, Mattheisen M, Muhleisen TW, Degenhardt F, Priebe L,
Maier W, et al: The complement control-related genes CSMD1 and
CSMD2 associate to schizophrenia. Biol Psychiatry. 70:35–42. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Shimizu A, Asakawa S, Sasaki T, Yamazaki
S, Yamagata H, Kudoh J, Minoshima S, Kondo I and Shimizu N: A novel
giant gene CSMD3 encoding a protein with CUB and sushi multiple
domains: A candidate gene for benign adult familial myoclonic
epilepsy on human chromosome 8q23.3-q24.1. Biochem Biophys Res
Commun. 309:143–154. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Trimborn M, Bell SM, Felix C, Rashid Y,
Jafri H, Griffiths PD, Neumann LM, Krebs A, Reis A, Sperling K, et
al: Mutations in microcephalin cause aberrant regulation of
chromosome condensation. Am J Hum Genet. 75:261–266. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wu N, Ming X, Xiao J, Wu Z, Chen X,
Shinawi M, Shen Y, Yu G, Liu J, Xie H, et al: TBX6 null variants
and a common hypomorphic allele in congenital scoliosis. N Engl J
Med. 372:341–350. 2015. View Article : Google Scholar : PubMed/NCBI
|