Introduction
Osteogenesis imperfecta (OI) is characterized by low
bone mass and increased bone fragility, and is observed in 1 in
15,000–20,000 births (1,2). According to the clinical and
radiographic features and mode of inheritance, the 1978 Sillence
classification (3) divided
osteogenesis imperfecta into 4 types: OI type I (MIM#166200, mild
nondeforming, with blue sclera), type II (MIM#166210, perinatal
lethal), type III (MIM#259420, progressive deforming) and type IV
(MIM#166220, moderately deforming, with normal sclera) (2,4,5). At
present, 11 new OI types (V–XV) (2) have been added to the pre-existing
types.
Although 15 types of OI have been identified, the
majority of the patients are diagnosed of type I–IV. The dominantly
inherited forms that account for ~90% patients with OI type I–IV
are autosomal dominant mutations in COL1A1 (MIM#120150) or
COL1A2 (MIM#120160), which encode two pro-α1 chains and one
pro-α2 chain of type I collagen (6). Type I collagen is the most abundant
protein of bone, skin, tendon, ligament, sclera and cornea tissues,
blood vessels and hollow organs (7). Both proα1 and proα2 chains are
composed of a triple helical domain of 1,014 amino acids where
glycine is invariantly in every third position, forming 338 Gly-X-Y
tripeptide motifs in the triple helical region, which is flanked by
globular carboxyl and amino terminal peptides (8,9). On
the basis of the University of Leicester's (Leicester, UK) database
(http://www.le.ac.uk/genetics/collagen/), hundreds of
mutations in the COL1A1 and COL1A2 genes have been
documented in the literature and recorded, which result in either
quantitative or qualitative protein defects.
To understand the association between genotype and
phenotype and extend the evidence for genetic and phenotypic
heterogeneity in OI type I, a RNA-splicing mutation in
COL1A1 gene in a large Chinese family was investigated. In
addition, the clinical features of the patients from this family
were summarized the clinical manifestations of OI type I varying
from mild to severe were reported.
Materials and methods
Case presentation and analysis
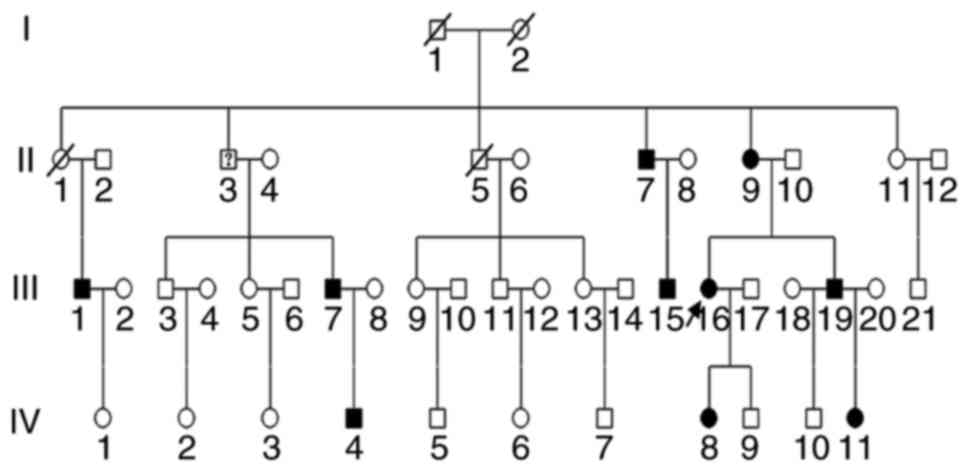
A large Chinese family was investigated, which
extended over four generations and comprised at least 46
individuals, of whom 4 had passed away, 1 had ambiguous diagnosis
and 10 had explicit clinical diagnosis of OI. The pedigree of the
family is presented in Fig. 1. The
autosomal dominant inheritance was identified. The proband
(III-16), a 46-year-old Chinese female, was referred for genetic
counseling of OI. Clinical data indicated that she had blue sclera
and mild hearing loss and her height, weight, vision and
intelligence were normal. Previously, she had experienced two
fractures. When she was 12-year-old, she suffered a femoral
fracture, and additionally suffered a pelvic fracture at 20 years
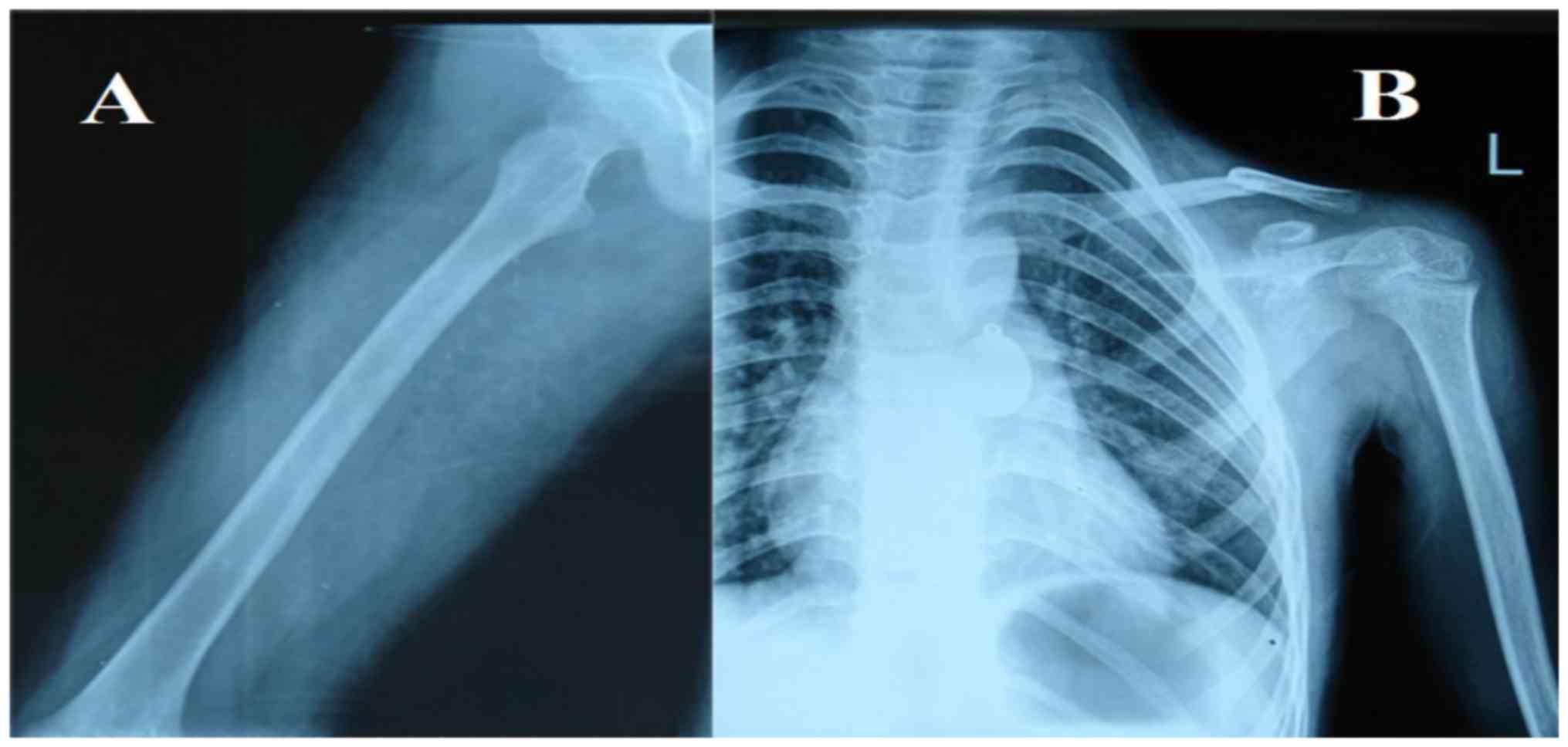
old. Radiographs of her daughter were presented in Fig. 2. Clinical characteristics of
certain patients in the family are listed in Table I.
 | Table I.Clinical information in osteogenesis
imperfecta type I patients from the family. |
Table I.
Clinical information in osteogenesis
imperfecta type I patients from the family.
| Patient | Age | Sex | Hearing loss | Blue sclerae | Dentinogenesis
imperfecta | Fracture rate | Severity |
|---|
| II-7 | 68 | M | + | + | − | + | Mild |
| II-9 | 70 | F | − | + | + | + | Mild |
| III-15 | 42 | M | − | + | − | + | Mild |
| III-16 | 46 | F | + | + | − | + | Mild |
| III-19 | 43 | M | − | + | − | ++ | Mild |
| IV-8 | 22 | F | − | + | − | + | Mild |
| IV-11 | 7 | F | − | ++ | − | ++ | Severe |
Laboratory studies
The current study was reviewed and approved by
Nanjing General Hospital Ethics Committee (Nanjing, China) prior to
initiation of the study. Blood samples were obtained with informed
consent from 10 affected and 31 unaffected individuals. Their ages
ranged between 7 and 69 and sex ratio was 19 (M):22 (F). In
addition to 10 patients suffering from OI, the other members in the
family were healthy. In addition, 200 normal healthy Chinese
individuals were recruited (aged 6–59 years old; sex ratio 1:1;
Nanjing General Hospital) as controls. Genomic DNA was isolated
from peripheral blood using a Genomic DNA Purification kit (Qiagen
GmbH, Hilden, Germany). NGS (10)
was used in COL1A1 and COL1A2 in 3 patients (the
proband, her daughter and her niece) of the family to screen the
mutation. Subsequently, it was confirmed in other members in the
family and 200 healthy controls by polymerase chain reaction (PCR).
Primers were designed using Primer Premier software (version 5;
Premier Biosoft India Pvt., Ltd., Indore, India) and the reaction
conditions for amplification were as follows: 1 cycle (5 min 96°C),
30 cycles (1 min 94°C, 1 min 58°C and 1 min 72°C), and 1 cycle (5
min 72°C) using 5′-CCGTGACTCCTCACAGTCCT-3′ and
5′-CGCAAAAGAGCCTGATGTTA-3′. PCR reactions were conducted directly
using an ABI Prism 3700 automated sequencer (Applied Biosystems;
Thermo Fisher Scientific, Inc., Waltham, MA, USA).
Results
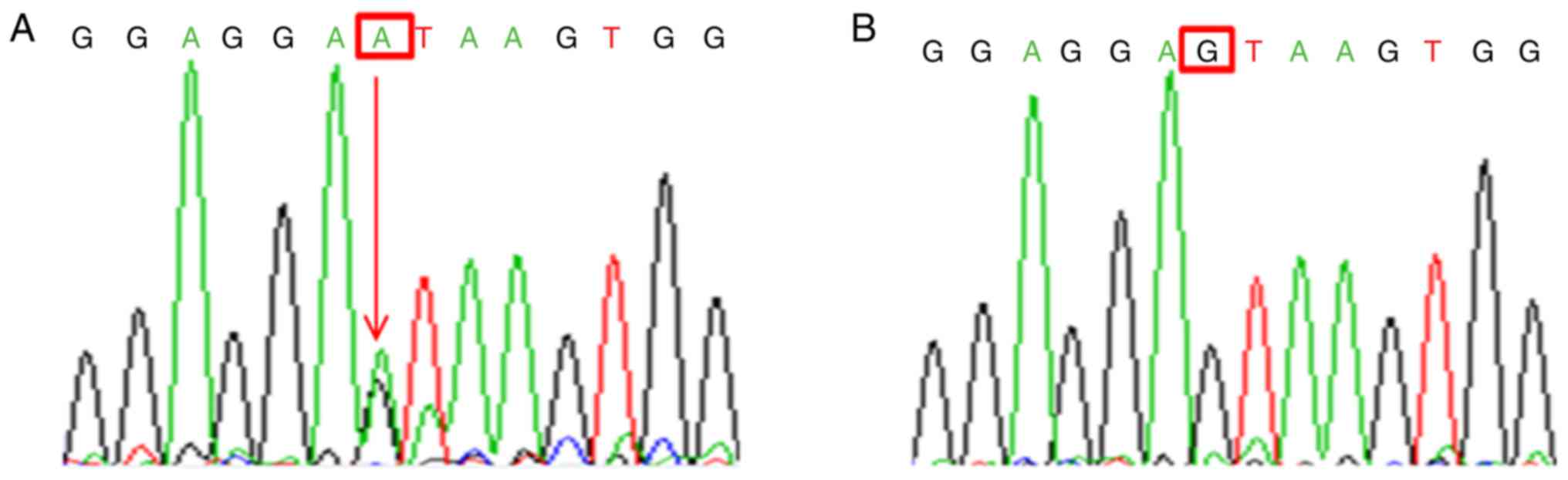
In the large pedigree, a splicing mutation was
identified (c.471+1G>A, also termed IVS5+1G>A) by sequence
analysis of the COL1A1 gene in affected persons (Fig. 3A). This mutation, however, was not
identified in 31 unaffected relatives and the 200 controls
(population-matched control individuals) (Fig. 3B), which suggested that this
mutation served a pathological role in patients with OI.
Among the 10 patients, notably, there was one person
from this family with severe clinical symptoms. This patient was a
7-year-old girl (IV11), and at present, had suffered greater than
10 fractures and presented with a severe spinal deformity. Other
conditions were normal. Although this patient had the same mutation
c.471+1G>A in COL1A1 with the patients as the rest of the
family, her phenotype was novel.
Discussion
In the past decade, a series of OI types have been
identified, and multiple genes responsible for it have been
established (11). However,
patients referred with the diagnosis of OI are predominantly
attributed to COL1A1 and COL1A2 mutations. Although
glycine substitutions have been observed to be the major mutation
sites of COL1A1 or COL1A2 in the majority of cases,
other mutation types exist in patients with OI (12–15).
Giving rise to OI types I–IV, there are two general categories of
type I collagen mutations. The first is caused by the substitution
of glycine in the Gly-X-Y triplet domain of the triple helix, which
involves the synthesis of collagen molecules with structural
abnormalities. The second type known as haploinsufficiency, results
in failure to synthesize the products of one COL1A1 allele,
which is caused by frameshift, nonsense and splice-site mutations
(6,16–18).
In the present study, a known splicing mutation (c.471+1G>A) was
identified in OI type I patients, which may be consistent with
previous studies that have reported that patients with OI with
haploinsufficiency mutations have milder phenotypes than glycine
mutations (19–21).
Notably, when we applied NGS to screen the mutation
in the proband at first, the pathogenic mutation was omitted, and
until it was confirmed it in other patients in the family, it has
been discovered. This indicates that mutations of exon/intron
boundaries are as important as mutations of exons, which result
from the sequences of exon/intron boundaries could be part of the
splice signal. Exon/GU-intron-AG/exon sequence in almost all
introns in the nuclear mRNA, made by Breathnach et al
(22), does not occur by chance
(4,23). When the exon/intron boundaries are
mutated, abnormal splicing can arise. In the current study,
c.471+1G>A in the patients was predicted to affect splicing due
to the fact that it alters the conserved splice donor sequence GT
at the 5′-end of intron 5, which was mutated to AT. The predicted
consequences (skipping of exon 5 or retention of intron 5) require
confirmation at the mRNA and protein levels. The blood samples in
the present study were too degraded therefore reverse transcription
PCR failed to verify the consequence of the sequence variant, and
patients were not willing to provide tissue samples.
In 2006, Pollitt et al (1) discovered 62 mutations by analysis of
COL1A1 and COL1A2 in a cohort of 83 unrelated
patients with OI type I–IV. Among the mutations, they identified
c.471+1G>A in an OI type I patient, similar to that of the
patient in the current study. This mutation may be more common in
OI type I patients than currently established. Although the
mutation was the same as that of the current study, regrettably,
they gave no depiction of the mutation and there was no clinical
information about the patient. There, however, had different
clinical phenotypes in the present study, and these intrafamilial
variations in patients' clinical and radiographic courses of
patients harboring identical mutations suggest that environmental
factors or other genetic factors may affect the outcome of
patients.
In conclusion, the present study indicated that a
recurrent mutation c.471+1G>A in COL1A1 served a
pathogenic role in a large Chinese family, and different phenotypes
of this variant were observed. The identification of the mutation
provides genetic counseling and prenatal diagnosis for other
members in the family, in addition to extending the evidence for
genetic and phenotypic heterogeneity in OI. Finally, NGS serves a
significant role in this congenital disorder as the clinical
manifestations of OI are changeable. To clarify the pathological
mechanisms of OI, further studies are required.
Acknowledgements
The current study was supported by Nanjing Science
and Technology Development Project (grant no. 201503010), Nanjing
Science and Technology Project (grant no. 2014020008), Foundation
of Nanjing General Hospital of Nanjing Military Command, PLA (grant
no. 2015046) and the Foundation of Nanjing General Hospital of
Nanjing Military Command, PLA (grant no. 2014044).
Glossary
Abbreviations
Abbreviations:
|
OI
|
osteogenesis imperfecta
|
|
NGS
|
next-generation sequencing
|
References
|
1
|
Pollitt R, Mcmahon R, Nunn J, Bamford R,
Afifi A, Bishop N and Dalton A: Mutation analysis of COL1A1 and
COL1A2 in patients diagnosed with osteogenesis imperfecta type
I–IV. Hum Mutat. 27:7162006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Forlino A and Marini JC: Osteogenesis
imperfecta. Lancet. 387:1657–1671. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Sillence DO and Rimoin DL: Classification
of osteogenesis imperfecta. Lancet. 1:1041–1042. 1978. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Xia XY, Cui YX, Huang YF, Pan LJ, Yang B,
Wang HY, Li XJ, Shi YC, Lu HY and Zhou YC: A novel RNA-splicing
mutation in COL1A1 gene causing osteogenesis imperfecta type I in a
Chinese family. Clin Chim Acta. 398:148–151. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Semler O, Garbes L, Keupp K, Swan D,
Zimmermann K, Becker J, Iden S, Wirth B, Eysel P, Koerber F, et al:
A mutation in the 5′-UTR of IFITM5 creates an in-frame start codon
and causes autosomal-dominant osteogenesis imperfecta type V with
hyperplastic callus. Am J Hum Genet. 91:349–357. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhang ZL, Zhang H, Ke YH, Yue H, Xiao WJ,
Yu JB, Gu JM, Hu WW, Wang C, He JW and Fu WZ: The identification of
novel mutations in COL1A1, COL1A2, and LEPRE1 genes in Chinese
patients with osteogenesis imperfect. J Bone Miner Metab. 30:69–77.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Chan TF, Poon A, Basu A, Addleman NR, Chen
J, Phong A, Byers PH, Klein TE and Kwok PY: Natural variation in
four human collagen genes across an ethnically diverse population.
Genomics. 91:307–314. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Hald JD, Folkestad L, Harsløf T, Lund AM,
Duno M, Jensen JB, Neghabat S, Brixen K and Langdahl B: Skeletal
phenotypes in adult patients with osteogenesis
imperfecta-correlations with COL1A1/COL1A2 genotype and collagen
structure. Osteoporos Int. 27:3331–3341. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rauch F and Glorieux FH: Osteogenesis
imperfect. Lancet. 363:1377–1385. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Bacchelli C and Williams HJ: Opportunities
and technical challenges in next-generation sequencing for
diagnosis of rare pediatric diseases. Expert Rev Mol Diagn.
16:1073–1082. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Hoornaert KP, Vereecke I, Dewinter C,
Rosenberg T, Beemer FA, Leroy JG, Bendix L, Björck E, Bonduelle M,
Boute O, et al: Stickler syndrome caused by COL2A1 mutations:
Genotype-phenotype correlation in a series of 100 patients. Eur J
Hum Genet. 18:872–880. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Pace JM, Wiese M, Drenguis AS, Kuznetsova
N, Leikin S, Schwarze U, Chen D, Mooney SH, Unger S and Byers PH:
Defective C-propeptides of the proalpha2(I) chain of type I
procollagen impede molecular assembly and result in osteogenesis
imperfecta. J Biol Chem. 283:16061–16067. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Martin E and Shapiro JR: Osteogenesis
imperfecta: Epidemiology and pathophysiology. Curr Osteoporos Rep.
5:91–97. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Malfait F, Symoens S, De Backer J,
Hermanns-Lê T, Sakalihasan N, Lapière CM, Coucke P and De Paepe A:
Three arginine to cysteine substitutions in the pro-alpha
(I)-collagen chain cause Ehlers-Danlos syndrome with a propensity
to arterial rupture in early adulthood. Hum Mutat. 28:387–395.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cabral WA, Makareeva E, Letocha AD,
Scribanu N, Fertala A, Steplewski A, Keene DR, Persikov AV, Leikin
S and Marini JC: Y-position cysteine substitution in type I
collagen (alpha1(I) R888C/p.R1066C) is associated with osteogenesis
imperfecta/Ehlers-Danlos syndrome phenotype. Hum Mutat. 28:396–405.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Rauch F, Lalic L, Roughley P and Glorieux
FH: Relationship between genotype and skeletal phenotype in
children and adolescents with osteogenesis imperfect. J Bone Miner
Res. 25:1367–1374. 2010.PubMed/NCBI
|
|
17
|
Ward LM, Lalic L, Roughley PJ and Glorieux
FH: Thirty-three novel COL1A1 and COL1A2 mutations in patients with
osteogenesis imperfecta types I–IV. Hum Mutat. 17:4342001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Benusiené E and Kucinskas V: COL1A1
Mutation analysis in Lithuanian patients with osteogenesis
imperfecta. J Appl Genet. 44:95–102. 2003.PubMed/NCBI
|
|
19
|
Byers PH: Haploinsufficiency for mutations
in type I collagen genes: Mechanisms and clinical effects. Elsevier
Inc; pp. 1–127. 2013
|
|
20
|
Ben Amor IM, Roughley P, Glorieux FH and
Rauch F: Skeletal clinical characteristics of osteogenesis
imperfecta caused by haploinsufficiency mutations in COL1A1. J Bone
Miner Res. 28:2001–2007. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang X, Pei Y, Dou J, Lu J, Li J and Lv Z:
Identification of a novel COL1A1 frameshift mutation, c.700delG, in
a Chinese osteogenesis imperfecta family. Genet Mol Biol. 38:1–7.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Breathnach R, Benoist C, O'Hare K, Gannon
F and Chambon P: Ovalbumin gene: Evidence for a leader sequence in
mRNA and DNA sequences at the exon-intron boundaries. Proc Natl
Acad Sci USA. 75:4853–4857. 1978; View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xia XY, Cui YX, Huang YF, Pan LJ, Yang B,
Wang HY, Li XJ, Shi YC, Lu HY and Zhou YC: A novel RNA-splicing
mutation in COL1A1 gene causing osteogenesis imperfecta type I in a
Chinese family. Clin Chim Acta. 398:148–151. 2008. View Article : Google Scholar : PubMed/NCBI
|