Introduction
Cardiac hypertrophy is a cardiac muscle disorder
that may eventually lead to heart failure, regardless of its
etiology. Cardiac hypertrophy is a risk factor for cardiovascular
mortality and morbidity independently or through interacting with
other cardiovascular risk factors. Hence, controlling hypertrophic
remodeling may offer the most promising novel therapeutic strategy
for reducing cardiovascular morbidity and mortality in cardiac
hypertrophy and heart failure. The vast majority of existing
therapeutic strategies exert antihypertrophic effects via targeting
extracellular receptors in cardiac cells, but their efficacy
appears to be limited. Attention has turned to targeting
intracellular signaling pathways in the last decade (1).
Coenzyme Q10 or ubiquinone-10 (CoQ10), the only
endogenously synthesized lipid-soluble antioxidant, has been
demonstrated to be important in maintaining normal cardiovascular
function in humans and a mouse disease model (2–4). For
example, a decreased expression level of CoQ10 was observed in
human cardiomyopathy (5) and CoQ10
significantly ameliorated cardiac function in children with dilated
cardiomyopathy (6). In a mouse
diabetic model, CoQ10 has been shown to efficiently diminish
cardiac hypertrophy and fibrosis (7). Furthermore, CoQ10 has been widely
used as a daily supplement to benefit cardiovascular function
(5). In addition, CoQ10 is
involved in NAD(P) H-oxidoreductase-dependent reactions, such as
the synthesis of nitric oxide (NO) (8), which is generated by endothelial NO
synthase (eNOS), a well-known mediator of cardiovascular
homeostasis (9,10). As in the case with
non-mitochondrial CoQ10, which is predominantly present in the
Golgi and plasma membrane (11,12),
eNOS is also specifically localized in the Golgi and plasma
membrane of heart and endothelial cells, and its function is
differentially regulated in these two cellular compartments
(13). Previous studies have shown
that CoQ10 improves endothelial dysfunction, in part, by
‘recoupling’ eNOS and modulating NO-associated signaling (14).
UbiA prenyltransferase domain containing 1 (UBIAD1;
also termed transitional epithelial response protein 1), belongs to
the UbiA superfamily of prenyltransferases (15). UBIAD1 was first identified as an
enzyme that catalyzes the synthesis of vitamin K2 (16,17),
a well-known factor involved in the maintenance of cardiovascular
homeostasis. Previously, UBIAD1 has been reported to synthesize
CoQ10 and exhibit anti-oxidant activity (18). Although UBIAD1 was first shown to
be potentially involved in the development of bladder and prostate
tumors (19), emerging evidence
indicates that UBIAD1 is essential in normal cardiovascular
development and function. For example, a UBIAD1 mutation has been
associated with cardiac edema and vascular phenotypes in zebrafish,
which were rescued by the re-expression of human UBIAD1 (20). In a mouse model, UBIAD1 knockout
led to embryonic death at E7.5-E10.5; however, whether there were
any cardiovascular phenotypes was not reported (21). Given that UBIAD1 contributes
significantly to the generation of non-mitochondrial CoQ (18), it is presumed that UBIAD1
contributes to mitochondrial heart disease. However, whether and
how UBIAD1 is involved in mediating cardiomyocyte hypertrophy in
response to agonists remains unknown.
Given the above findings that link UBIAD1 to CoQ10,
it was hypothesized in the present study that UBIAD1 may be
involved in mediating stressor-induced cardiac hypertrophy via
mediating CoQ10 expression levels. The current study aimed to
determine the role and potential underlying mechanism of UBIAD1 in
angiotensin II (Ang II)-induced myocardial hypertrophy in
vitro.
Materials and methods
Cell culture and treatments
AC16 human myocardial cells (Guangzhou Lede Kangkang
Biotechnology Co., Ltd., Guangzhou, Guangdong, China) were cultured
in Dulbecco's modified Eagle's medium supplemented with 10% fetal
bovine serum (HyClone; GE Healthcare Life Sciences, Chicago, IL,
USA), 2 mM glutamine, 100 U/ml penicillin, 100 µg/ml streptomycin,
and 1 mM pyruvate in humidified air (5% CO2) at 37°C.
The AC16 cells were transiently transfected with UBIAD1 siRNA
(Si-UBIAD1; Si-UBIAD1 was tagged with a carboxyfluorescein green
fluorophore) or a negative control siRNA (NC; Shanghai GenePharma
Co., Ltd., Shanghai, China) using Invitrogen Lipofectamine 2000
reagent (Thermo Fisher Scientific, Inc., Waltham, MA, USA),
according to the manufacturer's protocol. The sequences of the
UBIAD1-siRNA were as follows: Forward,
5′-CACUUGGCUCUUAUCUACUdTdT-3′ and reverse,
5′-AGUAGAUAAGAGCCAAGUGdTdT-3′. The sequences of the NC were as
follows: Forward, 5′-UUCUCCGAACGUGUCACGUTT-3′ and reverse,
5′-ACGUGACACGUUCGGAGAATT-3′.
Measurement of the surface area
Motic Images 1.3 software (Beijing Hanmengzixing
Instruments and Meters Co., Ltd., Beijing, China) was used to
measure the surface area of the AC16 cells. Five fields were
randomly selected for each treatment, and 15–25 cells/field were
measured.
3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT)
assay
The MTT assay was performed to analyze the viability
of the AC16 cells. Briefly, AC16 cells were plated at a density of
1×105/ml in a 96-well plate for 24 h, transfected with
Si-UBIAD1 or NC, followed by treatment with vehicle or Ang II for
24 h (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). The AC16
cells were incubated with 20 µl of 5 mg/ml MTT solution (HyClone;
GE Healthcare Life Sciences) for 4 h at 37°C. Thereafter, 150 µl
dimethyl sulfoxide (HyClone; GE Healthcare Life Sciences) was added
to each well to dissolve the crystal formazan dye, and the plate
was agitated for 10 min until all the crystals were dissolved. The
quantity of MTT formazan was determined by the absorbance at a
wavelength of 490 nm using a microplate reader.
Determination of apoptosis with flow
cytometry
The rate of cell apoptosis was analyzed with flow
cytometry (BD FACSCanto™ II system; BD Biosciences, Franklin Lakes,
NJ, USA) following FITC-Annexin V and propidium iodide (PI)
staining. The staining procedure was performed according to the
instructions of the Annexin V-FITC Apoptosis Detection kit
(BioVision, Inc., Milpitas, CA, USA). Briefly, AC16 cells were
resuspended following centrifugation to a concentration of
1×106 cells/ml. A total of 100 µl cells were transferred
to 5 ml tubes, followed by addition of 5 µl FITC-Annexin V and 5 µl
PI. The cell suspension was gently vortexed and incubated at room
temperature for 15 min, shielded from light, and then 400 µl
Annexin V binding buffer was added. Flow cytometry was performed
within 1 h. The following method was applied: The Annexin
V-negative/PI-negative fraction represented viable cells; the
Annexin V-positive/PI-negative fraction represented early apoptotic
cells; and the Annexin V-positive/PI-positive fraction represented
late apoptotic and dead cells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was isolated using the PureLink
Micro-to-Midi Total RNA Purification system (Invitrogen; Thermo
Fisher Scientific, Inc.) and cDNA was synthesized with an RT High
Capacity kit (Applied Biosystems; Thermo Fisher Scientific, Inc.)
according to the manufacturer's protocol. RT-qPCR was performed
with an ABI Prism 7300 Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) using Platinum Quantitative PCR
SuperMix-UDG with ROX (Invitrogen; Thermo Fisher Scientific, Inc.).
The primers used in the RT-qPCR were as follows: Forward,
5′-AACGACTGTCCCGAGCAA-3′ and reverse, 5′-CGGCACAACCCACCAA-3′ for
UBIAD1; forward, 5′-AGCCGAGACAGCAAACA-3′ and reverse,
5′-GCCTGGGAGCCAAAA-3′ for atrial natriuretic factor (ANF); forward,
5′-AGAGCTGGACTGCGGTATTGAG-3′ and reverse,
5′-GAACCATGACCCGTCCCTTG-3′ for caspase-3; forward,
5′-GACCATAATGCCTCACC-3′ and reverse, 5′-ATGCGTTCATCACCAA-3′ for
CoQ10; forward, 5′-GCAGAGGAGTCCAGCGAACA-3′ and reverse,
5′-TGGGTGCTGAGCTGACAGAGTA-3′ for eNOS; and forward,
5′-GAGGCTCTCTTCCAGCCTTC-3′ and reverse, 5′-AGGGTGTAAAAGCAGCTCA-3′
for GAPDH. The thermocycling conditions were as follows: 50°C for 2
min, then 10 min at 95°C, followed by 40 cycles of 95°C for 30 sec
and 55°C for 30 sec. The relative expression was assessed using the
2−ΔCq method, and converted to fold changes using the
2−ΔΔCq method (22).
Western blot analysis
Whole cell lysates were extracted from AC16 cells
using RIPA lysis buffer, according to the manufacturer's
instructions (Abcam, Cambridge, UK) and quantified using a
Bicinchoninic Acid Protein Assay kit (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). A total of 20 µg protein samples were separated
by 10% sodium dodecyl sulfate-polyacrylamide gel electrophoresis
and transferred (70 V, 90 min) onto a polyvinylidene difluoride
membrane (EMD Millipore, Billerica, MA, USA). The membrane was
blocked in Tris-buffered saline containing 5% nonfat milk and 0.1%
Tween-20 for 1 h at room temperature, followed by an incubation
with the primary antibody against GAPDH (1:5,000; cat no. 5174;
Cell Signaling Technology, Inc., Danvers, MA, USA), UBIAD1
(1:1,000; cat no. ab191691; Abcam), ANF (1:500; cat no. sc-80686),
and caspase-3 (1:1,000; cat no. sc-7272) (both from Santa Cruz
Biotechnology, Inc., Dallas, TX, USA), CoQ10 (1:1,000; cat no.
17812-1-AP) and eNOS (1:1,000; cat no. 20116-1-AP) (both from Wuhan
Sanying Biotechnology, Wuhan, China) at 4°C overnight. Following
washing with TBST (0.1% Tween-20), the membrane was incubated with
a peroxidase-conjugated secondary antibody (1:400; cat no. A0216;
Beyotime Institute of Biotechnology, Haimen, China) for 1 h at room
temperature. Protein bands were visualized using the ECL detection
system (GE Healthcare Life Sciences). Image analysis and
quantification were performed using a scanning densitometer.
Measurement of NO production
The accumulation of NO was assayed using the Griess
reagent. Briefly, the cells were transfected with Si-UBIAD1 or NC,
and then treated with Ang II for 24 h. Thereafter, 100 µl Griess
reagent was combined with an equal volume of cell supernatant and
incubated at room temperature for 5 min. The optical density at 540
nm was measured, and the concentration of nitrite was calculated
according to the standard curve generated from known concentrations
of sodium nitrite (14).
Statistical analysis
Data were expressed as the mean ± standard error of
the mean. Statistical differences were assessed by one way-analysis
of variance, followed by the Duncan test for comparison between
groups. P<0.05 was considered to indicate a statistically
significant difference.
Results
Ang II increased the expression of ANF
and caspase-3, and decreased UBIAD1 expression in AC16 cells
Western blot analysis was performed in cell lysates
purified from AC16 cells stimulated with either vehicle or ANG II
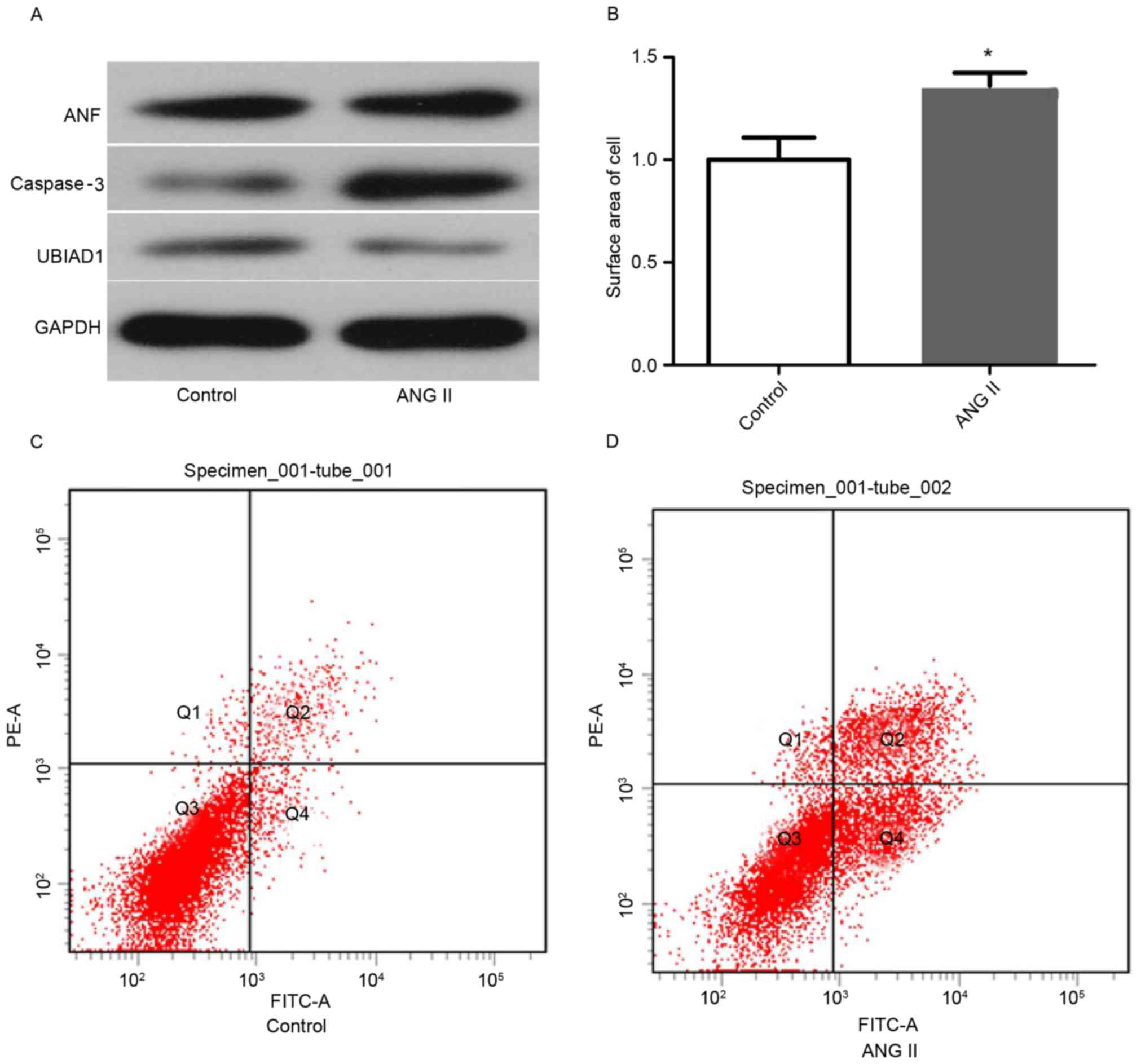
(100 nM) for 24 h. As shown in Fig.
1A, Ang II markedly increased ANF and caspase-3 expression,
compared with the vehicle group. In addition, the level of UBIAD1
in the AC16 cells of these two stimulation groups was measured and
UBIAD1 was observed to be markedly decreased in Ang II-stimulated
cells, compared with that in vehicle-treated cells. The surface
area of the AC16 cells stimulated by Ang II or vehicle was measured
using Motic Images 1.3. As shown in Fig. 1B, Ang II stimulation significantly
increased the cell size by 1.46 times, compared with the vehicle
treatment group (P<0.05). As caspase-3 expression was elevated
by Ang II stimulation, whether apoptosis also increased in Ang
II-stimulated AC16 cells was subsequently investigated. Indeed,
flow cytometry experiments revealed that more apoptotic cells were
observed in Ang II-treated AC16 cells than in vehicle-treated cells
(Fig. 1C and D).
Depletion of UBIAD1 increased ANG
II-stimulated expression of ANF and caspase-3 in AC16 cells
Next, whether UBIAD1 was involved in the Ang
II-induced elevated expression of ANP and caspase-3 in AC16 cells
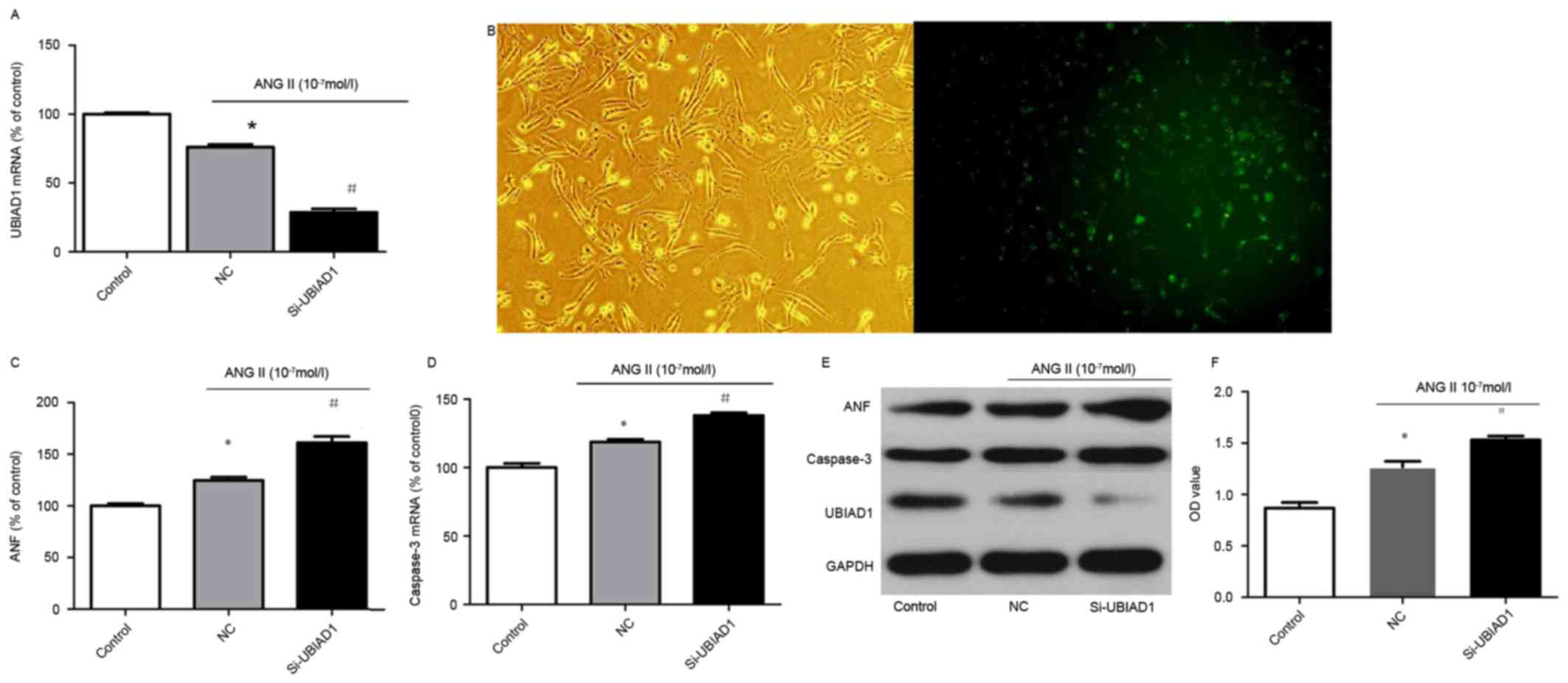
was investigated. The expression level of UBIAD1 in vehicle- and
Ang II-treated AC16 cells was quantitatively assessed by RT-qPCR.
Ang II stimulation significantly decreased UBIAD1 mRNA expression
(Fig. 2A).
Subsequently, to investigate whether UBIAD1
contributed to the hypertrophic response of AC16 cells to Ang II
stimulation, knock down of UBIAD1 in AC16 cells was performed with
Si-UBIAD1. Compared with AC16 cells transfected with control siRNA
(the NC), Si-UBIAD1 markedly diminished the expression of UBIAD1
with an efficacy of ~95% (Fig.
2B). Furthermore, downregulation of UBIAD1 further promoted the
increased expression of ANF and caspase-3 induced by Ang II at the
mRNA and protein expression levels (Fig. 2C-E). In addition, Si-UBIAD1
depletion significantly increased the number of viable cells in the
Ang II-stimulated group as measured by the MTT assay (Fig. 2F).
UBIAD1 knockdown potentiated the Ang
II-induced decrease in the expression of CoQ10 and eNOS
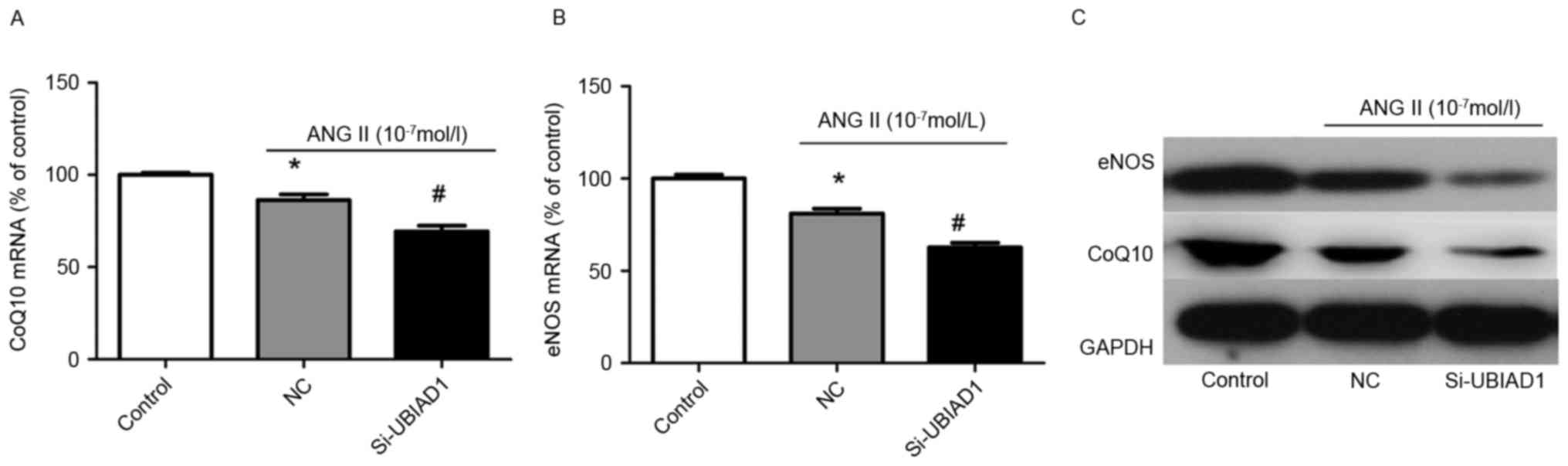
To understand the mechanisms by which knockdown of
UBIAD1 potentiated the molecular phenotypes induced by Ang II
stimulation in AC16 cells, the protein and mRNA expression levels
of CoQ10 and eNOS, which are key regulators of cardiovascular
homeostasis were analyzed. Fig. 3A and
B indicate that knockdown of UBIAD1 further attenuated the
decrease in mRNA expression of CoQ10 and eNOS in Ang II-stimulated
AC16 cells. Consistent with this finding, the protein expression
levels of CoQ10 and eNOS were also further decreased in Ang
II-treated cells transfected with NC (Fig. 3C).
UBIAD1 knockdown potentiated the Ang
II-induced decrease in NO production
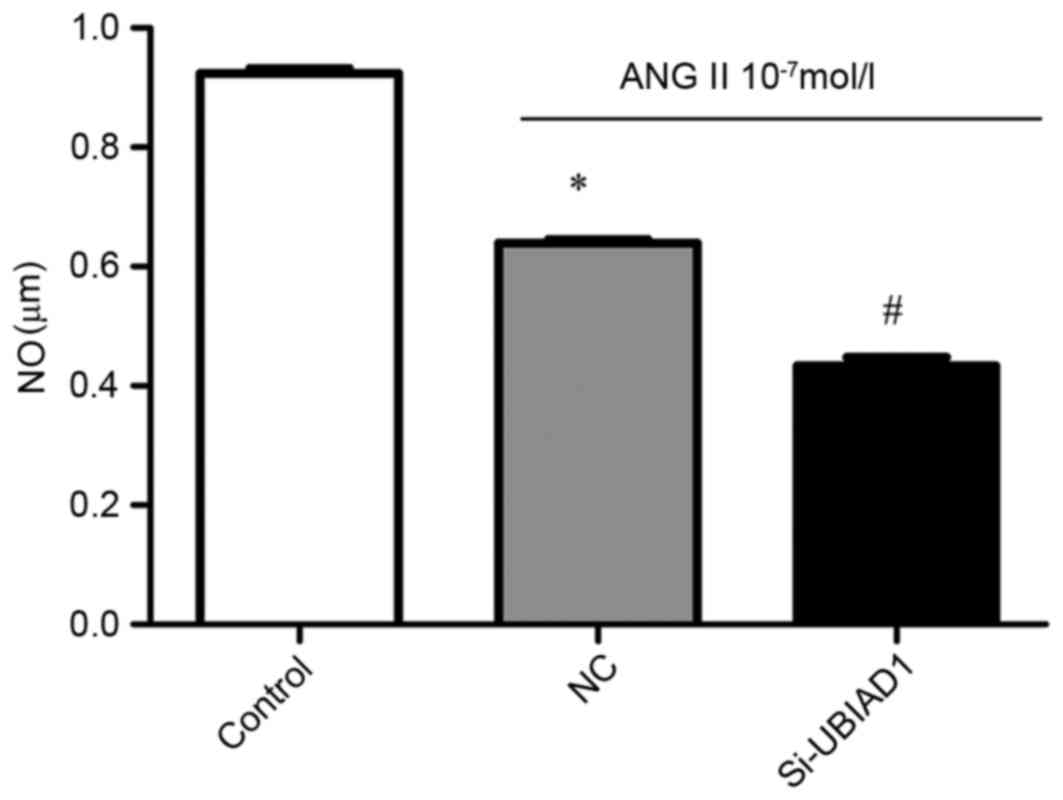
The levels of NO in vehicle- and Ang II-treated AC16
cells that were transfected with NC and Si-UBIAD1, respectively
were also measured. As shown in Fig.
4, the NO level was decreased in Ang II-stimulated AC16 cells,
compared with the vehicle-treated cells (P<0.05). UBIAD1
knockdown further attenuated this decrease (P<0.05).
Discussion
In the present study, the potential involvement of
UBIAD1 in Ang II-induced hypertrophy was investigated in AC16
cells, a human ventricular cardiac cell line. It was demonstrated
that Ang II stimulation increased the expression of ANF and
caspase-3, which coincided with the decreased expression of UBIAD1.
Knockdown of UBIAD1 further potentiated the Ang II-triggered
decrease in the levels of ANF and caspase-3. Mechanistically,
downregulation of UBIAD1 in Ang II-stimulated AC16 cells was
associated with a further decrease in the expression of CoQ10 and
eNOS, as well as the production of NO induced by Ang II.
Ang II is important in the pathophysiology of
cardiovascular diseases and metabolic disorders (23–25).
Evidence from clinical trials, animal models, and cellular studies
indicates that renin-angiotensin system inhibitors, including renin
inhibitors, Ang II type 1 receptor blockers, and Ang-converting
enzyme inhibitors, improve cardiac function and ameliorate cardiac
hypertrophy (26–29). eNOS and its product, NO are also
involved in Ang II-induced pathophysiological changes in the
cardiovascular system (30). In
addition, accumulating evidence indicates that reduced NO
bioavailability contributes to the pathogenesis of cardiac
hypertrophy and heart failure (31). In our model system, Ang II
stimulation inhibited the expression of eNOS and NO production,
which coincided with the decrease in UBIAD1 expression.
Additionally, depletion of UBIAD1 further enhanced the Ang
II-triggered inhibition of eNOS and NO, indicating that UBIAD1
potentially antagonizes the repressive impact of Ang II on eNOS and
NO expression/production. In addition, a significant decrease in
CoQ10 expression was observed in Ang II-treated cells, compared
with the vehicle-treated cells. Given that CoQ10 offers a variety
of benefits to the cardiovascular system against oxidative stress,
a decrease in CoQ10 level is detrimental to cellular function.
However, CoQ10 has been shown to ameliorate the clinical
manifestations of patients with hypertrophic cardiomyopathy
(32). Furthermore, depletion of
UBIAD1 further attenuated the decrease in CoQ10 by Ang II, which is
consistent with the recent finding that UBIAD1 is involved in the
synthesis of CoQ10 (18).
Caspase-3 and ANF are representative indicators for
cardiac hypertrophy and apoptosis, respectively. In the current
study, Ang II (100 nM) stimulation for 24 h induced hypertrophy and
apoptosis in AC16 cells, which was consistent with studies on
animal models and other cell lines (30,33).
Silencing of UBIAD1 by siRNA further potentiated the increase in
expression levels of ANF and caspase-3 elicited by Ang II
treatment, indicating that UBIAD1 is negatively implicated in the
Ang II-induced expression of ANF and caspase-3 in AC16 cells.
Furthermore, the MTT assay for cell viability demonstrated that the
optical density of silenced UBIAD1 myocardial cells was higher than
that of the negative control group.
Missense mutations of the UBIAD1 gene are
etiologically linked to the genetic disorder Schnyder corneal
dystrophy (SCD), in which the accumulation of cholesterol and
phospholipids occurs in the cornea of the eye, eventually leading
to blindness (34). However, no
cardiac phenotypes in patients with SCD have been reported. Given
that UBIAD1 is implicated in the generation of CoQ10 and eNOS,
which are important for cardiovascular homeostasis, it will be
interesting to clarify whether a mutation of the UBIAD1 gene
predisposes humans or mice to cardiac muscle disorders in response
to external stressors.
In conclusion, the present study demonstrated that
Ang II stimulation induces hypertrophy in AC16 cardiac cells,
accompanied by increased apoptosis and expression levels of the
cardiac disease marker ANF. In addition, Ang II treatment
suppresses the expression levels of eNOS and CoQ10, which have
beneficial effects on cardiomyocyte functions. Silencing UBIAD1
further aggravates the deleterious effects rendered by Ang II,
indicating that a normal or increased level of UBIAD1 may offer
protection against the Ang II-induced hypertrophic response and
apoptosis. Thus, the current findings provide novel insight into
the potential protection of UBIAD1 in cardiac hypertrophy and its
underlying molecular mechanisms.
References
|
1
|
McKinsey TA and Kass DA: Small-molecule
therapies for cardiac hypertrophy: Moving beneath the cell surface.
Nat Rev Drug Discov. 6:617–635. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
2
|
Kumar A, Kaur H, Devi P and Mohan V: Role
of coenzyme Q10 (CoQ10) in cardiac disease, hypertension and
Meniere-like syndrome. Pharmacol Ther. 124:259–268. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Crane FL: Discovery of ubiquinone
(coenzyme Q) and an overview of function. Mitochondrion. 7
Suppl:S2–S7. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bentinger M, Tekle M and Dallner G:
Coenzyme Q-biosynthesis and functions. Biochem Biophys Res Commun.
396:74–79. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Saha SP and Whayne TF Jr: Coenzyme Q-10 in
human health: Supporting evidence? South Med J. 109:17–21. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Kocharian A, Shabanian R, Rafiei-Khorgami
M, Kiani A and Heidari-Bateni G: Coenzyme Q10 improves diastolic
function in children with idiopathic dilated cardiomyopathy.
Cardiol Young. 19:501–506. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Huynh K, Kiriazis H, Du XJ, Love JE,
Jandeleit-Dahm KA, Forbes JM, McMullen JR and Ritchie RH: Coenzyme
Q10 attenuates diastolic dysfunction, cardiomyocyte hypertrophy and
cardiac fibrosis in the db/db mouse model of type 2 diabetes.
Diabetologia. 55:1544–1553. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Navas P, Villalba JM and de Cabo R: The
importance of plasma membrane coenzyme Q in aging and stress
responses. Mitochondrion. 7 Suppl:S34–S40. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Alp NJ and Channon KM: Regulation of
endothelial nitric oxide synthase by tetrahydrobiopterin in
vascular disease. Arterioscler Thromb Vasc Biol. 24:413–420. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Forsgren M, Attersand A, Lake S, Grünler
J, Swiezewska E, Dallner G and Climent I: Isolation and functional
expression of human COQ2, a gene encoding a polyprenyl transferase
involved in the synthesis of CoQ. Biochem J. 382:519–526. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Bentinger M, Tekle M, Brismar K, Chojnacki
T, Swiezewska E and Dallner G: Stimulation of coenzyme Q synthesis.
Biofactors. 32:99–111. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Kalén A, Appelkvist EL, Chojnacki T and
Dallner G: Nonaprenyl-4-hydroxybenzoate transferase, an enzyme
involved in ubiquinone biosynthesis, in the endoplasmic
reticulum-Golgi system of rat liver. J Biol Chem. 265:1158–1164.
1990.PubMed/NCBI
|
|
13
|
Fulton D, Fontana J, Sowa G, Gratton JP,
Lin M, Li KX, Michell B, Kemp BE, Rodman D and Sessa WC:
Localization of endothelial nitric-oxide synthase phosphorylated on
serine 1179 and nitric oxide in Golgi and plasma membrane defines
the existence of two pools of active enzyme. J Biol Chem.
277:4277–4284. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Tsai KL, Huang YH, Kao CL, Yang DM, Lee
HC, Chou HY, Chen YC, Chiou GY, Chen LH, Yang YP, et al: A novel
mechanism of coenzyme Q10 protects against human endothelial cells
from oxidative stress-induced injury by modulating NO-related
pathways. J Nutr Biochem. 23:458–468. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
McGarvey TW, Nguyen T, Puthiyaveettil R,
Tomaszewski JE and Malkowicz SB: TERE1, a novel gene affecting
growth regulation in prostate carcinoma. Prostate. 54:144–155.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nakagawa K, Hirota Y, Sawada N, Yuge N,
Watanabe M, Uchino Y, Okuda N, Shimomura Y, Suhara Y and Okano T:
Identification of UBIAD1 as a novel human menaquinone-4
biosynthetic enzyme. Nature. 468:117–121. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Hirota Y, Tsugawa N, Nakagawa K, Suhara Y,
Tanaka K, Uchino Y, Takeuchi A, Sawada N, Kamao M, Wada A, et al:
Menadione (vitamin K3) is a catabolic product of oral phylloquinone
(vitamin K1) in the intestine and a circulating precursor of tissue
menaquinone-4 (vitamin K2) in rats. J Biol Chem. 288:33071–33080.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mugoni V, Postel R, Catanzaro V, De Luca
E, Turco E, Digilio G, Silengo L, Murphy MP, Medana C, Stainier DY,
et al: Ubiad1 is an antioxidant enzyme that regulates eNOS activity
by CoQ10 synthesis. Cell. 152:504–518. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
McGarvey TW, Nguyen T, Tomaszewski JE,
Monson FC and Malkowicz SB: Isolation and characterization of the
TERE1 gene, a gene down-regulated in transitional cell carcinoma of
the bladder. Oncogene. 20:1042–1051. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Hegarty JM, Yang H and Chi NC:
UBIAD1-mediated vitamin K2 synthesis is required for vascular
endothelial cell survival and development. Development.
140:1713–1719. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Nakagawa K, Sawada N, Hirota Y, Uchino Y,
Suhara Y, Hasegawa T, Amizuka N, Okamoto T, Tsugawa N, Kamao M, et
al: Vitamin K2 biosynthetic enzyme, UBIAD1 is essential for
embryonic development of mice. PLoS One. 9:e1040782014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cooper SA, Whaley-Connell A, Habibi J, Wei
Y, Lastra G, Manrique C, Stas S and Sowers JR:
Renin-angiotensin-aldosterone system and oxidative stress in
cardiovascular insulin resistance. Am J Physiol Heart Circ Physiol.
293:H2009–H2023. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Nakashima H, Suzuki H, Ohtsu H, Chao JY,
Utsunomiya H, Frank GD and Eguchi S: Angiotensin II regulates
vascular and endothelial dysfunction: Recent topics of Angiotensin
II type-1 receptor signaling in the vasculature. Curr Vasc
Pharmacol. 4:67–78. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Olivares-Reyes JA, Arellano-Plancarte A
and Castillo-Hernandez JR: Angiotensin II and the development of
insulin resistance: Implications for diabetes. Mol Cell Endocrinol.
302:128–139. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Carvalheira JB, Calegari VC, Zecchin HG,
Nadruz W Jr, Guimarães RB, Ribeiro EB, Franchini KG, Velloso LA and
Saad MJ: The cross-talk between angiotensin and insulin
differentially affects phosphatidylinositol 3-kinase- and
mitogen-activated protein kinase-mediated signaling in rat heart:
Implications for insulin resistance. Endocrinology. 144:5604–5614.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Habibi J, Whaley-Connell A, Hayden MR,
DeMarco VG, Schneider R, Sowers SD, Karuparthi P, Ferrario CM and
Sowers JR: Renin inhibition attenuates insulin resistance,
oxidative stress, and pancreatic remodeling in the transgenic Ren2
rat. Endocrinology. 149:5643–5653. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Koh KK, Quon MJ, Lee Y, Han SH, Ahn JY,
Chung WJ, Kim JA and Shin EK: Additive beneficial cardiovascular
and metabolic effects of combination therapy with ramipril and
candesartan in hypertensive patients. Eur Heart J. 28:1440–1447.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Lastra G, Whaley-Connell A, Manrique C,
Habibi J, Gutweiler AA, Appesh L, Hayden MR, Wei Y, Ferrario C and
Sowers JR: Low-dose spironolactone reduces reactive oxygen species
generation and improves insulin-stimulated glucose transport in
skeletal muscle in the TG(mRen2)27 rat. Am J Physiol Endocrinol
Metab. 295:E110–E116. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Lin LY, Lin CY, Su TC and Liau CS:
Angiotensin II-induced apoptosis in human endothelial cells is
inhibited by adiponectin through restoration of the association
between endothelial nitric oxide synthase and heat shock protein
90. FEBS Lett. 574:106–110. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Landmesser U, Engberding N, Bahlmann FH,
Schaefer A, Wiencke A, Heineke A, Spiekermann S, Hilfiker-Kleiner
D, Templin C, Kotlarz D, et al: Statin-induced improvement of
endothelial progenitor cell mobilization, myocardial
neovascularization, left ventricular function, and survival after
experimental myocardial infarction requires endothelial nitric
oxide synthase. Circulation. 110:1933–1939. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Langsjoen PH, Langsjoen A, Willis R and
Folkers K: Treatment of hypertrophic cardiomyopathy with coenzyme
Q10. Mol Aspects Med. 18 Suppl:S145–S151. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Lakó-Futó Z, Szokodi I, Sármán B, Földes
G, Tokola H, Ilves M, Leskinen H, Vuolteenaho O, Skoumal R,
deChâtel R, et al: Evidence for a functional role of angiotensin II
type 2 receptor in the cardiac hypertrophic process in vivo in the
rat heart. Circulation. 108:2414–2422. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Orr A, Dubé MP, Marcadier J, Jiang H,
Federico A, George S, Seamone C, Andrews D, Dubord P, Holland S, et
al: Mutations in the UBIAD1 gene, encoding a potential
prenyltransferase, are causal for Schnyder crystalline corneal
dystrophy. PLoS One. 2:e6852007. View Article : Google Scholar : PubMed/NCBI
|