Introduction
A number of pathogenic factors, including
environmental risk factor exposure, aging, genetic defects and
metabolic disorders, are frequently associated with the development
of atherosclerosis. As atherosclerotic lesions primarily emerge and
develop locally at typical arterial regions, including branched,
bifurcated and curvature sites, the local hemodynamics of arteries
maybe taken into consideration when investigating the pathogenesis
of atherosclerosis (1). Generally,
two types of blood flow patterns characterize the local arterial
hemodynamics: The steady laminar blood flow (s-flow), and the
disturbed blood flow (d-flow) (2).
Vascular regions dominated by s-flow are relatively
atheroresistant; however, d-flow, comprising transient flow,
separated flow and reversal, frequently results in shear stress,
which makes the subjected vascular region atherosusceptible
(3).
Vascular endothelial cells (ECs) form the inner wall
of arterial vessels, and are therefore directly exposed to the
blood flow. Under mechanical stimuli, including d-flow, ECs
modulate various biological responses, including vasoconstriction
maladjustment, local inflammation, oxidative stress and cellular
apoptosis (4). Injury to the
arterial endothelium serves an important role in the development of
inflammation, redox and coagulation, making the vessel
atherosusceptible (5). A variety
of signal transduction pathways may lead to the apoptosis of ECs.
It has previously been demonstrated that the mitochondria-induced,
death receptor-induced and endoplasmic reticulum stress-induced
pathways are the three primary terminal apoptotic signaling
pathways (6–8).
The majority of newly-synthesized proteins are
folded and matured in the endoplasmic reticulum (ER). When the cell
is exposed to stressful conditions, the folding and maturation
processes cease and the unfolded protein response is initiated.
When the accumulation of unfolded/misfolded proteins exceeds the
capacity of the ER to correct the assembly of abnormally-folded
proteins, the ER stress response is triggered to provoke multiple
pathological alterations (9). As
one of the molecular chaperones of ER, 78 kDa glucose-regulated
protein (GRP78) binds to stress sensors of the ER (serine/threonine
protein kinase/endoribonuclease IRE1, eukaryotic translation
initiation factor 2-α kinase 3 and cyclic AMP-dependent
transcription factor ATF6-α) to keep the apoptotic signaling
inactive under normal physiological conditions. However, under
stressful conditions, GRP78 disassociates from the stress signal
transducers to trigger the activation of their signaling pathways.
Previous studies have demonstrated that ER stress may induce
cellular apoptosis through the DNA damage-inducible transcript 3
protein (CHOP) pathway (10).
Activated CHOP is able to initiate the translocation of caspase12
from the ER to the plasma in order to trigger the activation of the
caspase cascade (11). CHOP is
generally considered to be a pro-apoptotic factor (12). A previous study demonstrated that
exposure of cultured ECs to d-flow results in an increase in the
expression level of GRP78, indicating that d-flow is one of the
pathological initiators of ER stress. These previous results formed
the basis of the investigation into the involvement of ER stress in
the apoptosis of ECs exposed to d-flow (13).
In addition, it has been hypothesized that
inflammation may be important in accelerating and exacerbating the
formation and rupture of atherosclerotic plaques (14). Inflammatory cytokines, including
tumor necrosis factor-α (TNF-α) and interleukins (ILs) are
important transducers and amplifiers of these processes (15). It was reported that TNF-α may
further recruit and stimulate the expression of other ILs to
exacerbate inflammation (16). The
IL-1 family is associated with the occurrence and development of
numerous inflammatory diseases. A previous study demonstrated that
the use of antagonists, including interleukin-1 receptor antagonist
protein (IL-1Ra) and interleukin-36 receptor antagonist protein was
able to relieve local and systemic inflammation (17). A recent study observed that IL-1β
promoted ER stress-induced cellular apoptosis via interleukin-1
receptor associated kinase 2 (IRAK2) (18). In the present study, using an
artificial in vitro model to mimic d-flow, cultured human
aortic endothelial cells (HAECs) were exposed to shear stress. By
using an ER stress inhibitor and specific small interfering (si)RNA
against IRAK2, the involvement of IRAK2/CHOP signaling in ER
stress-mediated shear stress-induced cellular apoptosis was
investigated.
Materials and methods
Cell culture and treatment
HAECs were purchased from the Type Culture
Collection of the Chinese Academy of Sciences (Shanghai, China) and
cultured in Dulbecco's modified Eagle's medium (Gibco; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) supplemented with 10%
fetal bovine serum (Gibco; Thermo Fisher Scientific, Inc.).
L-glutamine (2 mmol/l; Invitrogen; Thermo Fisher Scientific, Inc.)
and antibiotics (1% penicillin/streptomycin; Invitrogen; Thermo
Fisher Scientific, Inc.). Cells were cultured in a cell incubator
providing a humidified environment, with 5% CO2 and 95%
air at 37°C. Equal amounts of cells were divided into 7 groups,
namely the control group (Control), 4-phenylbutyrate
(4-PBA)-treated group (4-PBA), d-flow-exposed group (d-flow),
siRNA-treated group (siRNA), IL-1Ra-treated d-flow-exposed group
(IL-1Ra+d-flow), 4-PBA-treated d-flow-exposed group (4-PBA+d-flow)
and siRNA treated d-flow-exposed group (d-flow+siRNA). Cell
treatments are listed in Table
I.
 | Table I.Cell grouping and treatments. |
Table I.
Cell grouping and treatments.
| Groups | Treatment
reagent | Description |
|---|
| Control | Medium | Treated with control
medium |
| 4-PBA | 4-PBA | Treated with 4-PBA at
concentration of 0.6 mmol/l |
| siRNA | siRNA | Specific siRNA
against IRAK2 |
| d-flow | d-flow | Artificial d-flow
model |
| d-flow+IL-1Ra | IL-1Ra and
d-flow | Treated with IL-1Ra
at concentration of 200 ng/ml when exposed to d-flow |
| d-flow+4PBA | 4-PBA and d-flow | Treated with 4-PBA
when exposed to d-flow |
| d-flow+siRNA | siRNA and d-flow | Pretreated with siRNA
against IRAK2 and subse quently exposed to d-flow |
Target gene silencing using siRNA
In accordance with a previous study, IRAK2 in HAECs
was silenced using siRNA (18).
The sequence of the siRNA against IRAK2 was
5′-CTTCGCCTCCTACGTGATCAC-3′, and was acquired from Shanghai
GenePharma Co., Ltd. (Shanghai, China). A scramble sequence,
5′-GAACAGACGACGTTGACAA-3′, was used as the scramble control.
According to the manufacturer's protocol, using HiPerFect siRNA
transfection reagent (Qiagen GmbH, Hilden, Germany), the siRNAs
were transfected into HAECs at final concentrations of 12.5 mmol/l.
Cells were used for subsequent experiments following 24 h
culturing.
Artificial d-flow model
The artificial d-flow model was created using a
cone-and-plate chamber according to the description in a previous
report (19). This apparatus
consists of a cone rotating about its center axis in a tissue
culture dish to produce stable laminar flow. The components of the
cone-and-late apparatus were produced at the College of Machinery
of Xi'an Jiaotong University (Xi'an, China). The HAECs were exposed
to d-flow for 0.5, 1, 2, 3 and 4 h.
Cellular apoptosis assay
Cellular apoptosis was detected using a terminal
deoxynucleotidyl transferase dUTP nick end labeling (TUNEL) assay.
The In Situ Cell Death Detection kit (Roche Diagnostics,
Basel, Switzerland) was used to perform the detection. Following
fixation with 4% paraformaldehyde for 12 h at room temperature
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), cultured HAECs
were permeabilized using 1% Triton X-100 (Sigma-Aldrich; Merck
KGaA). Cells were subsequently treated with TUNEL staining solution
for 1 h at 37°C in accordance with manufacturer's protocol. After
washing the slides five times in PBS and mounted with 3% hydrogen
peroxide in formaldehyde, a fluorescence microscope was used to
observe TUNEL positive cells. Five fields of view were
selected.
IL-1β concentration detection
IL-1β concentration in the cell culture supernatants
was measured using an ELISA assay, with a Quantkine Rat IL-1β ELISA
kit (catalog no. DLB50; R&D Systems, Inc., Minneapolis, MN,
USA). The measurements were carried out according to the
manufacturer's protocol. Standard curves and absorbance values were
used to calculate the concentrations.
Western blotting
Harvested HAECs were washed and lysed in
radioimmunoprecipitation assay buffer (Santa Cruz Biotechnology,
Inc., Dallas, TX, USA) supplemented with phenylmethylsulfonyl
fluoride (0.5 mmol/l; Santa CruzBiotechnology, Inc.),
dithiothreitol (1 mmol/l; Beyotime Institute of Biotechnology,
Haimen, China) and protease inhibitor (150 mmol/l; Invitrogen;
Thermo Fisher Scientific, Inc.). Protein concentrations were
determined using the colorimetric Bradford assay (Pierce; Thermo
Fisher Scientific, Inc.). A total of 10 µg protein was loaded onto
10% gels and subjected to SDS-PAGE, and the loaded proteins were
separated by vertical electrophoresis. Proteins were subsequently
electronically transferred to polyvinylidene fluoride or
nitrocellulose membranes (EMD Millipore, Billerica, MA, USA).
Following blocking for 1 h in 5% non-fat milk in TBST for 1 h at
25°C, specific antibodies against IL-1β (catalog no. sc-130323;
Santa Cruz Biotechnology, Inc.; dilution 1:2,000), GRP78 (catalog
no. 3183; 1:2,000; Cell Signaling Technology, Inc., Danvers, MA,
USA), IRAK2 (catalog no. ab62419; 1:1,000; Abcam, Cambridge, UK),
CHOP (catalog no. ab10444; 1:2,000; Abcam), caspase 12 (catalog no.
ab62484; 1:1,000; Abcam) and GAPDH (catalog no. ab8245; 1:5,000;
Abcam) were used to incubate the membranes for 12 h at 4°C.
Corresponding horseradish peroxidase-conjugated secondary
antibodies (catalog no. ab7010, Abcam; catalog no. sc2364 and
sc3747; Santa Cruz Biotechnology, Inc.) were used to incubate the
membranes for 1 h at room temperature, which were illuminated using
SuperSignal West Pico enhanced chemiluminescence reagents (Pierce;
Thermo Fisher Scientific, Inc.).
Statistical analysis
Data acquired in the present study are presented as
the mean ± standard deviation of three independent experiments. The
data were analyzed using SPSS software, version 16.0 (SPSS, Inc.,
Chicago, IL, USA). Differences between groups were analyzed using
one-way analysis of variance followed by Bonferroni's post hoc
test. P<0.05 was considered to indicate a statistically
significant difference.
Results
d-flow triggers the activation of ER
stress, which induces HAEC apoptosis in a time-dependent
manner
As presented in Fig.
1, following exposure to d-flow, ER stress-mediated apoptotic
signaling was observed to be activated in HAECs in a time-dependent
manner. The expression levels of the molecular markers of this
pathway, GRP78 and CHOP, were observed to be significantly
increased in a time-dependent manner. The actual time-dependent
d-flow induced apoptosis was observed using a TUNEL assay.
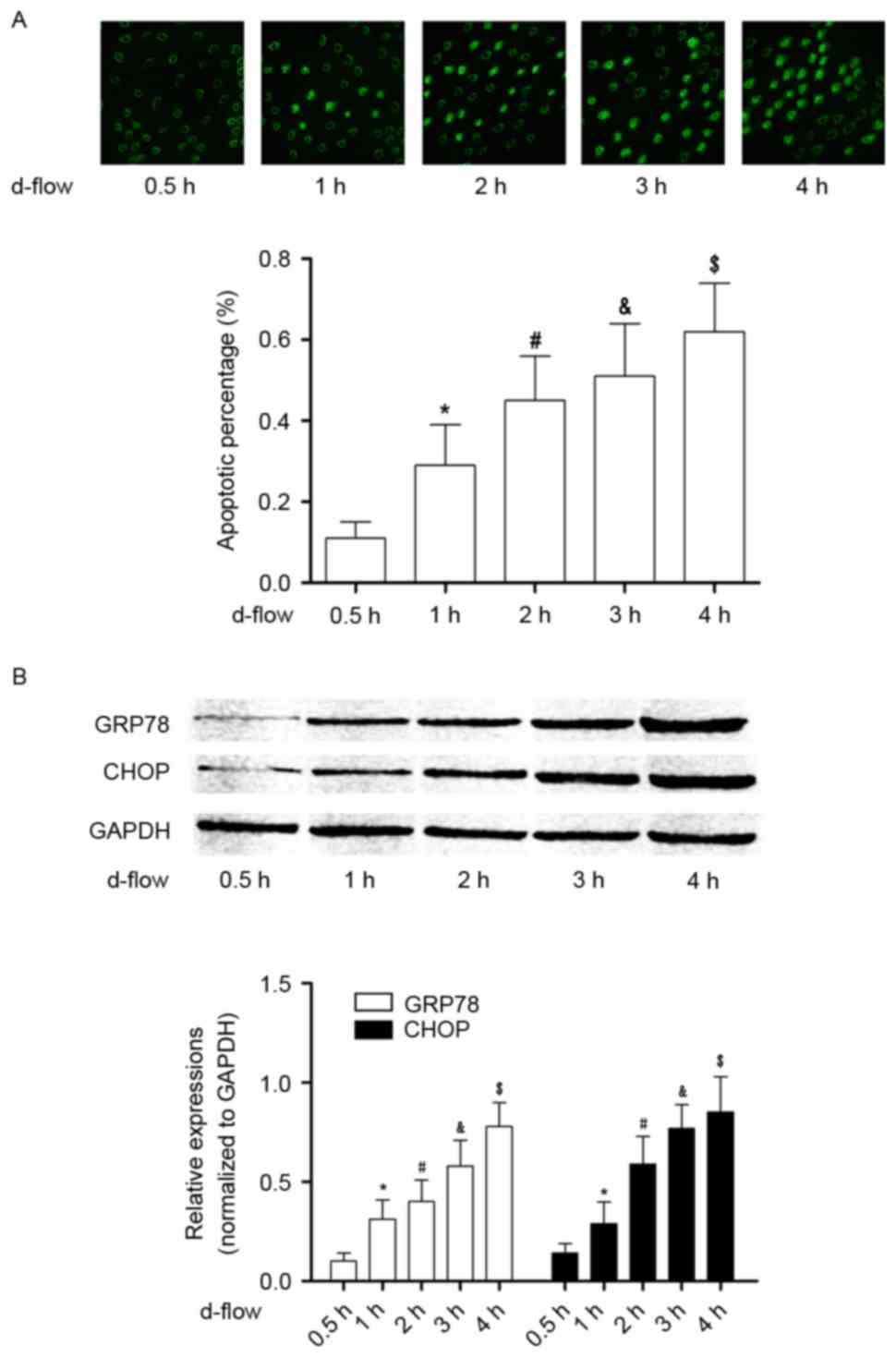 | Figure 1.Analysis of apoptosis in HAECs exposed
to d-flow. (A) The upper panel demonstrates the captured images
from the terminal deoxynucleotidyl transferase dUTP nick end
labeling assay of HAECs treated with d-flow for 0.5, 1, 2, 3 and 4
h. The graph on the lower panel indicates the detected apoptotic
percentage of HAECs. Magnification, ×200. (B) The upper panel
exhibits the immunoblotting analysis of GRP78, CHOP and GAPDH in
HAECs treated with d-flow for 0.5, 1, 2, 3 and 4 h. The graph on
the lower panel presents the relative expression levels of GRP78
(white columns) and CHOP (black columns), with GAPDH as the
internal reference. *P<0.05 vs. 0.5 h; #P<0.05 vs.
1 h; &P<0.05 vs. 2 h; $P<0.05 vs. 3
h. HAECs, human aortic endothelial cells; d-flow, disturbed blood
flow; GRP78, 78 kDa glucose-regulated protein; CHOP, DNA
damage-inducible transcript 3 protein; d-flow, disturbed blood
flow. |
Exposure to d-flow increases the
synthesis and secretion of IL-1β in cultured HAECs, in a
time-dependent manner
As demonstrated in Fig.
2, the synthesis of IL-1β was significantly increased in
d-flow-treated HAECs in a time-dependent manner, which was
evidenced by an increased protein expression level in harvested
HAECs. Additionally, the secretion of IL-1β in d-flow-treated HAECs
was observed to be increased in a time-dependent manner, evidenced
by increased protein expression in the supernatant of cultured
HAECs.
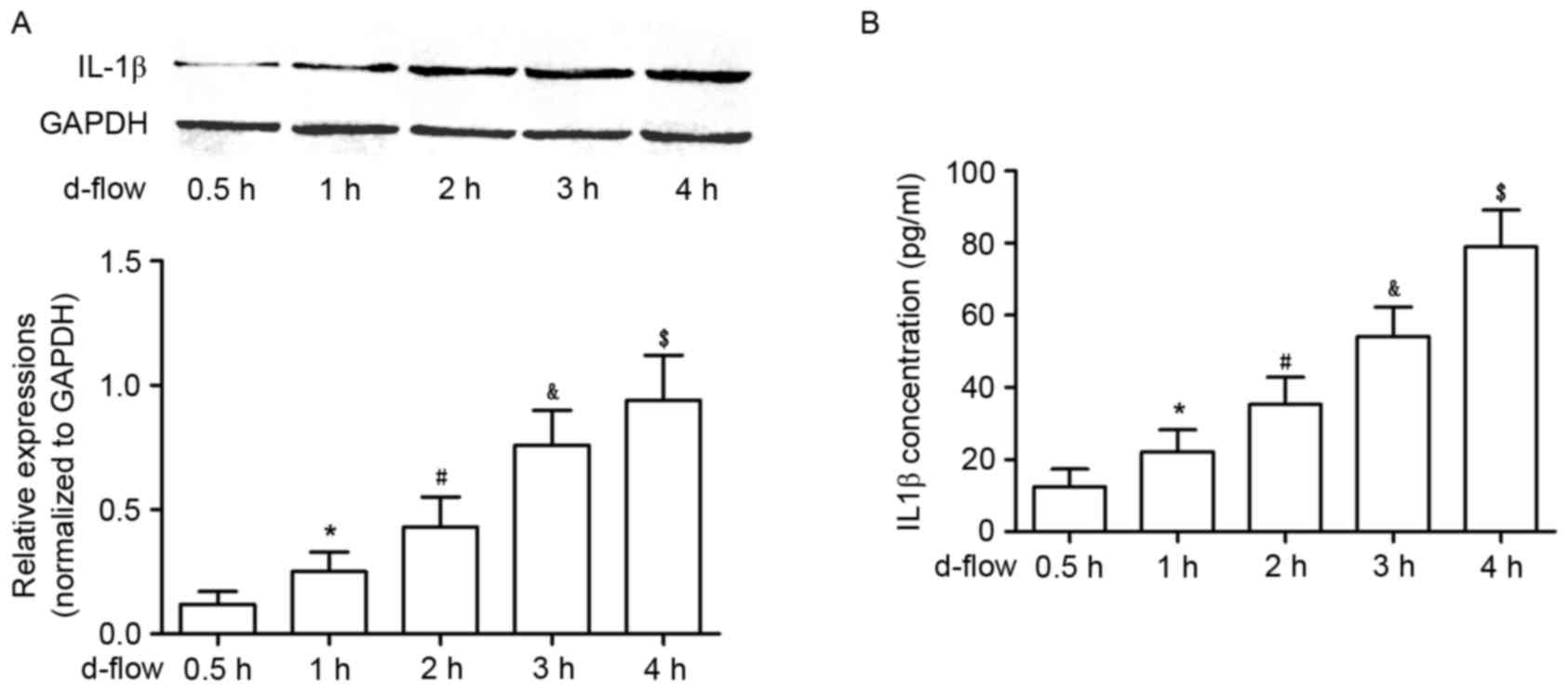 | Figure 2.Analysis of IL-1β expression in HAECs
exposed to d-flow. (A) The upper panel exhibits the representative
image of the western blotting of IL-1β and GAPDH in HAECs treated
with d-flow for 0.5, 1, 2, 3 and 4 h. The graph on the lower panel
indicates the relative expression of IL-1β in HAECs, with GAPDH as
the internal reference. (B) Results of the ELISA assay of IL-1β.
The graph exhibits the detected IL-1β concentrations in the
supernatant of cultured HAECs treated with d-flow for 0.5, 1, 2, 3
and 4 h. *P<0.05 vs. 0.5 h; #P<0.05 vs. 1 h;
&P<0.05 vs. 2 h; $P<0.05 vs. 3 h.
HAECs, human aortic endothelial cells; IL-1β, interleukin-1β;
d-flow, disturbed blood flow. |
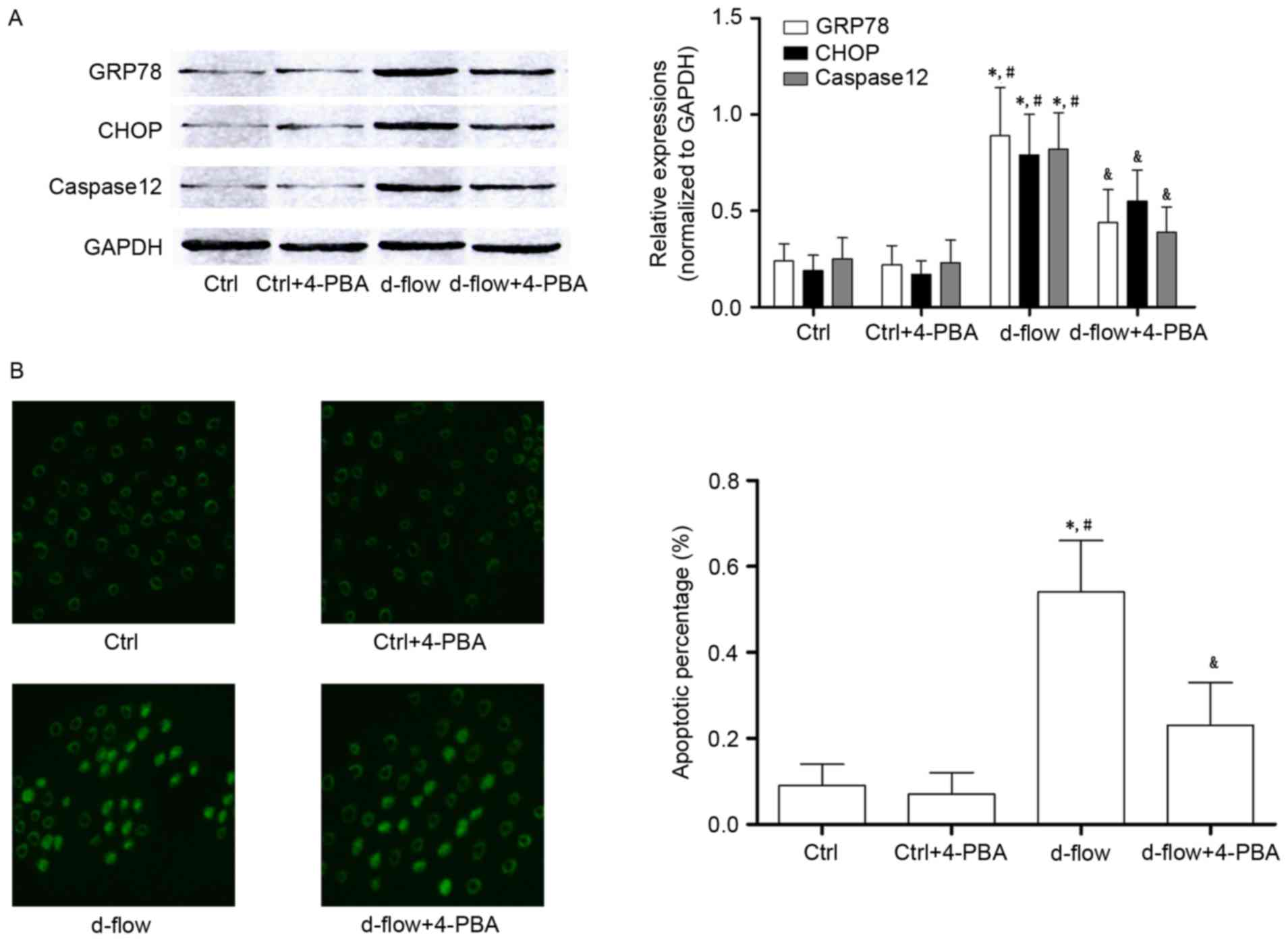
4-PBA incubation attenuates
d-flow-mediated HAEC death by suppressing ER stress
Treatment with 4-PBA treatment significantly
decreased the expression of GRP78 and CHOP, which were considered
to be molecular markers of ER stress-mediated apoptotic signaling.
Additionally, results from the TUNEL assay and the expression
levels of caspase12 demonstrated that the d-flow induced ER stress
mediated apoptosis was inhibited by 4-PBA in HAECs (Fig. 3).
IL-1Ra incubation lowers the apoptotic
rate of d-flow-exposed HAECs by inhibiting IRAK2/CHOP
signaling
The association between IL-1Ra and alterations in
d-flow-induced ER stress-mediated signal transduction and actual
apoptosis are demonstrated in Fig.
4. IL-1Ra did not affect cellular apoptosis (determined by
TUNEL assay and caspase12 expression level) in untreated HAECs.
However, the d-flow-induced apoptosis was significantly inhibited
by IL-1Ra administration. It was additionally observed that in
d-flow-treated HAECs, IL-1Ra treatment decreased the expression of
IRAK2 and CHOP without affecting the expression levels of GRP78 and
IL-1β. The results of the present study indicated that the IL-1Ra
inhibited apoptotic signal transduction during ER stress by
affecting the signal transduction of IRAK2/CHOP.
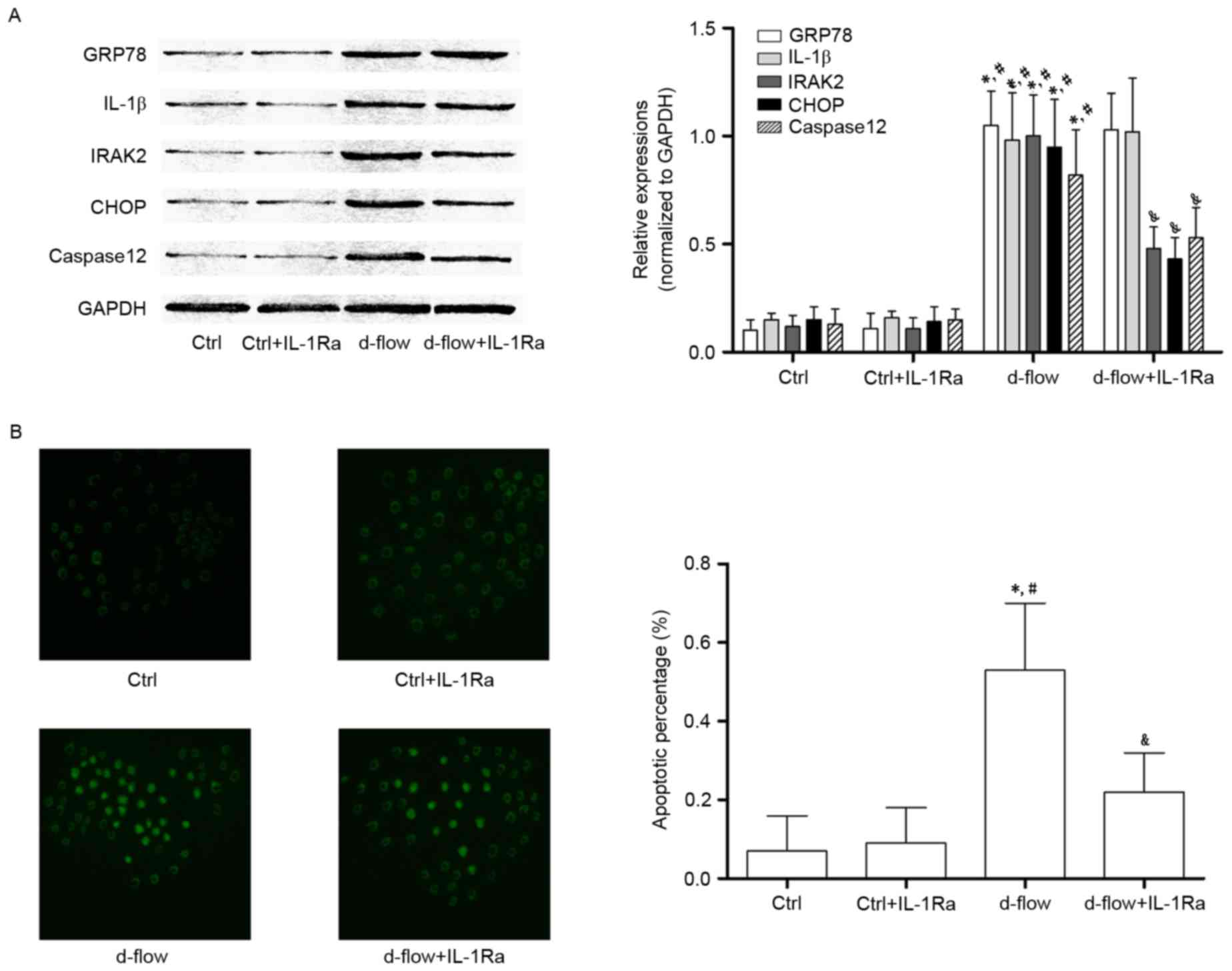 | Figure 4.Analysis of the effects of treatment
with IL-1Ra in HAECs exposed to d-flow. (A) Results of the western
blotting of GRP78, IL-1β, IRAK2, CHOP, caspase 12 and GAPDH in
HAECs treated with IL-1Ra and/or d-flow. The relative expressions
of GRP78 (white columns), IL-1β (light gray columns), IRAK2 (dark
gray columns) and CHOP (black columns) were normalized to GAPDH.
(B) Images were captured of the terminal deoxynucleotidyl
transferase dUTP nick end labeling assay of HAECs treated with
IL-1Ra and/or d-flow. The detected apoptotic percentages of HAECs
treated by IL-1Ra and/or d-flow were quantified. Magnification,
×200. *P<0.05 vs. Ctrl; #P<0.05 vs. Ctrl+IL-1Ra;
&P<0.05 vs. d-flow. IL-1Ra, interleukin-1
receptor antagonist protein; GRP78, 78 kDa glucose-regulated
protein; CHOP, DNA damage-inducible transcript 3 protein; d-flow,
disturbed blood flow; HAECs, human aortic endothelial cells; Ctrl,
control; IRAK2, interleukin-1 receptor associated kinase 2; IL-1β,
interleukin-1β. |
siRNA against IRAK2 attenuates
d-flow-mediated HAEC apoptosis by blocking IRAK2/CHOP
signaling
In order to further confirm the role of IRAK2/CHOP
signaling in mediating d-flow-induced HAEC apoptosis, RNA
interference was performed. siRNA against IRAK2 was transfected
into HAECs. As presented in Fig.
5, IRAK2 siRNA transfection exhibited apparent inhibitory
effects on cellular apoptosis (determined by TUNEL assay and
caspase12 expression level analysis) induced by d-flow. IRAK2 siRNA
transfection markedly inhibited the expression levels of CHOP in
d-flow treated HAECs. However, the expression levels of GRP78 and
IL-1β were not affected by IRAK2 siRNA transfection.
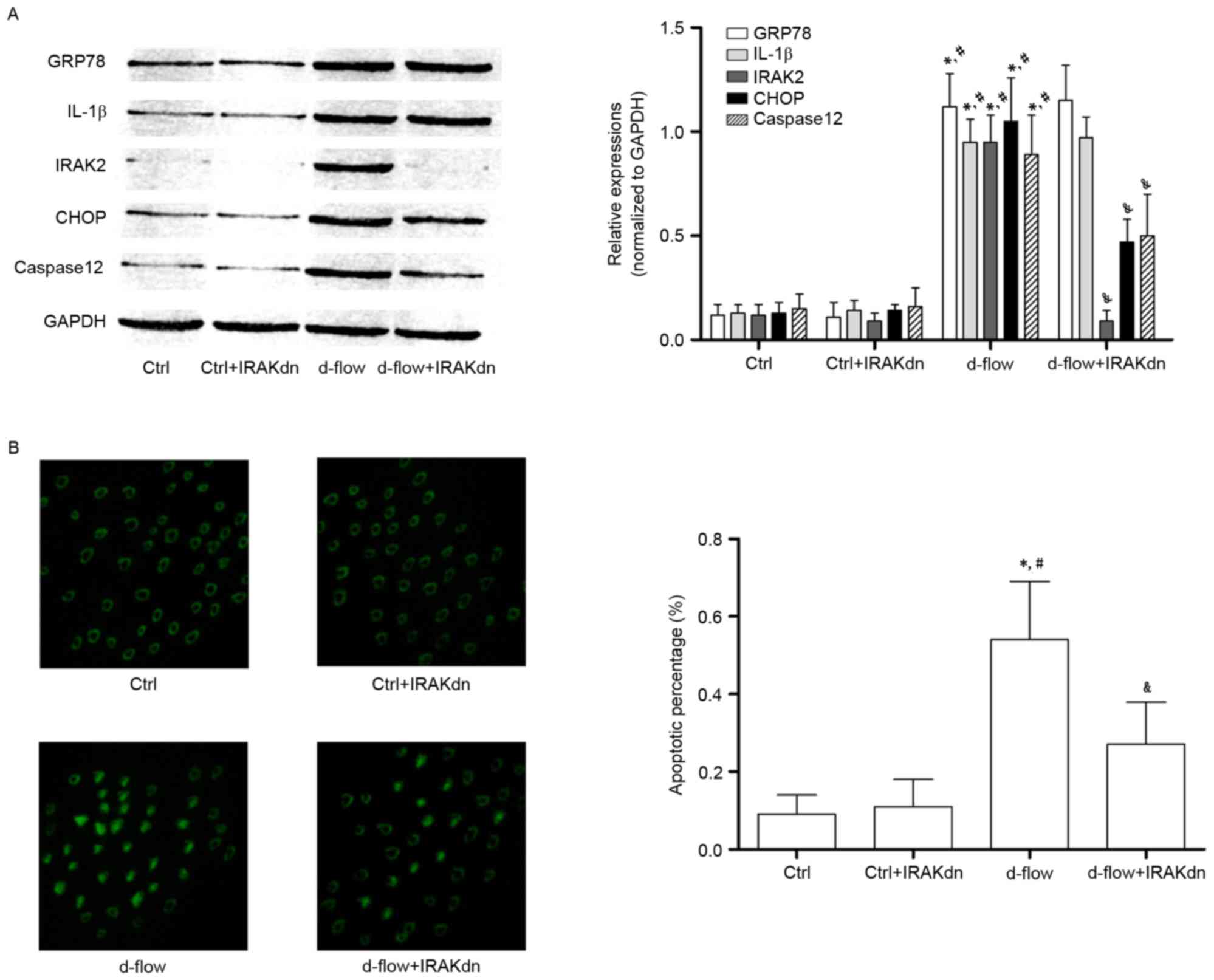 | Figure 5.Analysis of the effects of treatment
with IRAK2 siRNA in HAECs exposed to d-flow. (A) Results of the
immunoblotting of GRP78, IL-1β, IRAK2, CHOP, caspase 12 and GAPDH
in HAECs treated with IRAK2 siRNA and/or d-flow. The relative
expression of GRP78 (white columns), IL-1β (light gray columns),
IRAK2 (dark gray columns) and CHOP (black columns) was normalized
to GAPDH. (B) Images were captured of the terminal deoxynucleotidyl
transferase dUTP nick end labeling assay of HAECs treated with
IRAK2 siRNA and/or d-flow. The detected apoptotic percentages of
HAECs treated with IRAK2 siRNA and/or d-flow were quantified.
Magnification, ×200. *P<0.05 vs. Ctrl; #P<0.05 vs.
Ctrl+IRAKdn; &P<0.05 vs. d-flow. GRP78, 78 kDa
glucose-regulated protein; CHOP, DNA damage-inducible transcript 3
protein; d-flow, disturbed blood flow; HAECs, human aortic
endothelial cells; Ctrl, control; IRAK2, interleukin-1 receptor
associated kinase 2; IL-1β, interleukin-1β; IRAKdn, IRAK
downregulated; siRNA, small interfering RNA. |
Discussion
Atherosclerotic vascular disease-associated
mortality represents a large proportion of cerebrovascular and
cardiovascular mortalities worldwide. In the arterial tree,
distinct lesions may be observed in different regions due to
regional differences in shear stress forces produced by blood flow
(20). A previous study indicated
that exposure to d-flow triggers pathological alterations which
render ECs dysfunctional and proatherogenic (21). Apoptosis in the endothelium
modulates biological alterations within the vessel wall, including
deregulation of vascular wall remodeling, decreased production of
vasorelaxant substances and initiation of the coagulation cascade
and pro-inflammatory processes (22–24).
These alterations have been hypothesized to be the mechanisms
underlying atherosclerosis. In the present study, cultured HAECs
were exposed to d-flow generated by an artificial device. It was
observed that d-flow induced the apoptosis of HAECs in a
time-dependent manner.
The ER is an important cellular organelle, primarily
involved in directing protein folding, modification and maturation.
Under certain pathological circumstances, when ER stress is severe
and prolonged, signal transductions are activated to trigger
multiple biological effects (25).
In vivo and in vitro investigations have demonstrated
the association between ER stress and atherosclerosis (26). Administration of an exogenous ER
stress activator was observed to promote the formation of
atherosclerotic plaques (27).
Evidence of ER stress activation has additionally been observed
within the human atherosclerotic endothelium (28). In addition, ER stress was
demonstrated to induce apoptosis in endothelial cells (29). Therefore, it may be hypothesized
that d-flow-induced EC apoptosis is mediated by ER stress. The
present study demonstrated that the expression of GRP78, considered
to be the chaperone of ER stress, was increased significantly in
HAECs following exposure to d-flow. The expression of the
pro-apoptotic factor of ER stress, CHOP, was additionally observed
to be elevated in d-flow-treated HAECs. The results of the present
study indicated that d-flow-induced apoptosis was mediated by ER
stress. 4-PBA is one of the specific suppressors of ER stress. In
the present study, treatment with 4-PBA inhibited the increased
expression levels of GRP78 and CHOP. Therefore, 4-PBA impaired
d-flow-induced ER stress-mediated HAEC apoptosis. The results of
the present study further supported the hypothesis that
d-flow-induced HAEC apoptosis is mediated by ER stress.
It is widely accepted that atherosclerosis is
induced and promoted by molecular and cellular pathways of
inflammation. Numerous inflammatory cytokines, including TNF-α,
tumor growth factor-β and IL-1 provide mechanisms that may be able
to alter the arterial biology of the endothelium to favor plaque
formation and atherothrombotic events (30). In a previous study, inflammation
and ER stress were hypothesized to be associated as they share a
number of effectors and regulators (31). A ‘vicious cycle’ was proposed,
since inflammatory and ER stress signals were observed to
exacerbate cellular apoptosis via their interactions (32). In the present study, it was
demonstrated that, following d-flow exposure, expression level of
IL-1β in HAECs and their supernatant increased significantly. ER
stress and inflammatory signaling were simultaneously initiated.
However, following treatment with the IL-1β receptor antagonist
IL-1Ra, cellular apoptosis was attenuated. IL-1Ra treatment
decreased the expression level of CHOP in d-flow-treated cells. The
results of the present study indicated that IL-1β promoted ER
stress-mediated cellular apoptosis via the CHOP signaling
pathway.
By binding to its receptor IL-1R, IL-1β activates
downstream signaling transduction by forming the IL-1R-AcP complex
with an accessory protein (AcP). The C-terminal death domain of
this complex couples IRAKs and subsequently activates TNF receptor
associated factor-6 to initiate the apoptotic pathway (33,34).
The close association between ER stress-mediated apoptosis and
IRAK2 was identified by a previous study (18), the results of which demonstrated
that the expression of IRAK2 increases in conditions of ER stress.
In addition, increased IRAK2 promotes CHOP expression, which
transduces the death signal in ER stress-mediated apoptosis. In the
present study, RNA interference was used to knockdown the
expression of IRAK2. The results of the present study demonstrated
that the knockdown of IRAK2 significantly inhibited the
d-flow-induced apoptosis of HAECs. It was additionally observed
that the knockdown of IRAK2 suppressed the expression of CHOP
without an apparent affect on GRP78 and IL-1β. The results of the
present study indicated that the secreted IL-1β in d-flow-treated
HAECs was involved in exacerbating apoptosis through the IRAK2/CHOP
signaling pathway.
In conclusion, by activating ER stress, exposure to
d-flow induced HAEC apoptosis via the CHOP signaling pathway. The
inflammatory cytokine IL-1β was produced in d-flow-treated HAECs.
IL-1β exacerbated d-flow-induced cellular apoptosis. In addition,
the apoptotic signal was transduced via the IL-1β/IRAK2/CHOP
signaling pathway. This signaling pathway may be considered to be
crosstalk between ER stress and inflammation under conditions of
disturbed blood flow. The results of the present study may provide
a theoretical basis for the identification of potential molecular
targets for the treatment of atherosclerosis.
Acknowledgements
The present study was supported by the project fund
of Shaanxi Province Science and Technology Key Projects (grant no.
2016YFJH2-05).
References
|
1
|
Gimbrone MA Jr and García-Cardeña G:
Vascular endothelium, hemodynamics, and the pathobiology of
atherosclerosis. Cardiovasc Pathol. 22:9–15. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Heo KS, Le NT, Cushman HJ, Giancursio CJ,
Chang E, Woo CH, Sullivan MA, Taunton J, Yeh ET, Fujiwara K and Abe
J: Disturbed flow-activated p90RSK kinase accelerates
atherosclerosis by inhibiting SENP2 function. J Clin Invest.
125:1299–1310. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
Xu Q: Disturbed flow-enhanced endothelial
turnover in atherosclerosis. Trends Cardiovasc Med. 19:191–195.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Heo KS, Fujiwara K and Abe J:
Disturbed-flow-mediated vascular reactive oxygen species induce
endothelial dysfunction. Circ J. 75:2722–2730. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
van der Heiden K, Hierck BP, Krams R, de
Crom R, Cheng C, Baiker M, Pourquie MJ, Alkemade FE, DeRuiter MC,
Gittenberger-de Groot AC and Poelmann RE: Endothelial primary cilia
in areas of disturbed flow are at the base of atherosclerosis.
Atherosclerosis. 196:542–550. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hida A, Kawakami A, Miyashita T, Yamasaki
S, Nakashima K, Tanaka F, Izumi Y, Tamai M, Huang M, Ida H, et al:
Nitric oxide acts on the mitochondria and protects human
endothelial cells from apoptosis. J Lab Clin Med. 144:148–155.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qiu ZL, Zhang JP and Guo XC: Endoplasmic
reticulum stress and vascular endothelial cell apoptosis. Zhongguo
Yi Xue Ke Xue Yuan Xue Bao. 36:102–107. 2014.(In Chinese).
PubMed/NCBI
|
|
8
|
Skurk C and Walsh K: Death receptor
induced apoptosis: A new mechanism of homocysteine-mediated
endothelial cell cytotoxicity. Hypertension. 43:1168–1170. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Pluquet O, Pourtier A and Abbadie C: The
unfolded protein response and cellular senescence. A review in the
theme: Cellular mechanisms of endoplasmic reticulum stress
signaling in health and disease. Am J Physiol Cell Physiol.
308:C415–C425. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma J, Qiu Y, Yang L, Peng L, Xia Z, Hou
LN, Fang C, Qi H and Chen HZ: Desipramine induces apoptosis in rat
glioma cells via endoplasmic reticulum stress-dependent CHOP
pathway. J Neurooncol. 101:41–48. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Lakshmanan AP, Thandavarayan RA,
Palaniyandi SS, Sari FR, Meilei H, Giridharan VV, Soetikno V,
Suzuki K, Kodama M and Watanabe K: Modulation of
AT-1R/CHOP-JNK-Caspase12 pathway by olmesartan treatment attenuates
ER stress-induced renal apoptosis in streptozotocin-induced
diabetic mice. Eur J Pharm Sci. 44:627–634. 2011.PubMed/NCBI
|
|
12
|
Nakagawa T, Zhu H, Morishima N, Li E, Xu
J, Yankner BA and Yuan J: Caspase-12 mediates
endoplasmic-reticulum-specific apoptosis and cytotoxicity by
amyloid-beta. Nature. 403:98–103. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Davies PF and Civelek M: Endoplasmic
reticulum stress, redox, and a proinflammatory environment in
athero-susceptible endothelium in vivo at sites of complex
hemodynamic shear stress. Antioxid Redox Signal. 15:1427–1432.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Bäck M, Weber C and Lutgens E: Regulation
of atherosclerotic plaque inflammation. J Intern Med. 278:462–482.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dewberry R, Holden H, Crossman D and
Francis S: Interleukin-1 receptor antagonist expression in human
endothelial cells and atherosclerosis. Arterioscler Thromb Vasc
Biol. 20:2394–2400. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Balato A, Di Caprio R, Canta L, Mattii M,
Lembo S, Raimondo A, Schiattarella M, Balato N and Ayala F: IL-33
is regulated by TNF-α in normal and psoriatic skin. Arch Dermatol
Res. 306:299–304. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Mattii M, Ayala F, Balato N, Filotico R,
Lembo S, Schiattarella M, Patruno C, Marone G and Balato A: The
balance between pro- and anti-inflammatory cytokines is crucial in
human allergic contact dermatitis pathogenesis: The role of IL-1
family members. Exp Dermatol. 22:813–819. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Liu Z, Zhao N, Zhu H, Zhu S, Pan S, Xu J,
Zhang X, Zhang Y and Wang J: Circulating interleukin-1β promotes
endoplasmic reticulum stress-induced myocytes apoptosis in diabetic
cardiomyopathy via interleukin-1 receptor-associated kinase-2.
Cardiovasc Diabetol. 14:1252015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Reinhart-King CA, Fujiwara K and Berk BC:
Physiologic stress-mediated signaling in the endothelium. Methods
Enzymol. 443:25–44. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Heo KS, Fujiwara K and Abe J: Shear stress
and atherosclerosis. Mol Cells. 37:435–440. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Davies PF, Civelek M, Fang Y, Guerraty MA
and Passerini AG: Endothelial heterogeneity associated with
regional athero-susceptibility and adaptation to disturbed blood
flow in vivo. Semin Thromb Hemost. 36:265–275. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang T, Tian F, Wang J, Jing J, Zhou SS
and Chen YD: Atherosclerosis-associated endothelial cell apoptosis
by MiR-429-mediated down regulation of Bcl-2. Cell Physiol Biochem.
37:1421–1430. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Zeng L, Zampetaki A, Margariti A, Pepe AE,
Alam S, Martin D, Xiao Q, Wang W, Jin ZG, Cockerill G, et al:
Sustained activation of XBP1 splicing leads to endothelial
apoptosis and atherosclerosis development in response to disturbed
flow. Proc Natl Acad Sci USA. 106:8326–8331. 2009; View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Choy JC, Granville DJ, Hunt DW and McManus
BM: Endothelial cell apoptosis: Biochemical characteristics and
potential implications for atherosclerosis. J Mol Cell Cardiol.
33:1673–1690. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Cao SS and Kaufman RJ: Endoplasmic
reticulum stress and oxidative stress in cell fate decision and
human disease. Antioxid Redox Signal. 21:396–413. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Hotamisligil GS: Endoplasmic reticulum
stress and atherosclerosis. Nat Med. 16:396–399. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tabas I: The role of endoplasmic reticulum
stress in the progression of atherosclerosis. Circ Res.
107:839–850. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ivanova EA and Orekhov AN: The role of
endoplasmic reticulum stress and unfolded protein response in
atherosclerosis. Int J Mol Sci. 17:pii:E1932016. View Article : Google Scholar
|
|
29
|
Lu JP, Li X, Jin YL and Chen MX:
Endoplasmic reticulum stress-mediated aldosterone-induced apoptosis
in vascular endothelial cells. J Huazhong Univ Sci Technolog Med
Sci. 34:821–824. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Krams R, Segers D, Gourabi B Mousavi, Maat
W, Cheng C, van Pelt C, van Damme LC, de Feyter P, Van Der Steen T,
de Korte CL and Serruys PW: Inflammation and atherosclerosis:
Mechanisms underlying vulnerable plaque. J Interv Cardiol.
16:107–113. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Dong Z, Zhou J, Zhang Y, Chen Y, Yang Z,
Huang G, Chen Y, Yuan Z, Peng Y and Cao T: Astragaloside-IV
alleviates Heat-induced inflammation by inhibiting endoplasmic
reticulum stress and autophagy. Cell Physiol Biochem. 42:824–837.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Benosman S, Ravanan P, Correa RG, Hou YC,
Yu M, Gulen MF, Li X, Thomas J, Cuddy M, Matsuzawa Y, et al:
Interleukin-1 receptor-associated kinase-2 (IRAK2) is a critical
mediator of endoplasmic reticulum (ER) stress signaling. PLoS One.
8:e642562013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Cenni V, Sirri A, De Pol A, Maraldi NM and
Marmiroli S: Interleukin-1-receptor-associated kinase 2
(IRAK2)-mediated interleukin-1-dependent nuclear factor kappaB
transactivation in Saos2 cells requires the Akt/protein kinase B
kinase. Biochem J. 376:303–311. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Meng Q, Zheng M, Liu H, Song C, Zhang W,
Yan J, Qin L and Liu X: TRAF6 regulates proliferation, apoptosis,
and invasion of osteosarcoma cell. Mol Cell Biochem. 371:177–186.
2012. View Article : Google Scholar : PubMed/NCBI
|