Introduction
Cardiac fibrosis, which may be induced by various
factors in cardiac tissue, is a result of excessive deposition of
extracellular matrix (ECM), and eventually leads to the destruction
of the physiological cardiac tissue architecture and heart failure
(1). Furthermore, as the fibrous
tissue increases, myocardial stiffness increases, which may
theoretically lead to decreased myocardial shortening. Systolic and
diastolic cardiac functions are associated with the degree of
cardiac fibrosis (2,3). Fibroblasts are the primary producers
of ECM and contribute substantially to myocardial fibrosis
(4). During cardiac remodeling,
fibroblasts differentiate into myofibroblasts, which are the
primary cell type involved in the reorganization of the ECM.
Myofibroblasts exhibit important contractile and secretory
functions, and are characterized by rapid proliferation and are
α-smooth muscle actin (α-SMA)-positive (5). Various growth factors may participate
in this process, including angiogenic factors, proteolytic enzymes
and fibrogenic cytokines (6). As
one of the major mediators of cardiac inflammation, it has been
demonstrated that Angiotensin II (Ang II) regulates vascular
constriction and influence cardiac function, particularly in the
process of cardiac remodeling (1,7).
MicroRNAs (miRNAs/miRs) are non-protein-encoding
small RNAs that are encoded in the genome, and negatively regulate
target gene expression at the post-transcriptional level (8,9). A
number of studies have demonstrated that the importance of these
small RNAs in disease initiation and progression was dependent on
the regulation of distinct disease-specific signal transduction
pathways (10,11). Increasing evidence has indicated
that several miRNAs act as a novel class of regulators in certain
cardiovascular diseases (12).
Notably, the dysregulation of certain miRNAs regulates various
fibrosis-associated proteins to influence the development and
progression of cardiac fibrosis (13,14).
It has been reported that miR-155, one member of the small RNA
family, has an extensive and close association with circulatory
system diseases (15). A previous
report indicated that miR-155 acts as a key mediator of cardiac
injury and inflammation in atherosclerosis by repressing B-cell
CLL/lymphoma 6 in macrophages (16). Additionally, inhibition of miR-155
in mouse cardiomyocytes was associated with protection from cardiac
hypertrophy, and the repression of endogenous jumonji and AT-rich
interaction domain containing 2 (Jarid2) in isolated cardiomyocytes
partially rescued the effect of miR-155 loss, as previously
reported (17). In addition,
miR-155 expression in macrophages also promotes cardiac
inflammation, hypertrophy and failure in response to Ang II by
regulating suppressor of cytokine signaling 1 (SOCS1) (18). Furthermore, a recent study
demonstrated the proinflammatory effects of miR-155 in the
promotion of liver fibrosis induced by alcohol or carbon
tetrachloride and alcohol-induced steatohepatitis (19).
However, data concerning the effect of miR-155 on
cardiac fibrosis, particularly in cardiac fibroblasts, remains
limited. The present study reported that miR-155, a frequently
upregulated miRNA in hypertrophic hearts, was able to increase the
excessive deposition of ECM proteins during the process of cardiac
remodeling. The results indicated that manipulating the expression
of miR-155 may be a promising therapeutic strategy for cardiac
fibrosis.
Materials and methods
Animal studies
Male miR-155−/− mice from a C57BL/6
background were purchased from Jackson Laboratory (Ben Harbor, ME,
USA). Male wild-type (WT) C57BL/6 mice were purchased from Beijing
HFK Bioscience Co., Ltd (Beijing, China). All experiments were
conducted with the approval of the Animal Care Committee of the
Union Hospital of Tongji Medical College, Huazhong University of
Science and Technology (Wuhan, China). Healthy adult male
miR-155−/− mice (n=80) and C57BL/6 mice (n=120) used in
the present study were kept under standard animal room conditions
(temperature, 21±1°C; humidity, 50–60%; 0.03% CO2; 12 h
for light and 12 h for dark) with food and water prior to
experiments. The cardiac hypertrophy model was induced in male
miR-155−/− and WT C57Bl/6J mice (age, 10–12 weeks;
weight, 20–25 g) by Ang II (1.5 µg/g/day; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) infusion for 4 consecutive weeks using
subcutaneously implanted ALZET® 2004 minipumps. Sham
groups (control) received an equivalent dose of PBS instead of Ang
II. Animal models were divided into different groups as described
in Table I, and mice (n=6–8) were
randomly assigned to each of the indicated treatment groups. Food
and water were available ad libitum throughout the duration of
experiment. When the drug was depleted after 4 weeks, some mice
were sacrificed, however, others continued to receive treatment for
an additional 4 weeks. All animals were anaesthetized
intraperitoneally with sodium pentobarbital (50 mg/kg) and
exsanguinated via a retro-orbital venous puncture after 4 or 8
weeks of treatment. Hearts and spleens were harvested rapidly from
the euthanized mice following rapid cervical dislocation.
 | Table I.Characteristics of mice following 4
and 8 weeks of Ang II infusion. |
Table I.
Characteristics of mice following 4
and 8 weeks of Ang II infusion.
| A, Characteristics
of mice following 4 weeks of Ang II infusion |
|---|
|
|---|
| Group | Number | Body weight, g | H/B, mg/g |
|---|
| WT-sham | 8 | 26.5±1.2 | 4.5±0.4 |
| KO-sham | 6 | 26.1±1.3 | 4.3±0.6 |
| WT-Ang II | 8 | 28.6±1.6 |
6.5±0.6a |
| KO-Ang II | 6 | 28.3±1.6 |
5.8±0.3b,c |
|
| B, Characteristics
of mice following 8 weeks of Ang II infusion |
|
| Group | Number | Body weight, g | H/B, mg/g |
|
| WT-sham | 8 | 29.3±1.1 | 4.6±0.8 |
| KO-sham | 6 | 28.9±1.9 | 4.5±0.7 |
| WT-Ang II | 8 | 30.5±1.7 |
6.7±0.4a |
| KO-Ang II | 6 | 30.1±1.4 |
6.5±0.5b |
1-3-day-old neonatal male WT C57BL/6 mice (n=100)
were purchased from Experimental Animal Center of Wuhan University
(Wuhan, China) and were kept in the pathogen-free room in the
experimental animal center (Tongji Medical College of Huazhong
University of Science and Technology) under the same housing
conditions as aforementioned.
Assessment of cardiac function
The cardiac function of mice was evaluated
noninvasively by echocardiography performed with a Vevo 1100
imaging system (FUJIFILM VisualSonics, Inc., Toronto, ON, Canada)
equipped with an MS400 (18–38 MHz) phased-array transducer after 4
and 8 weeks of Ang II treatment, as previously described (20). Left ventricular diastole diameter
(LVDd) and interventricular septum diastolic thickness (IVS) were
measured, and left ventricular ejection fraction (LVEF) and left
ventricular fractional shortening (LVFS) were calculated from
measured recordings. The sonographer was blinded to the
randomization of mice. Every 2 weeks, the systolic blood pressure
was determined via tail-cuff plethysmography by using a
non-invasive blood pressure controller and PowerLab system.
Obtaining peripheral blood mononuclear
cells (PBMCs)
Peripheral blood samples were collected via a
retro-orbital venous puncture following anesthetizing the mice, as
aforementioned. PBMCs were separated from erythrocytes by density
centrifugation at 700 × g, 18°C for 20 min, using
Ficoll® PM 400 Histopaque®-1077
(Sigma-Aldrich; Merck KGaA). Isolated PBMCs were prepared for RNA
analysis.
Cardiac fibroblast isolation and
culture
Mouse neonatal cardiac fibroblasts were prepared
from the hearts of 1–3-day-old neonatal male WT C57BL/6 mice, which
were finely minced and placed together in 0.25% trypsin, as
described previously (21,22). The resuspension was plated onto
culture flasks with Dulbecco's modified Eagle's medium (DMEM;
Sigma-Aldrich; Merck KGaA) containing 10% fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) for 60
min at 37°C in humidified air with 5% CO2, which allowed
for preferential attachment of fibroblasts to the bottom of the
culture flasks. Flasks were washed twice with PBS to detach the
weakly attached and non-adherent cells and the medium was changed.
Cardiac fibroblasts were cultured in Dulbecco's modified Eagle's
medium (DMEM; Sigma-Aldrich; Merck KGaA) containing 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.) at 37°C in
humidified air with 5% CO2. Cells were removed into 24-
or 6-well plates (1×106 cells/ml) for experimental
preparation with DMEM containing 1% fetal bovine serum for 12 h at
37°C. Subsequently, the cells were treated with Ang II (1 µM) and
controls were treated with the same dose of PBS for 48 h at 37°C in
a humidified 5% CO2 atmosphere (22).
Cell transfection
Cardiac fibroblasts were cultured in 24-well plates
until they reached 50–80% confluence, as previously described
(23). Cell transfection was
performed using a riboFEC CP Transfection kit (Guangzhou RiboBio
Co., Ltd., Guangzhou, China), according to the manufacturer's
protocol. Cardiac fibroblasts were transfected with double-chain
agomiR-155 (mature sequence, UUA AUG CUA AUU GUG AUA GGG GU;
complementary sequence, AAU UAC GAU UAA CAC UAU CCC CA) or agomir
negative control for miR-155 overexpression, and single-chain
antagomiR-155 (complementary sequence, AAU UAC GAU UAA CAC UAU CCC
CA) or antagomir negative control for miR-155 inhibition. The
agomir and antagomir negative control were the same (UUU GUA CUA
CAC AAA AGU ACU G). Cardiac fibroblasts were transfected with
agomiR-155 or an agomir negative control at a concentration of 20
nM, and antagomiR-155 or an antagomir negative control at a final
concentration of 100 nM. Subsequently, cells were incubated for 6 h
at 37°C, the medium was changed and cells were cultured with Ang II
stimulation (100 nmol/l) for 48 h in DMEM containing 5% fetal
bovine serum at 37°C in humidified air with 5% CO2.
Cells were harvested after 48 h. Second passage cells were used in
all experiments. All agents for transfection were purchased from
Guangzhou RiboBio Co., Ltd.
Western blot analysis
Total protein was isolated from heart tissue and
cultured cardiac fibroblasts, as previously described (24). Protein concentration was quantified
using a BCA protein assay kit (Pierce; Thermo Fisher Scientific,
Inc.) according to the manufacturer's protocol. Briefly, protein
samples (50 µg/lane) were subjected to SDS-PAGE (10% gel) and
transferred to polyvinylidene difluoride membranes. The blots were
blocked with 5% nonfat milk in TBS containing 0.05% Tween-20 (TBST)
for 2 h at room temperature, and subsequently probed with specific
primary antibodies against α-SMA (1:1,000; YM3364; ImmunoWay
Biotechnology Company, Plano, TX, USA), type I collagen (1:1,000;
YT6135 ImmunoWay Biotechnology Company) and GAPDH (1:1,000; YM1038;
ImmunoWay Biotechnology Company) at 4°C overnight. The membranes
were washed with TBST and incubated with secondary horseradish
peroxidase-conjugated antibodies (1:3,000; 14708S; Cell Signaling
Technology, Inc., Danvers, MA, USA) for 2 h at room temperature.
Finally, the protein bands were washed and developed with enhanced
chemiluminescence (ECL) kit (Thermo Fisher Scientific, Inc.) and
semiquantitatively analyzed using densitometric methods in each
group with Quantity One software (version 4.62; Bio-Rad
Laboratories, Inc., Hercules, CA, USA). The results were expressed
as fold changes by normalizing the data to the control values.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA from cardiac fibroblasts, PBMCs and heart
and spleen tissues was isolated using TRIzol reagent (Invitrogen;
Thermo Fisher Scientific, Inc.), and was reverse transcribed into
cDNA by using a PrimeScript RT reagent kit at 37°C for 15 min
(Takara Bio, Inc., Otsu, Japan), according to the manufacturer's
protocol. The 10 µl PCR mixture contained 1 µg total cDNA and 5
pmol each of the primers. The primers for mRNAs are listed in
Table II. GAPDH was used as an
internal standard. All reactions were performed with SYBR Premix Ex
Taq II (Takara Bio, Inc.) and incubated in a 7500 Real-Time PCR
system (Applied Biosystems; Thermo Fisher Scientific, Inc.) in a
96-well plate, according to the manufacturer's protocol. For
miR-155 detection, the following thermocycling conditions were
used: An initial predenaturation step at 50°C for 2 min, followed
by 40 cycles of denaturation at 95°C for 10 min and annealing at
60°C for 1 min. For other factor detection, the thermocycling
conditions were as follows: An initial predenaturation step at 94°C
for 5 min, followed by 40 cycles of denaturation at 95°C for 30
sec, annealing at 60°C for 30 sec and extension at 72°C for 20 sec.
Each sample was repeated at least three times. The quantitative
assessment of specific miR-155 level was measured using standard
protocols, as previously described (23). The expression levels of target
genes relative to endogenous controls was quantified by comparative
quantitation cycle method (24)
The primers used in the present study were purchased from Guangzhou
RiboBio Co., Ltd. The primer sequences used are presented in
Table II. Data were expressed as
the fold change compared with the control.
| Table II.Primer sequences for reverse
transcription-quantitative polymerase chain reaction. |
Table II.
Primer sequences for reverse
transcription-quantitative polymerase chain reaction.
| Gene | Forward primer,
5′-3′ | Reverse primer,
5′-3′ |
|---|
| SOCS1 |
CTGCGGGCTTCTATTGGGGAC |
AAAAGGCAGTCGAAGGTCTCG |
| Collagen I |
GACTGGCAACCTCAAGAAGG |
GACTGTCTTGCCCCAAGTTC |
| TGF-β1 |
CTCCCGTGGCTTCTAGTGC |
GCCTTAGTTTGGACAGGATCTG |
| GAPDH |
AGGTCGGTGTGAACGGATTTG |
TGTAGACCATGTAGTTGAGGTCA |
| α-SMA |
GTCCCAGACATCAGGGAGTAA |
TCGGATACTTCAGCGTCAGGA |
| SHIP1 |
GCCCCTGCATGGGAAATCAA |
TGGGTAGCTGGTCATAACTCC |
| SMAD3 |
CACGCAGAACGTGAACACC |
GGCAGTAGATAACGTGAGGGA |
| U6 |
CTCGCTTCGGCAGCACA |
AACGCTTCACGAATTTGCGT |
| MiR-155 |
TGCCTCCAACTGACTCCTAC |
GCCAGCAGAATAATACGAC |
Histopathological experiments
The left ventricle was fixed in 4% formaldehyde for
48 h at 4°C and embedded in paraffin. Adjacent paraffin-embedded
left ventricle sections (5 µm) were cut for hematoxylin and eosin
(HE) and Masson's trichrome staining. In brief, paraffin-embedded
tissue sections were stained with dimethylbenzene for 15 min at
room temperature, then stained with hematoxylin for 10–15 min and
washed with water prior to being stained with 0.5% eosin for 30
sec-1 min then washed with water at room temperature to demonstrate
distinct colors in the nucleus and cytoplasm. For Masson's
trichrome, paraffin-embedded tissue sections were stained with
Regaud hematoxylin for 5–10 min at room temperature, then stained
with masson acid complex red liquid for 5 min and 0.2% glacial
acetic acid aqueous solution for 30 sec at room temperature then
washed in water, and subsequently stained with toluidine blue for 5
min at room temperature. The HE Staining kit and Masson's trichrome
Staining kit used in this study were purchased from Wuhan Goodbio
Technology Co., Ltd. (Wuhan, China), according to manufacturer's
protocol. Heart sections were stained with HE and Masson's
trichrome for the assessment of inflammatory cell areas and
collagen volume fraction with HE and Masson procedures,
respectively (25). A BX51TF
microscope (magnification, ×40, 100, 200 and 400; Olympus
Corporation, Tokyo, Japan) was used to observe pathological change
areas. Image-Pro Plus 6.703 software (Media Cybernetics, Inc.,
Rockville, MD, USA) was used to calculate fibrosis areas and the
statistical analysis.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. All analyses were performed using GraphPad Prism software
(version 5.0; GraphPad Software, Inc., La Jolla, CA, USA).
Student's test was performed for comparisons between two groups and
one-way analysis of variance followed by Bonferroni's post-hoc test
was performed among multiple groups. P<0.05 was considered to
indicate a statistically significant difference.
Results
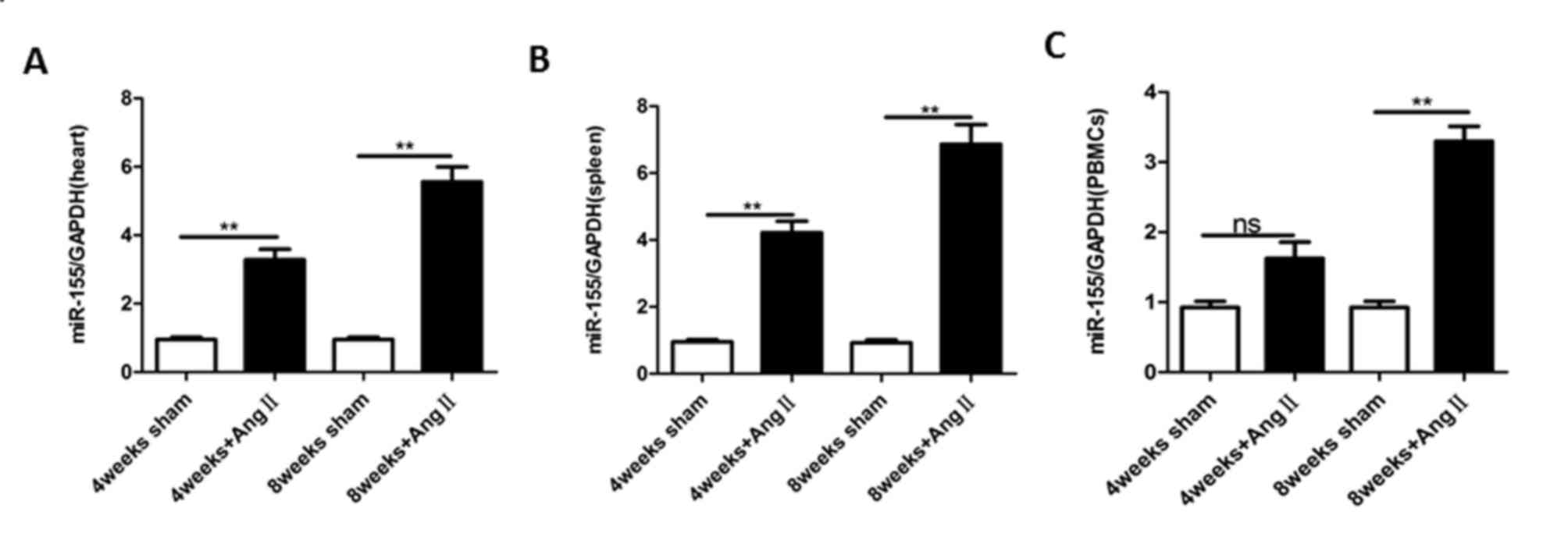
miR-155 is expressed in mouse hearts
and its expression is induced in cardiac remodeling
Previous studies have reported that the expression
of miR-155 is ubiquitous in adult mouse tissues, and is enriched in
T lymphocytes and infiltrating macrophages (26–28).
The present study investigated the expression of miR-155 in the
heart, PBMCs and spleen of adult mice. As demonstrated in Fig. 1A-C, miR-155 was expressed in adult
mouse tissues. In addition, the expression of miR-155 was increased
during the process of cardiac remodeling induced by treatment with
Ang II for 4 and 8 weeks. These results indicate that miR-155 is
expressed in the hearts of adult mice and that its expression is
regulated during cardiac remodeling.
miR-155 deficiency improves cardiac
function in mice
As miR-155 was significantly upregulated in mouse
hearts during cardiac remodeling induced by Ang II,
miR-155−/− and WT mice were subjected to Ang II infusion
for 4 and 8 weeks to investigate whether the absence of miR-155
influences Ang II-induced cardiac remodeling. As demonstrated in
Fig. 2A, Ang II induced heart
hypertrophy in both miR-155−/− and WT mice, as evidenced
by an increase in the size of hearts compared with sham mice.
However, the size of the heart in miR-155−/− mice
appeared substantially smaller compared with WT mice following Ang
II infusion. The development of cardiac hypertrophy in the hearts
of WT mice and the repression of hypertrophic growth in
miR-155−/− mice following Ang II infusion are supported
by differences in the heart weight/body weight ratio (Table I). The mean arterial pressure (MAP)
of mice in each group at different time points was also measured,
and similar changes in the MAP were observed in WT compared with
miR-155−/− mice, with marginally higher MAP in the WT
group throughout treatment (Fig.
2B).
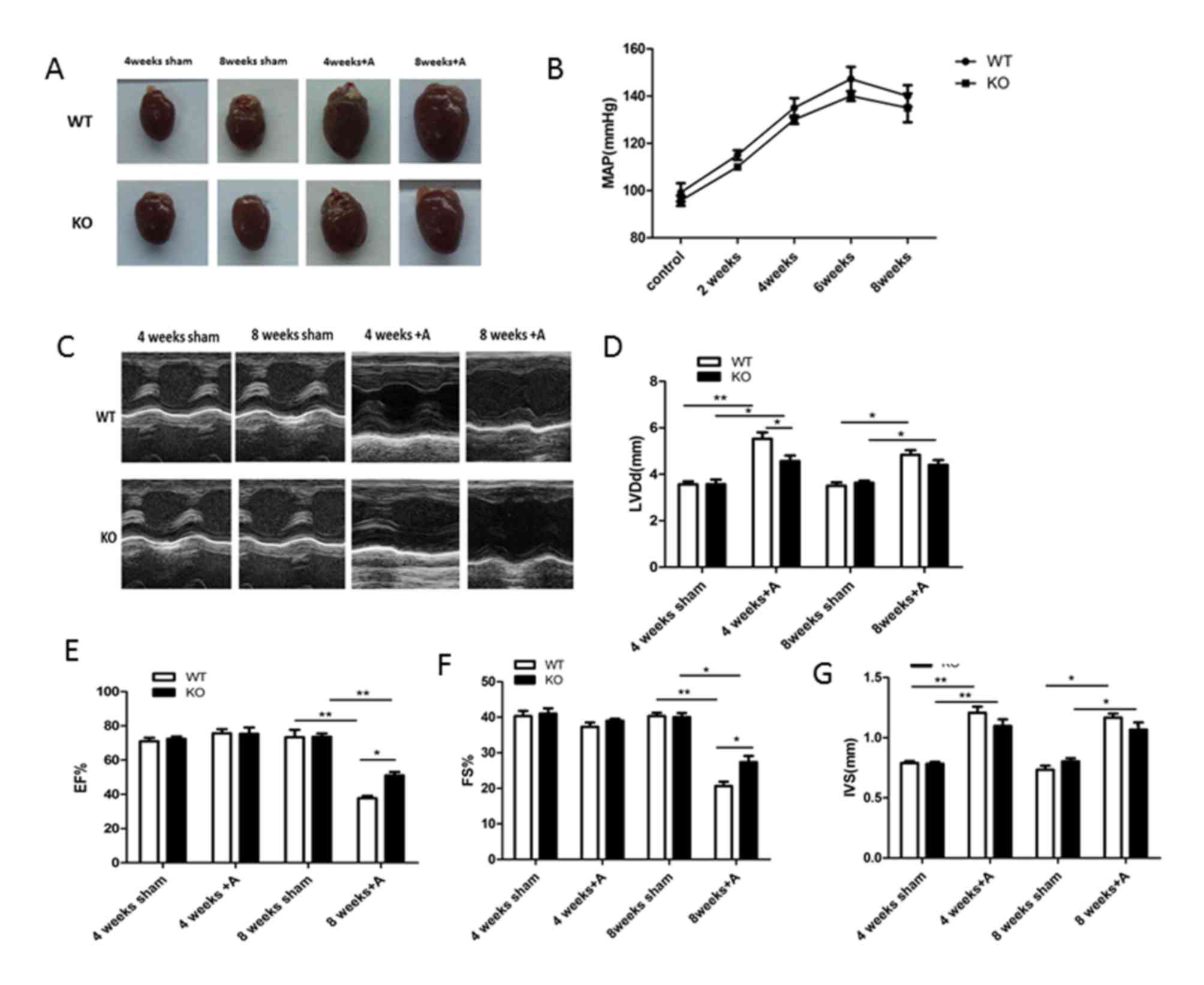 | Figure 2.Absence of miR-155 ameliorates the
cardiac function following Ang II treatment. (A) Representative
images of whole hearts in WT and miR-155 KO groups with or without
Ang II treatment. (B) MAP of mice in WT and miR-155 KO groups at
different time points during Ang II treatment. (C) Representative
M-mode echocardiograms obtained from WT-sham, KO-sham, WT-Ang II
and KO-Ang II mice. Echocardiographic measurements of (D) LVDd, (E)
LVEF, (F) LVFS and (G) IVS were recorded in WT and KO-sham and Ang
II mice. Data are presented as the mean ± standard error of the
mean. *P<0.05 and **P<0.01, as indicated. miR, microRNA; Ang,
angiotensin; WT, wild-type; KO, knockout; MAP, mean arterial
pressure; LVDd, left ventricular diastole diameter; LVEF, left
ventricular ejection fraction; LVFS, left ventricular fractional
shortening; IVS, interventricular septum diastolic thickness; A,
Ang II. |
Echocardiography was performed to determine cardiac
function at different time points in each group (Fig. 2C). As demonstrated in Fig. 2D-G, cardiac dysfunction, as
demonstrated by significantly decreased LVEF and LVFS compared with
the sham group at 8 weeks, was observed in the Ang II-treated WT
group. However, LVEF and LVFS were significantly higher at 8 weeks
in the miR-155−/− Ang II-treated group compared with the
WT Ang II-treated group. In addition, increases in LVDd and IVS
were more pronounced in in the Ang II-treated WT group compared
with the Ang II-treated miR-155−/− mice at 4 and 8
weeks. Combined, these results indicate that the absence of miR-155
in mice markedly improved cardiac function during the process of
Ang II-induced cardiac remodeling.
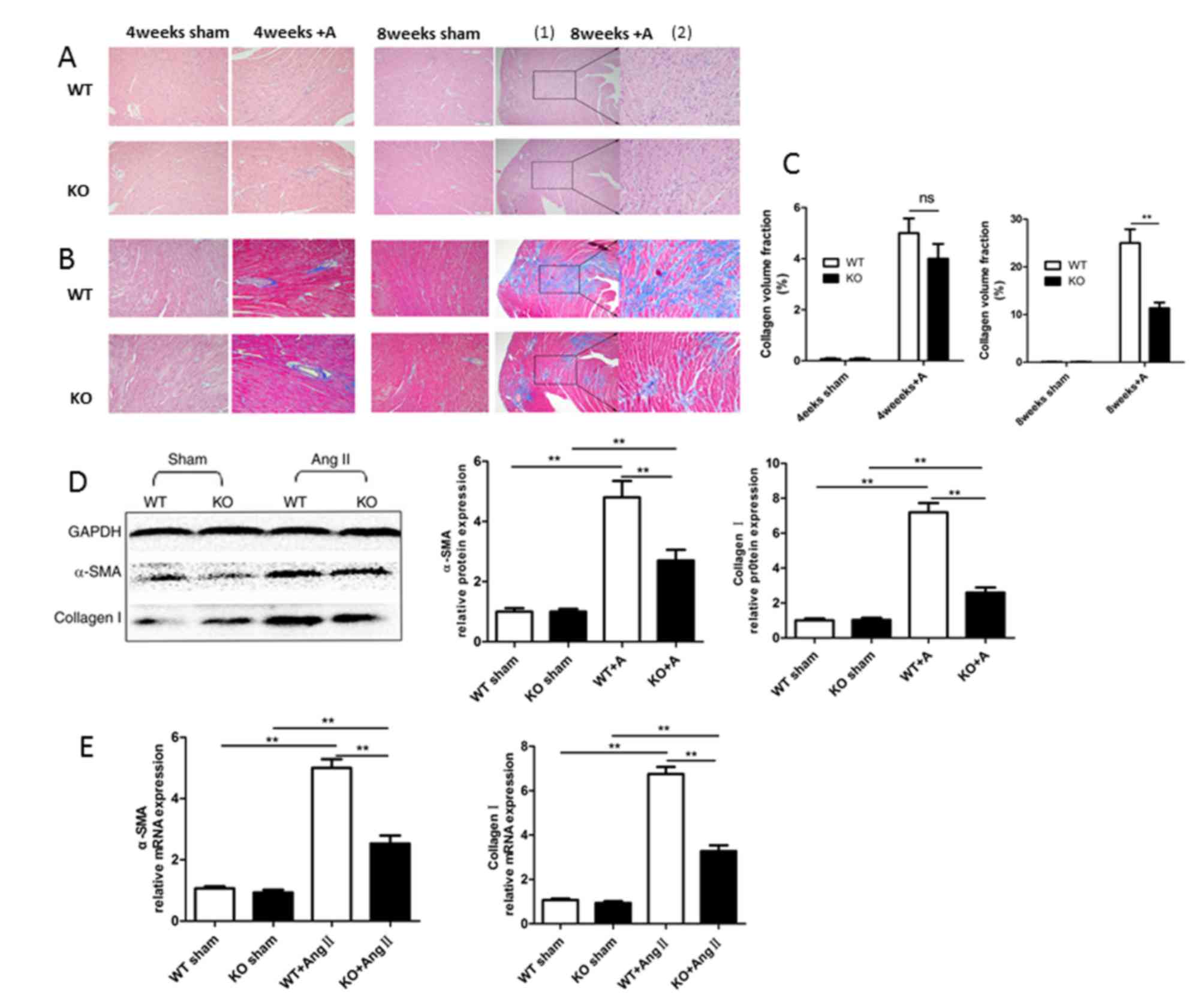
miR-155 deficiency alleviates cardiac
fibrosis
To investigate the potential effects of miR-155 on
cardiac fibrosis, differences in the severity of fibrosis based on
the infiltration of inflammatory cells and fibrosis area of the
left ventricle in miR-155−/− mice and WT mice were
examined. HE and Masson's trichrome staining were performed to
assess inflammatory cell areas and collagen volume fractions,
respectively. As demonstrated in Fig.
3A and B, inflammatory cell infiltration was markedly reduced
in miR-155−/− mice with Ang II treatment compared with
Ang II-treated WT mice, and the interstitial fibrotic response was
also reduced in miR-155−/− compared with WT mice,
particularly at 8 weeks. In addition, the fibrosis areas in
miR-155−/− and WT groups were calculated as the collagen
volume fraction using Image-Pro Plus 6.703 software, as
demonstrated in Fig. 3C.
Myofibroblasts are the primary source of ECM and
have a critical role in cardiac fibrosis, they are characterized by
the presence of a microfilamentous contractile apparatus enriched
with α-SMA. Therefore, the expression of α-SMA and collagen I was
determined to clarify whether miR-155 has a direct role in cardiac
fibrosis. As demonstrated in Fig. 3D
and E, the protein and mRNA expression levels of α-SMA and
collagen I were decreased in miR-155−/− mice compared
with WT mice following 8 weeks of Ang II treatment. These results
indicate that miR-155 may be involved in cardiac fibrotic
remodeling, and may increase collagen production in Ang II-induced
cardiac remodeling.
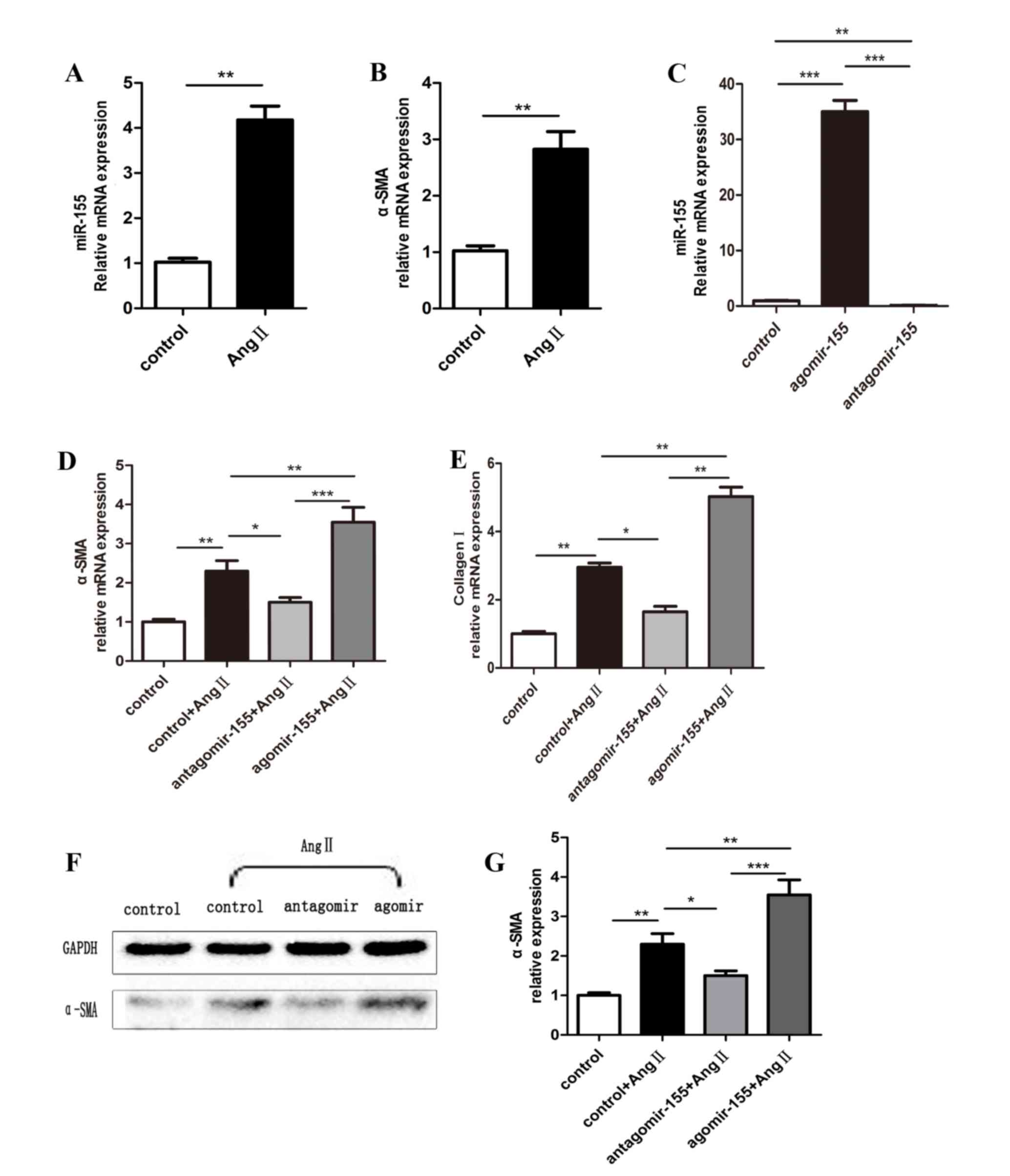
miR-155 induces a profibrotic
myofibroblast phenotype in isolated cardiac fibroblasts
Cardiac fibroblasts have important functions in
reparative and detrimental fibrotic responses during the process of
cardiac remodeling. To determine whether miR-155 is involved in
cardiac fibroblasts, mouse neonatal cardiac fibroblasts were
prepared from neonatal C57BL/6 mice. The expression of miR-155 was
low in cardiac fibroblasts, however, the expression was
significantly induced by Ang II stimulation for 48 h (Fig. 4A). Similarly, cultured mouse
neonatal cardiac fibroblasts treated with Ang II also exhibited
increased expression of α-SMA compared with control cells (Fig. 4B). To further confirm whether
miR-155 was involved in the expression of α-SMA, gain- and
loss-of-function studies on the expression of miR-155 were
performed by transfection with agomiR-155 and antagomiR-155,
respectively. miR-155 expression was significantly elevated in
cardiac fibroblasts transfected with agomiR-155, whereas levels
were suppressed in antagomiR-155-treated cells, compared with the
control-transfected cells (Fig.
4C). Notably, as demonstrated in Fig. 4D-G, forced expression of miR-155,
using agomiR-155, increased the expression levels of α-SMA and
collagen I in Ang II-treated fibroblasts compared with the
control-transfected group, while the opposite effects were observed
in antagomiR-155-treated cells. In conclusion, these results
indicate that miR-155 may contribute to cardiac fibrotic remodeling
by inducing fibroblast to myofibroblast transformation.
SOCS1 is involved in the process of
miR-155-regulated fibrosis
Numerous miR-155 target genes have previously been
described (29). Direct
anti-inflammatory miR-155 targets include SOCS1 and Src homology
2-containing inositol phosphatase-1 (SHIP1). The present study
demonstrated that, in the absence of miR-155, SOCS1 expression was
elevated in Ang II-treated mice compared with WT Ang II-treated
mice (Fig. 5A), whereas SHIP1 was
not significantly induced in miR-155−/− mice with Ang II
treatment compared with WT mice with Ang II treatment (Fig. 5B). Therefore, we hypothesized that
SOCS1, which functions as a negative regulator of cytokine
signaling, may be involved in miR-155-regulated cardiac fibrosis.
The results of the current study also indicated that increased
SOCS1 levels in Ang II-treated miR-155−/− mice were
associated with diminished profibrotic transforming growth factor
(TGF)-β1/SMAD3 signaling, compared with WT Ang II-treated mice
(Fig. 5C and D). In addition, the
results in Fig. 3E indicate that a
reduction in the expression of collagen I may also be associated
with increased SOCS1 expression, as collagen expression was reduced
in Ang II-treated miR-155−/− mice compared with Ang
II-treated WT mice, while SOCS1 expression was increased. These
results indicate that miR-155 deficiency may inhibit profibrotic
TGF-β1/SMAD-3 signaling via interactions with SOCS1.
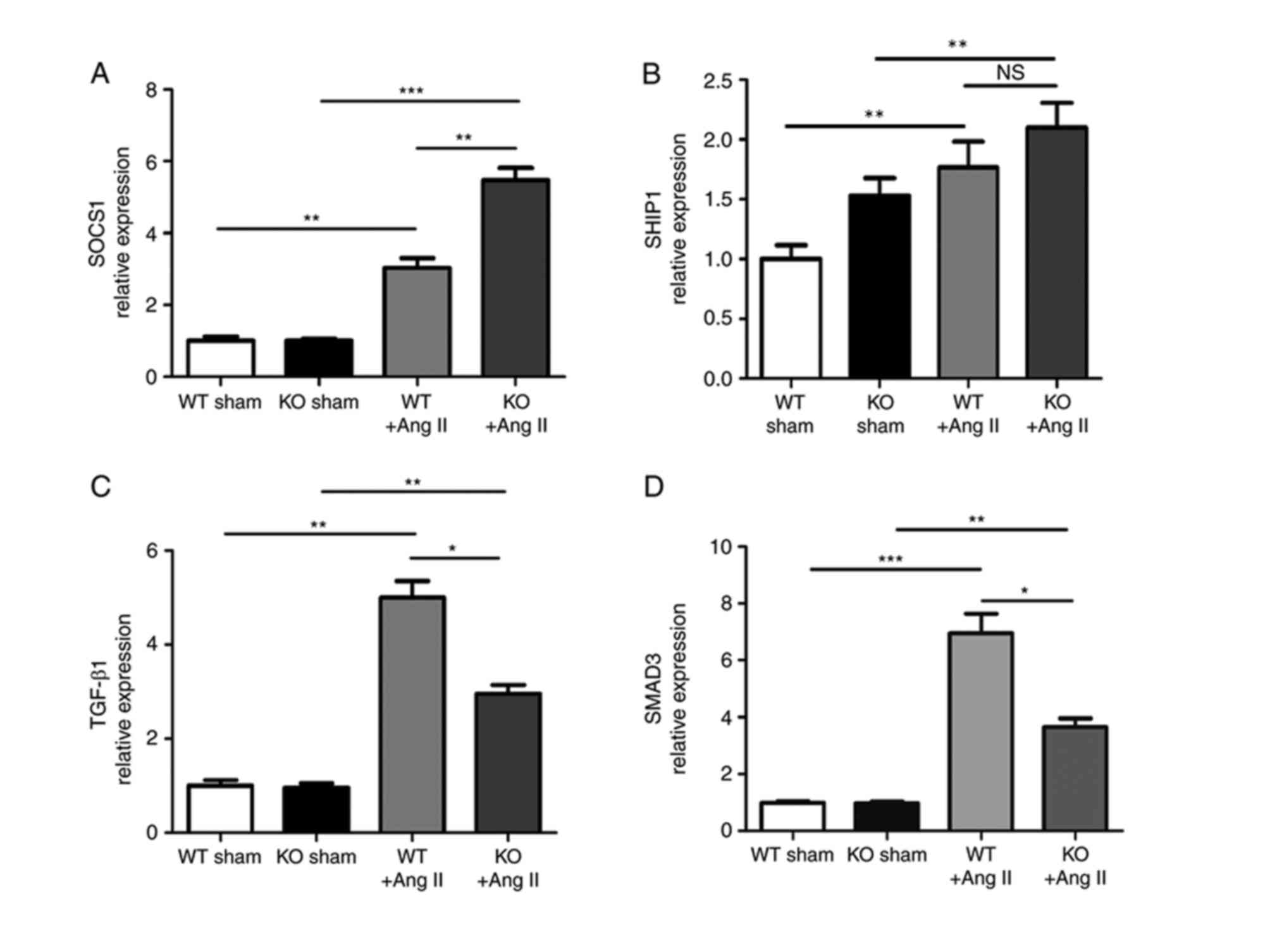 | Figure 5.SOCS1 was involved in
miR-155-regulated cardiac fibrosis. mRNA expression levels of (A)
SOCS1 and (B) SHIP1, which are direct target miR-155 targets, in KO
and WT mice heart tissues at 8 weeks. (C and D) mRNA expression
levels of TGF-β1 and SMAD3 in heart tissues of KO and WT mice. Data
are presented as the mean + standard error of the mean. *P<0.05,
**P<0.01 and ***P<0.001, as indicated. SOCS1, suppressor of
cytokine signaling 1; miR, microRNA; SHIP1, Src homology
2-containing inositol phosphatase; KO, knockout; WT, wild-type;
TGF, transforming growth factor. |
Discussion
It is evident that cardiac fibrosis, characterized
by excessive ECM deposition, seriously limits cardiac systolic and
diastolic function (30). Various
growth and inflammatory factors are reported to be involved in this
process, including certain miRNAs (13,14,31).
Previous studies have reported that miR-155 has an important role
in various inflammatory heart diseases, including cardiac
hypertrophy, myocarditis, atherosclerosis and heart failure
(15,16,18).
Despite this, it is generally accepted that fibroblasts are
critically involved in the reparative response and the pathogenesis
of cardiac remodeling. However, direct evidence for a role of
miR-155 in Ang II-induced cardiac fibrotic remodeling is limited.
The present study used miR-155 knockout (miR-155−/−)
mice to demonstrate that miR-155 enhances fibrosis in Ang
II-induced cardiac remodeling. Furthermore, gain- and
loss-of-function studies on the expression of miR-155 were
performed in cardiac fibroblasts to demonstrate that miR-155 is
involved in the induction of fibroblast to myofibroblast
transformation.
It is established that certain miRNAs are expressed
abnormally in a variety of diseases (32). As certain miRNAs have important
roles in response to stress signals, it may be hypothesized that
miRNAs may be involved in modulating the progression from adaptive
to pathological cardiac remodeling by acting alone or in
combination. The results of the present study demonstrated that the
expression of miR-155 was upregulated in the damaged heart tissue
of mice, and miR-155−/− mice exhibited relatively mild
heart damage compared with WT mice in response to Ang II treatment.
This result is consistent with the results of previous studies,
which demonstrated that miR-155 expression promoted cardiac
inflammation, hypertrophy and failure in response to pressure
overload (17,18). However, the existing literature
concerning the role of miR-155 in cardiac fibrotic remodeling is
conflicting. Heymans et al (18) and confirmed that absence of miR-155
reduced pressure overload-induced cardiac hypertrophy and
inflammation, while the cardiac fibrotic response remained intact.
In addition, their results indicated that pressure overload-induced
fibrotic remodeling may be independent of macrophage miR-155
function, or fibrotic remodeling in mouse hearts may not be solely
dependent on macrophage signaling. Furthermore, another study
concluded that loss of miR-155 substantially eliminated the
increase in cardiomyocyte size in transverse aortic
constriction-induced hypertrophic mice, and cardiac fibrosis was
markedly suppressed in the hearts of miR-155−/− mice.
They reported that endogenous miR-155 may suppress cardiomyocyte
hypertrophy by targeting Jarid2 in isolated cardiomyocytes,
however, the mechanism of miR-155-induced cardiac fibrosis was not
reported (17). However, despite
these results, the mechanisms of miR-155-regulated fibrotic
remodeling, particularly in cardiac fibroblasts, remain unclear. By
contrast, the present study overexpressed and inhibited miR-155, by
using miR-155 agomir and antagomirs, in cardiac fibroblasts to
demonstrate that miR-155 may partially affect cardiac fibrosis by
inducing fibroblast to myofibroblast transformation. The results of
the current study also demonstrated that the expression of α-SMA
increased with increasing miR-155 expression in Ang II-treated
hearts and cardiac fibroblasts, indicating that miR-155 may be
involved in fibroblast to myofibroblast transformation.
Unfortunately, persistent myofibroblast activation and the
resultant increase in fibrous tissue produced may cause progressive
adverse myocardial remodeling.
The mechanisms by which miRNAs regulate cardiac
fibrosis have been attributed to the alteration of certain
signaling pathways in the pathological process of fibrotic growth
(33). It is established that
certain inflammatory factors have important functions in fibrotic
remodeling. The present study focused on SOCS1, a target gene of
miR-155 that is elevated and suppresses macrophages during
inflammatory responses. Previous studies have reported that SOCS1
activates associated inflammatory molecules as a potent inhibitor
of the production and release of cytokines (34–36).
It is well established that miR-155 confers competitive fitness to
regulatory T cells by targeting SOCS1 (37). In addition, it was previously
confirmed that miR-155 targets SOCS1 to activate the
interleukin-6/Janus kinase/signal transducer and activator of
transcription 3 signaling pathway for T helper 17 cell
differentiation (38).
Furthermore, another study demonstrated that the expression of
miR-155 in macrophages was a potent contributor to cardiac
hypertrophy and failure in pressure overload conditions, and SOCS1
knockdown restored the hypertrophy-stimulating potency in miR-155
knockout macrophages (18). It has
also been reported that overexpression of miR-155 promoted the
proliferation and invasion of human laryngeal squamous cell
carcinoma by targeting SOCS1 (39). In the current study, the results
demonstrated that the expression of SOCS1 was induced in heart
tissue and cardiac fibroblasts by Ang II; the expression of SOCS1
was lower in miR-155 KO mice compared with WT mice. The results
indicate that the absence of miR-155 may inhibit profibrotic
remodeling by targeting SOCS1. TGF-β1, the most potent inducer of
ECM production, may promote fibroblast to myofibroblast
differentiation. The TGF-β1/SMAD3-mediated signaling pathway is
reported to have a pivotal role in ECM metabolism (40). Overexpression of miR-155 in the
present study led to reduced levels of SOCS1 and hyperactivation of
profibrotic TGF-β1/SMAD3 signaling, which results in excessive
fibrosis and adverse ventricular remodeling. However, a limitation
of the present study was that the derepression of SOCS1 in miR-155
KO mice was not prevented. Therefore, deletion of SOCS1 in heart
and isolated cardiac fibroblasts should be performed to validate
the role of SOCS1 in miR-155-regulated cardiac fibrosis. In future
studies, we aim to further investigate the associations between and
among miR-155, SOCS1 and TGF-β1/SMAD3 signaling. In addition, other
limitations of the present study exist; the number of the mice in
each group is relatively small and, considering that there appears
to be inflammatory cell infiltration in the Ang II hearts, it is
not easy to determine whether intrinsic cells of the heart
(myocytes or fibroblasts) or inflammatory cells are the most
important source of cells for modulation of SOCS1 by miR-155. The
mechanisms underlying miR-155-induced cardiac fibrotic remodeling
therefore require further investigation.
The current study addressed the role of miR-155 and
its potential regulatory mechanism in cardiac fibrosis induced by
Ang II. Both genetic inactivation and pharmacological inhibition of
miR-155 reduced the production of collagen, which indicates that
miR-155 may be an important contributor to cardiac fibrosis induced
by Ang II. Furthermore, it was demonstrated that miR-155
facilitated fibroblast to myofibroblast transformation in isolated
cardiac fibroblasts. Clinically, pharmacological inhibitors
targeting miR-155 may become a useful adjunct in the treatment of
Ang II-dependent cardiac remodeling. However, the mechanisms that
drive cardiac fibrotic remodeling in response to pressure overload
require further investigation. Further studies concerning miR-155,
particularly human studies, are required. In conclusion, inhibition
of miR-155 reduced fibroblast to myofibroblast transformation and
ameliorated the progression of cardiac fibrotic remodeling induced
by Ang II. Therefore, inhibition of endogenous miR-155 may have
clinical potential in alleviating cardiac fibrosis in heart
failure.
Acknowledgements
The present study was supported by a grant from the
National Natural Science Foundation of China (grant no.
81170205).
References
|
1
|
Lorenzen JM, Schauerte C, Hübner A,
Kölling M, Martino F, Scherf K, Batkai S, Zimmer K, Foinquinos A,
Kaucsar T, et al: Osteopontin is indispensible for AP1-mediated
angiotensin II-related miR-21 transcription during cardiac
fibrosis. Eur Heart J. 36:2184–2196. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
González A, Ravassa S, Beaumont J, López B
and Díez J: New targets to treat the structural remodeling of the
myocardium. J Am Coll Cardiol. 58:1833–1843. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Díez J, Querejeta R, López B, González A,
Larman M and Ubago JL Martinez: Losartan-dependent regression of
myocardial fibrosis is associated with reduction of left
ventricular chamber stiffness in hypertensive patients.
Circulation. 105:2512–2517. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Chen W and Frangogiannis NG: Fibroblasts
in post-infarction inflammation and cardiac repair. Biochim Biophys
Acta. 1833:945–953. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Melchior-Becker A, Dai G, Ding Z, Schäfer
L, Schrader J, Young MF and Fischer JW: Deficiency of biglycan
causes cardiac fibroblasts to differentiate into a myofibroblast
phenotype. J Biol Chem. 286:17365–17375. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Thum T and Lorenzen JM: Cardiac fibrosis
revisited by microRNA therapeutics. Circulation. 126:800–802. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Kvakan H, Luft FC and Muller DN: Role of
the immune system in hypertensive target organ damage. Trends
Cardiovasc Med. 19:242–246. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lee RC and Ambros V: An extensive class of
small RNAs in Caenorhabditis elegans. Science. 294:862–864. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Small EM and Olson EN: Pervasive roles of
microRNAs in cardiovascular biology. Nature. 469:336–342. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Rawal S, Manning P and Katare R:
Cardiovascular microRNAs: As modulators and diagnostic biomarkers
of diabetic heart disease. Cardiovasc Diabetol. 13:442014.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Blahna MT and Hata A: Regulation of miRNA
biogenesis as an integrated component of growth factor signaling.
Curr Opin Cell Biol. 25:233–240. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Papageorgiou N, Tsalamandris S, Giolis A
and Tousoulis D: MicroRNAs in cardiovascular disease: Perspectives
and reality. Cardiol Rev. 24:110–118. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Duisters RF, Tijsen AJ, Schroen B,
Leenders JJ, Lentink V, Van Der Made I, Herias V, van Leeuwen RE,
Schellings MW, Barenbrug P, et al: miR-133 and miR-30 regulate
connective tissue growth factor: Implications for a role of
microRNAs in myocardial matrix remodeling. Circ Res. 104:170–178.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Thum T, Gross C, Fiedler J, Fischer T,
Kissler S, Bussen M, Galuppo P, Just S, Rottbauer W, Frantz S, et
al: MicroRNA-21 contributes to myocardial disease by stimulating
MAP kinase signalling in fibroblasts. Nature. 456:980–984. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Elton TS, Selemon H, Elton SM and
Parinandi NL: Regulation of the MIR155 host gene in physiological
and pathological processes. Gene. 532:1–12. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Nazari-Jahantigh M, Wei Y, Noels H, Akhtar
S, Zhou Z, Koenen RR, Heyll K, Gremse F, Kiessling F, Grommes J, et
al: MicroRNA-155 promotes atherosclerosis by repressing Bcl6 in
macrophages. J Clin Invest. 122:4190–4202. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Seok HY, Chen J, Kataoka M, Huang ZP, Ding
J, Yan J, Hu X and Wang DZ: Loss of MicroRNA-155 protects the heart
from pathological cardiac hypertrophy. Circ Res. 114:1585–1595.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Heymans S, Corsten MF, Verhesen W, Carai
P, van Leeuwen RE, Custers K, Peters T, Hazebroek M, Stöger L,
Wijnands E, et al: Macrophage microRNA-155 promotes cardiac
hypertrophy and failure. Circulation. 128:1420–1432. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bala S, Csak T, Saha B, Zatsiorsky J,
Kodys K, Catalano D, Satishchandran A and Szabo G: The
pro-inflammatory effects of miR-155 promote liver fibrosis and
alcohol-induced steatohepatitis. J Hepatol. 64:1378–1387. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Shyu KG, Lu MJ, Chang H, Sun HY, Wang BW
and Kuan P: Carvedilol modulates the expression of
hypoxia-inducible factor-1alpha and vascular endothelial growth
factor in a rat model of volume-overload heart failure. J Card
Fail. 11:152–159. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sreejit P, Kumar S and Verma RS: An
improved protocol for primary culture of cardiomyocyte from
neonatal mice. In Vitro Cell Dev Biol Anim. 44:45–50. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Tanaka A, Ide T, Fujino T, Onitsuka K,
Ikeda M, Takehara T, Hata Y, Ylikallio E, Tyynismaa H, Suomalainen
A and Sunagawa K: The overexpression of Twinkle helicase
ameliorates the progression of cardiac fibrosis and heart failure
in pressure overload model in mice. PLoS One. 8:e676422013.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Weber M, Kim S, Patterson N, Rooney K and
Searles CD: MiRNA-155 targets myosin light chain kinase and
modulates actin cytoskeleton organization in endothelial cells. Am
J Physiol Heart Circ Physiol. 306:H1192–H1203. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shyu KG, Ko WH, Yang WS, Wang BW and Kuan
P: Insulin-like growth factor-1 mediates stretch-induced
upregulation of myostatin expression in neonatal rat
cardiomyocytes. Cardiovasc Res. 68:405–414. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Matsusaka H, Ide T, Matsushima S, Ikeuchi
M, Kubota T, Sunagawa K, Kinugawa S and Tsutsui H: Targeted
deletion of matrix metalloproteinase 2 ameliorates myocardial
remodeling in mice with chronic pressure overload. Hypertension.
47:711–717. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Tili E, Croce CM and Michaille JJ:
miR-155: On the crosstalk between inflammation and cancer. Int Rev
Immunol. 28:264–284. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Faraoni I, Antonetti FR, Cardone J and
Bonmassar E: miR-155 gene: A typical multifunctional microRNA.
Biochim Biophys Acta. 1792:497–505. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Corsten MF, Papageorgiou A, Verhesen W,
Carai P, Lindow M, Obad S, Summer G, Coort SL, Hazebroek M, van
Leeuwen R, et al: MicroRNA profiling identifies microRNA-155 as an
adverse mediator of cardiac injury and dysfunction during acute
viral myocarditis. Circ Res. 111:415–425. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Roy S and Sen CK: MiRNA in innate immune
responses: Novel players in wound inflammation. Physiol Genomics.
43:557–565. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wu D, Lei H, Wang JY, Zhang CL, Feng H, Fu
FY, Li L and Wu LL: CTRP3 attenuates post-infarct cardiac fibrosis
by targeting Smad3 activation and inhibiting myofibroblast
differentiation. J Mol Med (Berl). 93:1311–1325. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Pan Z, Sun X, Shan H, Wang N, Wang J, Ren
J, Feng S, Xie L, Lu C, Yuan Y, et al: MicroRNA-101 inhibited
postinfarct cardiac fibrosis and improved left ventricular
compliance via the FBJ osteosarcoma oncogene/transforming growth
factor-β1 pathway. Circulation. 126:840–850. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Divakaran V and Mann DL: The emerging role
of microRNAs in cardiac remodeling and heart failure. Circ Res.
103:1072–1083. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Oury C, Servais L, Bouznad N, Hego A,
Nchimi A and Lancellotti P: MicroRNAs in valvular heart diseases:
Potential role as markers and actors of valvular and cardiac
remodeling. Int J Mol Sci. 17(pii): E11202016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Heinrich PC, Behrmann I, Haan S, Hermanns
HM, Müller-Newen G and Schaper F: Principles of interleukin
(IL)-6-type cytokine signalling and its regulation. Biochem J.
374:1–20. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Cittadini A, Monti MG, Iaccarino G,
Castiello MC, Baldi A, Bossone E, Longobardi S, Marra AM, Petrillo
V, Saldamarco L, et al: SOCS1 gene transfer accelerates the
transition to heart failure through the inhibition of the
gp130/JAK/STAT pathway. Cardiovasc Res. 96:381–390. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Xu HF, Fang XY, Zhu SH, Xu XH, Zhang ZX,
Wang ZF, Zhao ZQ, Ding YJ and Tao LY: Glucocorticoid treatment
inhibits intracerebral hemorrhage-induced inflammation by targeting
the microRNA-155/SOCS1 signaling pathway. Mol Med Rep.
14:3798–3804. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lu LF, Thai TH, Calado DP, Chaudhry A,
Kubo M, Tanaka K, Loeb GB, Lee H, Yoshimura A, Rajewsky K and
Rudensky AY: Foxp3-dependent microRNA155 confers competitive
fitness to regulatory T cells by targeting SOCS1 protein. Immunity.
30:80–91. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yao R, Ma YL, Liang W, Li HH, Ma ZJ, Yu X
and Liao YH: MicroRNA-155 modulates Treg and Th17 cells
differentiation and Th17 cell function by targeting SOCS1. PLoS
One. 7:e460822012. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhao XD, Zhang W, Liang HJ and Ji WY:
Overexpression of miR-155 promotes proliferation and invasion of
human laryngeal squamous cell carcinoma via targeting SOCS1 and
STAT3. PLoS One. 8:e563952013. View Article : Google Scholar : PubMed/NCBI
|