Introduction
Marfan syndrome (MFS) is an autosomal dominant
hereditary disease comprising a disorder of fibrous connective
tissue involving the ocular, skeletal and cardiovascular systems
(1). According to the Ghent
criteria, patients with malfunctions of at least two organ systems
could be diagnosed with MFS (2).
Aortic root dilatation/dissection and lens dislocation were two
cardinal manifestations to establish an unequivocal diagnosis of
MFS in patients with positive family history. Due to the large
clinical variability of MFS, and several other connective tissue
disorders with comparable clinical features, distinguishing MFS
from those similar syndromes is still challenging.
Increasing evidence indicates that heredity holds a
key role in the development of MFS. It has been reported that MFS
generally results from mutations in the human fibrillin-1
(FBN1) gene (3,4). At present, >3,000 mutations have
been identified in relation to MFS. Most mutations are specific to
a family with MFS, whereas ~10% of FBN1 mutations are shared
by different families (5). Located
at chromosome 15q-21.1 with 65 exons, the FBN1 gene encodes
a secreted 350 kDa glycoprotein (6). Human FBN1 protein shares conserved
sequences with other species. FBN1 protein constitutes
extracellular microfibrils and controls the stability, as well as
the microfibril assembly. Mutations within the FBN1 gene may
disrupt microfibril formation, leading to abnormalities of
fibrillin and eventually weakening the connective tissue (7).
In the present study, the entire coding region of
FBN1 was analyzed, and a novel mutation in exon 14 of
FBN1 was identified in all affected members. The newly
identified FBN1 mutation in a Chinese family with MFS
further emphasizes the important role of FBN1 in the
mechanism of MFS development. The present study not only expanded
the mutation spectrum of FBN1 resulting in MFS development
in a Chinese family, but is also likely to aid understanding of the
molecular pathogenesis and clinical diagnosis of
FBN1-associated MFS.
Materials and methods
Subjects
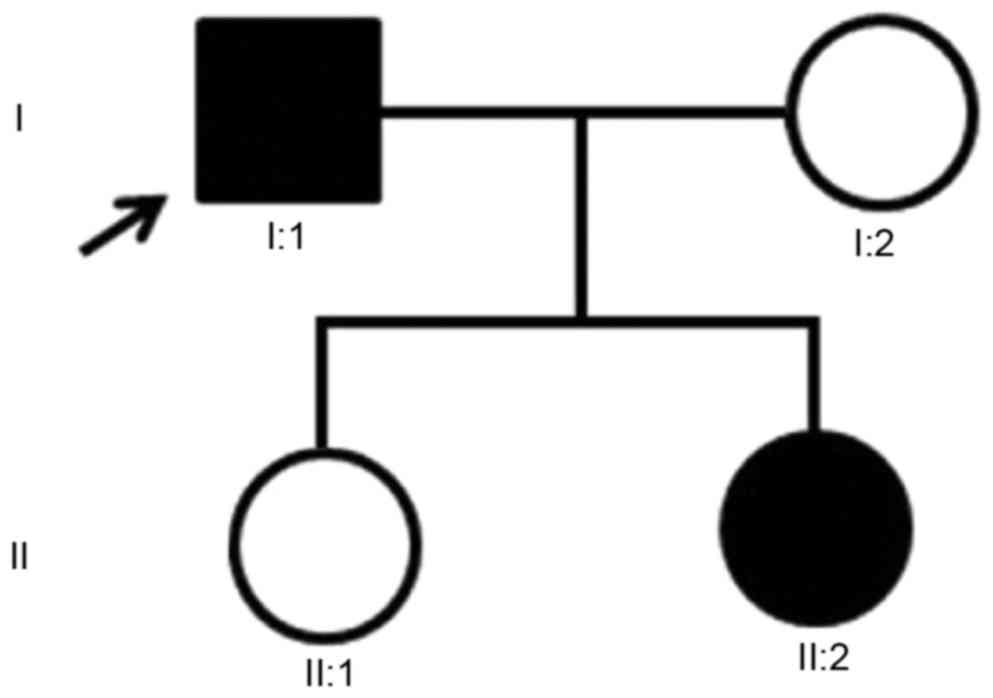
A family with MFS was recruited from the Shandong
Provincial Hospital Affiliated to Shandong University (Jinan,
China) (Fig. 1). This study was
conducted in accordance to the tenets of The Declaration of
Helsinki and was approved by the Institutional Review Boards of the
Hospital of University of Electronic Science and Technology of
China and Sichuan Provincial People's Hospital (Chengdu, China),
and the Shandong Provincial Hospital Affiliated to Shandong
University. A total of 383 ethnically matched, unrelated and normal
healthy individuals were recruited from the Hospital of University
of Electronic Science and Technology of China & Sichuan
Provincial People's Hospital (255 males and 128 females; mean age
at recruitment 55.26±8.78 years). These control individuals had no
medical history associated with any related diseases. Written
informed consent was obtained from all participants prior to the
study.
Clinical diagnosis
Two of the family members were diagnosed with MFS
according to the revised Ghent criteria (2). Non-consanguineous marriages were
found in the family; clinical information of the affected family
members is summarized in Table I.
All members of this family underwent complete physical,
cardiovascular and ophthalmologic examinations. Unrelated healthy
individuals also underwent the same examinations.
 | Table I.Clinical details of the patients with
Marfan syndrome in the family. |
Table I.
Clinical details of the patients with
Marfan syndrome in the family.
| Characteristic | Proband (I:1) | Proband's daughter
(II:2) |
|---|
| Age (years) | 44 | 8 |
| Gender | M | F |
| Ectopialentis | + | + |
| Myopia | + | + |
| Strabismus | +, exotropia | +, exotropia |
| Glaucoma | − | − |
| Retinal
detachment | + | − |
| Height (cm) | 184 | 134 |
| Arm span (cm) | 186 | 137 |
| AS/H | 1.01 | 1.02 |
| Overgrowth of the
long bones | + | + |
| Arachnodactyly | + | + |
| Scoliosis | − | − |
| Pectus excavatum | − | − |
| Pectus carinatum | + | − |
| Flatfeet | + | + |
| Mitral valve
prolapse | − | − |
| Aortic aneurysm | + (ruptured 5 years
ago then formed aortic dissection; Bentall surgery was performed at
that time) | − |
| Aortic root dimension
(mm) | 25.0 (artificial
vessel diameter) | 29.1 |
Mutation screening
Genomic DNA samples were extracted from peripheral
blood using a Blood DNA extraction kit (Thermo Fisher Scientific,
Inc., Waltham, MA, USA). The whole coding region of FBN1
(NM_000138.4) was amplified by polymerase chain reaction (PCR) with
35 cycles (30 sec at 95°C for initial denaturation, 30 sec for
annealing at different temperatures as shown in Table II, and 30 sec at 72°C for
extension), using a GeneAmp® PCR system 9700 (Applied
Biosystems; Thermo Scientific Inc.). Sequencing primers of all the
exons were designed using Primer 5.0 (Premier Biosoft
International, Palo Alto, CA, USA; Table II). Amplified PCR products were
purified and sequenced directly (BigDye Terminators Sequencing kit)
with an Automated Genetic Analysis system 3130 (both from Applied
Biosystems; Thermo Fisher Scientific, Inc.). Comparative amino acid
sequence analysis of the human FBN1 protein was performed across
different species using HomoloGene (https://www.ncbi.nlm.nih.gov/homologene/?term=FBN1).
The potentially damaging effects of the mutation on the structure
and function of FBN1 was predicted using SIFT (http://sift.jcvi.org) and PolyPhen-2 (http://genetics.bwh.harvard.edu/pph2/).
| Table II.Primers used for mutation screening of
the FBN1 gene. |
Table II.
Primers used for mutation screening of
the FBN1 gene.
| Primer name | Primer sequence
(5′-3′) | Product size
(bp) | Annealing temperature
(°C) |
|---|
|
FBN11&2F |
TCGGGGATTTGTCTCTGTGT | 434 | 59 |
|
FBN11&2R |
GCCCGTTGTTCTGGATCTTG |
|
|
| FBN13F |
ACCAACCCAGCATTGAGTCT | 308 | 60 |
| FBN13R |
TTCTAAGGCTCCCCATGCAA |
|
|
| FBN14F |
TTGTGAGGGACCTGAGAACC | 296 | 59 |
| FBN14R |
TTGCAGGAAAGAGGAAAGCC |
|
|
| FBN15F |
CAACTCCTGTGAGCTGTTGC | 278 | 60 |
| FBN15R |
AAACATGCTGTGTCCCAGGT |
|
|
| FBN16F |
GTCCTTCCAGAGGACCACAA | 228 | 60 |
| FBN16R |
CAGCTTTAGGTACCAGCATGTC |
|
|
| FBN17F |
GCATGATGGTTCCTGCTTTT | 380 | 60 |
| FBN17R |
GCAGTCAGCGAAATTGTGAA |
|
|
| FBN18F |
TTCCAAATATTGTGATGGACAAA | 448 | 60 |
| FBN18R |
ACAGGGTTTTTCTGGTCCAA |
|
|
| FBN19F |
GCTGTTTCCAGGGACATGAT | 441 | 60 |
| FBN19R |
TTTATGGGAGGCAAAACGTC |
|
|
| FBN110F |
AGCCCCAGTGTGAAGTATGG | 396 | 60 |
| FBN110R |
TTCCCTGGACGTCATCTCTT |
|
|
| FBN111F |
TGACTTCTGTGGGCCTATGA | 300 | 59 |
| FBN111R |
TTAACTTGAACAATGCAAGAAAAA |
|
|
| FBN112F |
TTGTCACCAGACGACCTTTG | 383 | 60 |
| FBN112R |
CCACCAAGTTTGGGGTAAGTT |
|
|
| FBN113F |
AAAAGGAACCCAGAAAGTCTTAGAA | 295 | 60 |
| FBN113R |
CTTCCGGCATGGGTTATTTA |
|
|
| FBN114F |
GGAGGGAGGGGGAAATAAA | 244 | 60 |
| FBN114R |
ACTGCAATGGAAGGAGAGGA |
|
|
| FBN115F |
GATCTTATTTGGATGAAAGTTAGCC | 400 | 59 |
| FBN115R |
AGTCAGGTTTCCCAAACCAA |
|
|
| FBN116F |
TTCCCCATTTTCAAGGGTTA | 294 | 61 |
| FBN116R |
CGTTTGTTACCATTGGGCTTT |
|
|
| FBN117F |
GGGGGTTCTCATCTGTTTGA | 242 | 60 |
| FBN117R |
CAGTACGAGGGCATCTCCAT |
|
|
| FBN118F |
ACCAAGGGCAGGATCTACCT | 188 | 60 |
| FBN118R |
ACCCACAAGAAAGCCTGATG |
|
|
| FBN119F |
CCTGTAGCTCCTAAGGTCATTACA | 300 | 60 |
| FBN119R |
CTCCCAGCAATGAAAGAAGG |
|
|
| FBN120F |
CAAAGTTTGGGCCCTTTTTA | 226 | 59 |
| FBN120R |
TGGCATTCCAAAAGATAGCA |
|
|
| FBN121F |
GGCCCAAGACTAGATTTTAGCA | 243 | 60 |
| FBN121R |
TTTTGCAGGAAAAGCTGACA |
|
|
| FBN122F |
AATGTCAGCTTTTCCTGCAA | 368 | 59 |
| FBN122R |
TGAAATACTAGGCTTCCCCTTT |
|
|
| FBN123F |
TGTCAGAACTGCAAAGTCTGG | 204 | 60 |
| FBN123R |
GACAGCTTTATCCAGTCCGAGT |
|
|
| FBN124F |
TGCTATTCAGGCACCCTAGA | 400 | 59 |
| FBN124R |
TGGAGTGTGTGTCTGTACCTGA |
|
|
| FBN125F |
AACAGAGTGTTGGCAGTTTGG | 373 | 60 |
| FBN125R |
CTGAGATCATGAAAATGCATCC |
|
|
|
FBN126&27F |
GACCTCCTGACTGCTTGCTC | 494 | 60 |
|
FBN126&27R |
CAAAGCTTCATGGAATCCTTCT |
|
|
|
FBN128&29F |
GAGTGCTTGGTCTGGTGGAG | 564 | 61 |
|
FBN128&29R |
AGCGATGAAAACAAAACTCAGA |
|
|
| FBN130F |
GGGACAGACATCCAAACCAT | 249 | 62 |
| FBN130R |
CAAAGCCTGGGCCCTAAAC |
|
|
| FBN131F |
CTCACTGAACAGTGGAACCAA | 280 | 59 |
| FBN131R |
GCTCTCTTTGGAATGCTGGT | 280 | 59 |
| FBN132F |
GAATCTTTCTATCACTGACCCAAAC |
|
|
| FBN132R |
TCGAGGGGAAAGTACTCAATG | 325 | 59 |
|
FBN133&34F |
CATTTGTGCTGAGCCTTTTTC | 495 | 60 |
|
FBN133&34R |
GAATGCCTGGCTTCTCTGAC |
|
|
| FBN135F |
TGCTGCACTGGAAAGTTGAT | 231 | 60 |
| FBN135R |
AGTGGCTTCCCCATCAGTTA |
|
|
| FBN136F |
TGCCCAGATTGGTGTTAGAT | 400 | 59 |
| FBN136R |
CAGGTCTGAGAAAAGGTATCTGTG |
|
|
|
FBN137&38F |
AGATTGGGCCCTGTTCTTTT | 819 | 60 |
|
FBN137&38R |
TTGGGAATAAGGTCCCCTCT |
|
|
|
FBN139&40F |
TCAGACGGGCAGAGTAACAA | 496 | 59 |
|
FBN139&40R |
CCATATTCTGGTTTTGCAGGT |
|
|
| FBN141F |
AGGCCATTCCAAAATGTGAA | 249 | 60 |
| FBN141R |
TTGTGAGCTCTCTTCCTCTTTGT |
|
|
| FBN142F |
ATTTCCCACATGGCATCAC | 300 | 60 |
| FBN142R |
TGCTTCCTTCGCTAAGACTGA |
|
|
| FBN143F |
CTATCCTCCCATCCCACCTT | 273 | 60 |
| FBN143R |
CAGGGTGTTTGCACAGTTTG |
|
|
| FBN144F |
CACAGGGATCATGTGCTGTC | 315 | 60 |
| FBN144R |
TCCACACCATGCCCTTTACT |
|
|
| FBN145F |
GGCTTTGTTGACTGGACACC | 218 | 62 |
| FBN145R |
GTAGGCATGTCCAGCCTGTG |
|
|
| FBN146F |
GAGCTAGGATTACTCCTGAGAATGA | 398 | 59 |
| FBN146R |
TCATGTTCAGATTGCCAAAGA |
|
|
| FBN147F |
GGCCTGGTGAACCCTAAAAT | 247 | 60 |
| FBN147R |
TTCCTTTGCTGATGCACAAT |
|
|
| FBN148F |
TGCTGGGATTATGACATCTTTG | 292 | 60 |
| FBN148R |
TTTTCCTCCAGGTTTCCAGA |
|
|
| FBN149F |
CCAGTGGGAACCTCTTCCTT | 205 | 60 |
| FBN149R |
GACACCCGACACTCCTCATT |
|
|
| FBN150F |
TGATGTCTCCATCGTGTTTTG | 208 | 61 |
| FBN150R |
ATTGAAAGCCCAAAGCCTTC |
|
|
| FBN151F |
GGAAAGCAACTGAAGGGTGT | 263 | 590 |
| FBN151R |
GCCTACAGTCTTACTTACATCATGG |
|
|
|
FBN152&53F |
GGAGAAGCTTGTAATGAATTGCT | 594 | 60 |
|
FBN152&53R |
AACTTATTTCAGTGCCATCTTGG |
|
|
| FBN154F |
TTTGGACACATTCCTGGTTTC | 207 | 60 |
| FBN154R |
CAACCAATTGTTCCCAGGAT |
|
|
| FBN155F |
CCTTTTGTTGCTGTCCATGAT | 249 | 60 |
| FBN155R |
AGGGAAGCTTTGAGGGACAT |
|
|
| FBN156F |
TCATACTCAACAGAGCAGAAGGA | 363 | 59 |
| FBN156R |
CAAGAACTCAGAGCCCAGGT |
|
|
| FBN157F |
AAGGAACAAAGGGAGGGAAG | 392 | 60 |
| FBN157R |
CAGTCATTACGGCATCTCCA |
|
|
| FBN158F |
CTGACATCCCCTTTGCCATA | 277 | 61 |
| FBN158R |
TCCCTGCAAGTATTTTTGGAC |
|
|
|
FBN159&60F |
CACTGAAGTGACCCCCTACA | 600 | 60 |
|
FBN159&60R |
TGAGGGGCAATGGTCAAT |
|
|
|
FBN161&62F |
TGTTGGCTTGACTCAAATGC | 600 | 61 |
|
FBN161&62R |
CCTCCACAAGGATTCACCAG |
|
|
| FBN163F |
TGGTGGCTCTGCTTCTTTTT | 178 | 60 |
| FBN163R |
GCCATGCATCTTGAGAGTGA |
|
|
| FBN164F |
AAGTGGCCAGATCCAATGTC | 334 | 60 |
| FBN164R |
ACCATGACCAGGAAGAGCAC |
|
|
| FBN165F |
CATCTATGCTCCCCTTCTGC | 243 | 60 |
| FBN165R |
TTCCACCACAGGAGACATCA |
|
|
| FBN166F |
GCAGCATAAGGCAGAAAATTG | 583 | 60 |
| FBN166R |
TGATTCTGATTGGGGGAAAA |
|
|
Results
Clinical findings
The parents and two daughters of a family from
Shandong, China, were included in the present study (Fig. 1). Other relatives of this family
were not willing to be tested and so additional clinical details
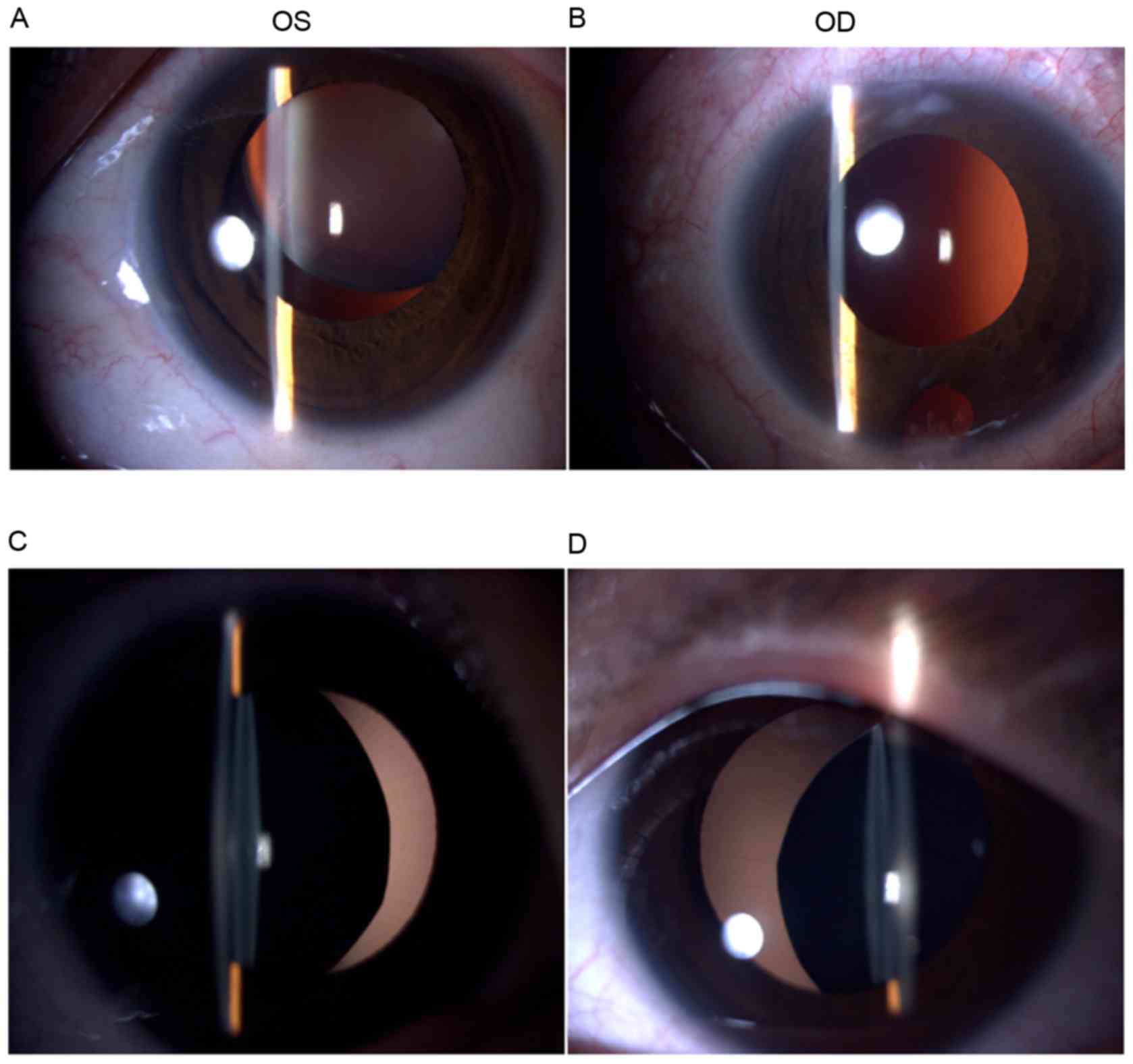
were unattainable. Two affected patients (I:1 and II:2) exhibited
similar clinical symptoms, including ectopialentis, myopia and
strabismus (Fig. 2 and Table I). The left eye of the proband
(I:1) underwent refractive lensectomy and vitrectomy combined with
silicone oil tamponade after retinal detachment 2 years prior to
the current study; following retinal re-attachment, silicone oil
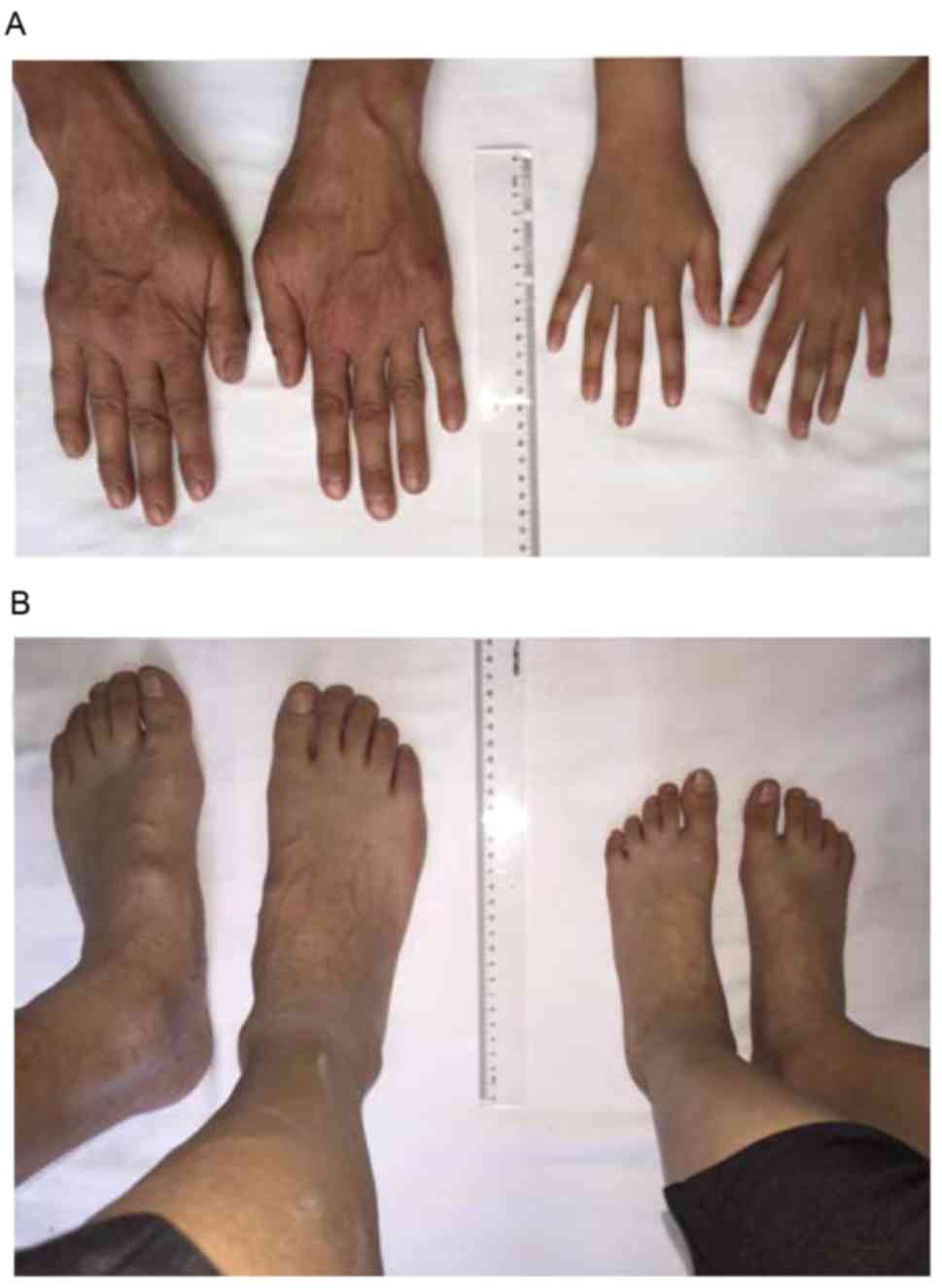
was removed 3 months later. The two patients both had the same
facial and skeletal features, including arachnodactyly, flat feet
and dilation of the aortic root (Fig.
3 and Table I). The proband
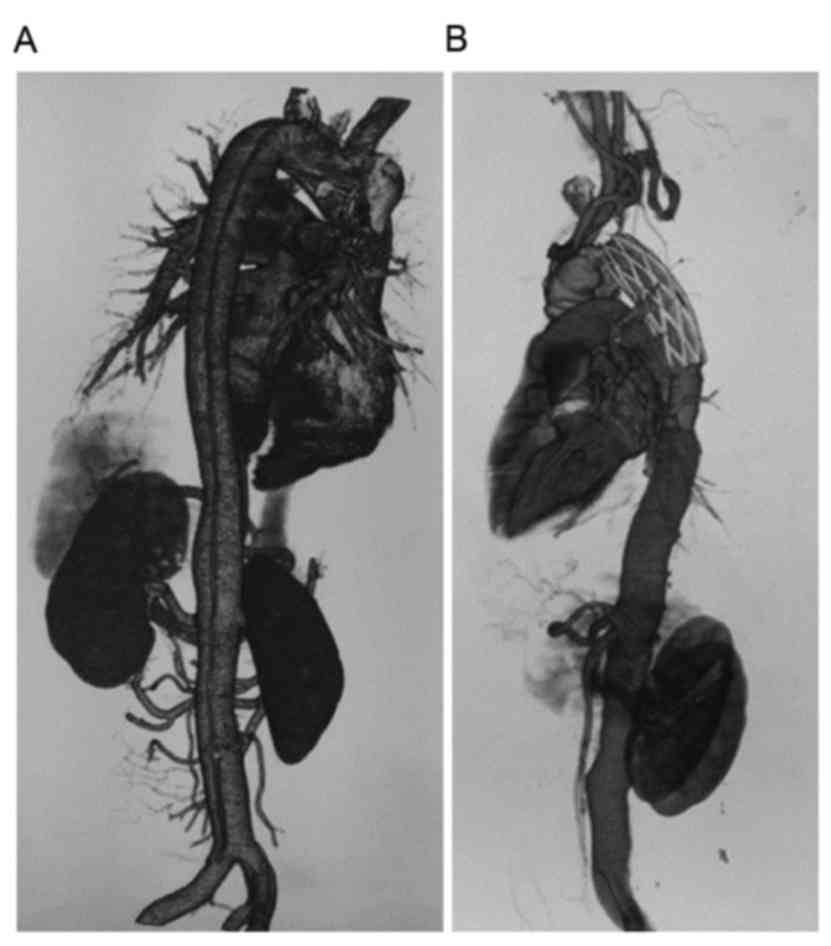
had pectus carinatum and aortic aneurysm. The patient received
Bentall surgery and underwent aortic arch replacement 5 years prior
to the current study, as their aortic aneurysm ruptured and formed
aortic dissection (Fig. 4). The
other two members of the family had no features of MFS.
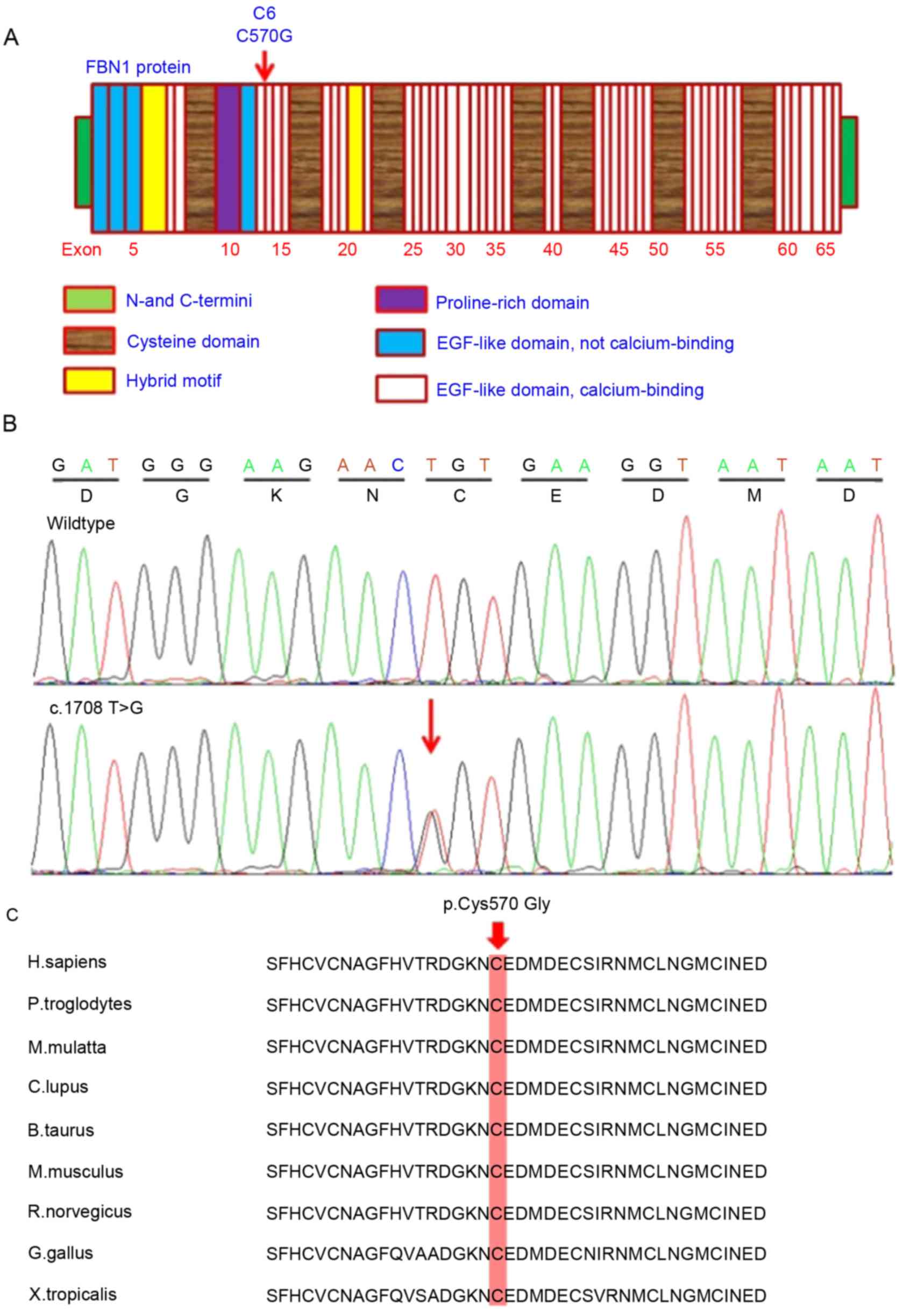
Mutation screening of FBN1
Direct sequencing of the whole coding region of
FBN1 detected a novel missense mutation c.1708 T>G
(p.C570G), situated at nucleotide 570 in exon 14 of the coding
region (Fig. 5A). This
heterozygous mutation was detected in the two affected patients
(I:1 and II:2) but was not found in the unaffected mother and
daughter (I:2 and II:1) of the family and in the 383 ethnically
matched healthy subjects. Therefore, c.1708 T>G (p.C570G)
cosegregated to the patients with MFS in this family. Multiple
sequencing alignment of human FBN1 protein with various species
revealed that the novel mutation occurred within a highly conserved
region of the calcium binding epidermal growth factor-like (cbEGF)
domain (Fig. 5C). This mutation is
a T>G transition, converting cysteine to glycine at amino acid
570 (p.C570G). This amino acid substitution in the FBN1 protein was
predicted to be damaging by SIFT and PolyPhen-2.
Discussion
It has been reported that MFS is mainly caused by
mutations in the FBN1 gene, which was the first gene
identified to cause MFS (8). Of
all the identified mutations in the FBN1 gene, 38.6% result
in a truncated FBN1 protein and 60.3% represent missense mutations
across different ethnic groups (9). FBN1 mutations may cause
abnormalities in the formation of microfibrils and fibrillin. As a
result, connective tissues weaken (10). A novel FBN1 heterozygous
missense mutation, c.1708 T>G (p.C570G) was identified within a
Chinese family associated with MFS in the present study.
FBN1 is an important component of microfibrils and
is expressed in many human tissues, including in zonules, the
cardiovascular system, cartilage, tendon and cornea. The protein
serves a role in the formation of zonules and is secreted from
ciliary bodies of non-pigmented cells (11). FBN1 protein is composed of repeated
modules, including cbEGF and transforming growth factor-1 binding
protein-like domains, and is responsible for maintaining
microfibers in an ordered arrangement (12,13).
The majority of identified missense mutations in FBN1 are
localized in cbEGF (14). The
mutated monomer of FBN1 could interfere with the polymerization of
fibrillin and microfiber aggregation (15). FBN1 mutations within cbEGF
modules may disrupt the stability of elastic fibers and render FBN1
susceptible to proteolysis. As a result, the transforming growth
factor-β signaling activity that affects extracellular matrix
formation may malfunction (4,16).
In the present study, a novel c.1708 T>G
(p.C570G) heterozygous missense mutation of the FBN1 gene
was reported in a Chinese family with MFS. Three similar missense
mutations: c.1709G>A (p.C570Y) (17), c.1709G>C (p.C570S) (18) and c.1709G>C (p.C570R) (19) have been reported in sporadic cases;
however, clinical data in these studies were not obtained. In this
pedigree, c.1708 T>G (p.C570G) in FBN1 was detected in
the two patients with MFS (I:1 and II:2). The proband (I:1)
initially came to Shandong Provincial Hospital to see an
ophthalmologist and was found to suffer from ectopialentis, myopia
and strabismus in both eyes. The proband and the affected daughter
(II:2) had similar facial and skeletal features of MFS, including
arachnodactyly, flat feet and dilation of aortic root. In addition,
pectus carinatum, aortic dissection and retinal detachment were
also detected in the proband. These findings suggested that the
clinical manifestations of the patient with MFS became more evident
with age. This mutation was not included in the Exome Aggregation
Consortium dataset; c.1708 T>G (p.C570G) of FBN1 was not
detected in the mother (I:2) and another daughter (II:1) of this
family, or in the 383 unrelated normal controls during the mutation
screening in the present study. This indicated that c.1708 T>G
(p.C570G) of FBN1 cosegregated with affected MFS patients
and may serve an important role in the pathogenesis of MFS
development in this pedigree.
The p.C570G mutation of FBN1 identified in
this family with MFS resulted in a substitution of a highly
conserved cysteine residue for glycine in a cbEGF domain of
FBN1. This mutation is predicted to abolish one disulfide
bond and thus affect the sixth conserved cysteine (C6) of the cbEGF
domain; disulfide bonds are essential for the correct EGF-like
domain structure. SIFT and PolyPhen-2 predictions indicated that
this mutation is critical to protein function, supporting a
possible pathogenic effect of this mutation. Evidence has revealed
that most FBN1 mutations are clustered in exons 24–32, a hot
spot region associated with classic and severe forms of MFS
(17,20); mutations in exons 12–15 encoding
cbEGF-like domains (C3-C6) cause a mild phenotype of MFS with
possible late cardiovascular involvement (21). Evidence from the present study
consistently indicated that the identified heterozygous mutation,
c.1708T>G, is located at exon 14 and that this cysteine
substitution detected in the proband resulted in pectus carinatum
and aortic dissection. These two factors correlated with increasing
age. However, evident symptoms were not detected in the young
affected daughter (II:2), even though significant dilation of the
aortic root was identified. Nevertheless, further functional
analyses are required to confirm the role of FBN1 and its
underlying mechanisms in MFS.
In conclusion, a novel heterozygous mutation, c.1708
T>G (p.C570G), in the FBN1 gene was identified in a
Chinese family associated with MFS. The results from the present
study enrich the spectrum of MFS-associated mutations of
FBN1 and may aid presymptomatic molecular diagnosis of
undetermined cases of MFS.
Acknowledgements
The present study was supported by grants from the
Natural Science Foundation of China [grant nos. 81670853 (B.G) and
81371048 (B.G)], the Department of Science and Technology of
Sichuan Province [grant no. 2015HH0031 (B.G.)] and the Health and
Family Planning Commission of Sichuan Province of China [grant no.
16ZD028 (B.G.)].
References
|
1
|
Judge DP and Dietz HC: Marfan's syndrome.
Lancet. 366:1965–1976. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Radonic T, de Witte P, Groenink M, de
Bruin-Bon RA, Timmermans J, Scholte AJ, van den Berg MP, Baars MJ,
van Tintelen JP, Kempers M, et al: Critical appraisal of the
revised Ghent criteria for diagnosis of Marfan syndrome. Clin
Genet. 80:346–353. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Zhao F, Pan X, Zhao K and Zhao C: Novel
mutations of fibrillin-1 gene correlate with different phenotypes
of Marfan syndrome in Chinese families. Mol Vis. 19:751–758.
2013.PubMed/NCBI
|
|
4
|
Boileau C, Jondeau G, Mizuguchi T and
Matsumoto N: Molecular genetics of Marfan syndrome. Curr Opin
Cardiol. 20:194–200. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Baetens M, Van Laer L, De Leeneer K,
Hellemans J, De Schrijver J, Van De Voorde H, Renard M, Dietz H,
Lacro RV, Menten B, et al: Applying massive parallel sequencing to
molecular diagnosis of Marfan and Loeys-Dietz syndromes. Hum Mutat.
32:1053–1062. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Faivre L, Collod-Beroud G, Child A,
Callewaert B, Loeys BL, Binquet C, Gautier E, Arbustini E, Mayer K,
Arslan-Kirchner M, et al: Contribution of molecular analyses in
diagnosing Marfan syndrome and type I fibrillinopathies: An
international study of 1009 probands. J Med Genet. 45:384–390.
2008. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Vollbrandt T, Tiedemann K, El-Hallous E,
Lin G, Brinckmann J, John H, Bätge B, Notbohm H and Reinhardt DP:
Consequences of cysteine mutations in calcium-binding epidermal
growth factor modules of fibrillin-1. J Biol Chem. 279:32924–32931.
2004. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Dietz HC, Cutting GR, Pyeritz RE, Maslen
CL, Sakai LY, Corson GM, Puffenberger EG, Hamosh A, Nanthakumar EJ,
Curristin SM, et al: Marfan syndrome caused by a recurrent de novo
missense mutation in the fibrillin gene. Nature. 352:337–339. 1991.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Faivre L, Collod-Beroud G, Loeys BL, Child
A, Binquet C, Gautier E, Callewaert B, Arbustini E, Mayer K,
Arslan-Kirchner M, et al: Effect of mutation type and location on
clinical outcome in 1,013 probands with Marfan syndrome or related
phenotypes and FBN1 mutations: An international study. Am J Hum
Genet. 81:454–466. 2007. View
Article : Google Scholar : PubMed/NCBI
|
|
10
|
Cañadas V, Vilacosta I, Bruna I and Fuster
V: Marfan syndrome. Part 1: Pathophysiology and diagnosis. Nat Rev
Cardiol. 7:256–265. 2010.PubMed/NCBI
|
|
11
|
Dureau P: Pathophysiology of zonular
diseases. Curr Opin Ophthalmol. 19:27–30. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Whiteman P and Handford PA: Defective
secretion of recombinant fragments of fibrillin-1: Implications of
protein misfolding for the pathogenesis of Marfan syndrome and
related disorders. Hum Mol Genet. 12:727–737. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Werner JM, Knott V, Handford PA, Campbell
ID and Downing AK: Backbone dynamics of a cbEGF domain pair in the
presence of calcium. J Mol Biol. 296:1065–1078. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dietz HC, Saraiva JM, Pyeritz RE, Cutting
GR and Francomano CA: Clustering of fibrillin (FBN1) missense
mutations in Marfan syndrome patients at cysteine residues in
EGF-like domains. Hum Mutat. 1:366–374. 1992. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Dietz HC, McIntosh I, Sakai LY, Corson GM,
Chalberg SC, Pyeritz RE and Francomano CA: Four novel FBN1
mutations: Significance for mutant transcript level and EGF-like
domain calcium binding in the pathogenesis of Marfan syndrome.
Genomics. 17:468–475. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mizuguchi T, Collod-Beroud G, Akiyama T,
Abifadel M, Harada N, Morisaki T, Allard D, Varret M, Claustres M,
Morisaki H, et al: Heterozygous TGFBR2 mutations in Marfan
syndrome. Nat Genet. 36:855–860. 2004. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Loeys B, Nuytinck L, Delvaux I, De Bie S
and De Paepe A: Genotype and phenotype analysis of 171 patients
referred for molecular study of the fibrillin-1 gene FBN1 because
of suspected Marfan syndrome. Arch Intern Med. 161:2447–2454. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Ogawa N, Imai Y, Takahashi Y, Nawata K,
Hara K, Nishimura H, Kato M, Takeda N, Kohro T, Morita H, et al:
Evaluating Japanese patients with the Marfan syndrome using
high-throughput microarray-based mutational analysis of fibrillin-1
gene. Am J Cardiol. 108:1801–1807. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schrijver I, Liu W, Brenn T, Furthmayr H
and Francke U: Cysteine substitutions in epidermal growth
factor-like domains of fibrillin-1: Distinct effects on biochemical
and clinical phenotypes. Am J Hum Genet. 65:1007–1020. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tiecke F, Katzke S, Booms P, Robinson PN,
Neumann L, Godfrey M, Mathews KR, Scheuner M, Hinkel GK, Brenner
RE, et al: Classic, atypically severe and neonatal Marfan syndrome:
Twelve mutations and genotype-phenotype correlations in FBN1 exons
24–40. Eur J Hum Genet. 9:13–21. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Pepe G, Lapini I, Evangelisti L, Attanasio
M, Giusti B, Lucarini L, Fattori R, Pellicanò G, Scrivanti M,
Porciani MC, et al: Is ectopia lentis in some cases a mild
phenotypic expression of Marfan syndrome? Need for a long-term
follow-up. Mol Vis. 13:2242–2247. 2007.PubMed/NCBI
|