Introduction
Acute lung injury (ALI) and the more severe form,
acute respiratory distress syndrome (ARDS), are the primary cause
of mortality and morbidity in intensive care (1,2). ALI
and ARDS are associated with similar characteristics, including
increased alveolar epithelial and pulmonary microvascular
endothelial permeability, pulmonary edema and fibrosis (3). However, the pathogenesis of these
diseases has not been fully elucidated, and no specific and
effective pharmacological intervention for ALI/ARDS is currently
available (4). Lipopolysaccharide
(LPS), a glycoprotein that is present on the outer membrane of
gram-negative bacilli, is a potent proinflammatory molecule that
leads to strong inflammatory responses. LPS triggers ALI/ARDS by
directly or indirectly damaging pulmonary microvascular endothelial
cells (PMVECS), resulting in increased alveolar capillary membrane
permeability and subsequent pulmonary edema, refractory hypoxemia
and pulmonary hypertension (5).
Vitamin D3, also termed cholecalciferol, is
primarily produced in the skin following sunlight exposure. It is
subsequently hydroxylated in the liver to produce 25-hydroxyvitamin
D3 [25(OH)D3], and again in the kidney to generate
1,25-dihydroxyvitamin D3 [1,25(OH)2D3] (6). 1,25-(OH)2D3 generation is an
indicator of local paracrine/autocrine action in various tissues,
such as pulmonary epithelial cells (7). Together, 1,25(OH)2D3 and
interleukin-2 inhibit the production of inflammatory cytokines by T
cells, and stimulate the development of Treg cells (8), which have been reported to exhibit
important functions in the treatment of experimental ALI (9). In addition, the vitamin D receptor
(VDR), which mediates the activities of 1,25(OH)2D3, protects
against sepsis-induced lung injury by inhibiting the
angiopoietin-2-TEK receptor tyrosine kinase-myosin light-chain
kinase pathway (10). These
results indicate a potentially beneficial effect of vitamin D on
ALI. However, the specific direct effect of vitamin D on
LPS-induced ALI/ARDS and the underlying mechanism are yet to be
fully investigated.
The renin-angiotensin (Ang) system (RAS), which
includes Ang I-converting enzyme (ACE) and ACE2, is a complex
network that has a major role in various biological functions,
including blood pressure regulation and water balance. ACE cleaves
Ang I into Ang II, while ACE2, a homologue of ACE, functions as an
endogenous counter-regulator of ACE by hydrolyzing Ang II into
Ang-(1–7) (11).
Upon binding to the Ang II type 1 receptor (AT1R), Ang II causes
vasoconstriction, inflammation and apoptosis, and Ang-(1–7)
opposes the effects of Ang II by interacting with its own receptor,
Mas (12). Therefore, the balance
between ACE and ACE2 levels affect the endogenous ratio of Ang II:
Ang-(1–7). ACE2 is closely associated with ARDS.
An animal model of ARDS, ACE2 knockout mice were reported to
exhibit severe lung disease, which was determined by increases in
vascular permeability and lung edema when compared with wild-type
mice (13). In addition,
recombinant ACE2 was previously reported to improve pulmonary blood
flow and oxygenation in LPS-induced lung injury in piglets
(14). Furthermore, overexpression
of ACE2 attenuated LPS-induced ARDS via the Ang-(1–7)/Mas
pathway by inhibiting extracellular signal-regulated kinase/nuclear
factor-κB (NF-κB) activation (15). Therefore, ACE2 may protect against
ALI. Vitamin D negatively regulatory blood pressure by inhibiting
the expression of RAS (16),
1α-hydroxylase-deficient mice exhibit increased activity of the
intrarenal RAS that is downregulated with the administration of
1,25(OH)2D (17), chronic vitamin
D deficiency may induce RAS activation lung fibrosis through
activation of the RAS (18);
therefore, increasing evidence indicates that 1,25(OH)2D3 may also
be a negative endocrine regulator of the RAS. However, whether
vitamin D may affect ALI by regulation of the RAS remains to be
investigated.
The aim of the present study was to investigate the
effect of a vitamin D agonist, calcitriol, on LPS-induced ALI in
vivo and in vitro, and to assess the effect of vitamin D
treatment on RAS activity. The results indicated that calcitriol
exhibits a protective role against ALI, and calcitriol may exhibit
this function, at least partially, by regulating the RAS.
Materials and methods
Animals and treatment
The ethics committee of Anhui Medical University of
Animal Welfare approved these experiments. Wistar rats (30 healthy
males; weight, 200±20 g; 3–4 months old; food and water available
ad libitum) were purchased from Shanghai SLAC Laboratory Animal
Co., Ltd. (Shanghai, China) and bred in a modified specific
pathogen free facility (temperature 22±2°C; humidity 55±2%) with a
12 h light/dark cycle). To induce lung injury, 3–4-month-old rats
(male; weight, 200±20 g; 6 groups of 5 rats per group) were
injected by caudal vein with one dose (5 mg/kg) of LPS
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). No treatment
control (NT) rats were treated with physiological saline. After 24
h, bronchoalveolar lavage (BAL) fluid was collected and the lung
was harvested to assess injury. In certain groups, rats received
supplementation of 1, 5 or 25 mg/kg calcitriol (1086301;
Sigma-Aldrich; Merck KGaA) by means of an intragastric injection 3
days prior to being challenged with LPS, while the control rats
were administered physiological saline.
Assessment of extravasation of Evans
blue
Pulmonary barrier permeability was assessed by using
Evans blue dye. Evans blue dye (20 mg/kg) was injected via the
caudal vein into rats 30 min prior to anesthesia, and was extracted
from the lung lobes by incubation at room temperature for 24 h in
formamide (3 ml/100 mg). The density of the supernatant was
assessed spectrophotometrically at 620 nm. The total amount of
Evans blue was determined against standard absorbance curves.
Total bronchoalveolar lavage fluid
cell count
Suture clamps were placed on the right main bronchus
resulting in the alveolus lavage being forced into the left lung. A
tracheal injection of 3 ml physiological saline was injected and
the lavage fluid was recovered; this was repeated three times. The
lavage fluid was centrifuged for 10 min at 100 × g in 4°C to form a
precipitant. The supernatant was removed and the precipitant was
diluted to 1 ml by PBS containing 1% bovine serum albumin (Cat.
#5003; TBD Science, Tianjin, China) and 0.1 µl transferred to a
blood cell count plate. The number of cells were then counted under
light microscopy at ×100 magnification.
PMVEC isolation and maintenance
Isolation of microvascular endothelial cells was
performed according to methodology described in our previous study
(19). Briefly, the fresh lungs,
isolated from the sacrificed rats, were washed with 50 ml
serum-free DMEM (FI101-01; Beijing Transgen Biotech Co., Ltd.,
Beijing, China). The pleura was carefully excised and discarded.
The outer edges of the remaining lung tissue, which did not contain
large blood vessels, were harvested and again rinsed in serum-free
DMEM. This tissue was decreased further, using scissors, and was
inserted into a glass pellet (Costar, Cambridge, MA) and washed in
DMEM containing 20% fetal calf serum (SH40007-10; GE Healthcare
Life Sciences. Logan, UT, USA), 100 U/ml penicillin/streptomycin
and 2.5 g/ml amphotericin B. Tissue cultures were incubated at 37°C
in saturated air containing 5% CO2. After 60 h, the
tissue was removed and thereafter, the culture medium was replaced
every 3 days. Following reaching confluence, the cells were
harvested by treating with a 0.25% trypsin. Cells were identified
as endothelial cells by cobblestone morphology. A total of 2–3
drops of cell suspension were added to a coverslip and were allowed
time to adhere. Coverslips were then washed with PBS 3 times prior
to being air dried and fixed in cold acetone for 5–10 min.
Coverslips were dried again and 0.5 ml fluorescein isothiocyanate
labeled phytohemagglutinin (FITC-PHA; 25 mg/l; Sigma-Aldrich; Merck
KGaA) was added and incubated for 30 min at 37°C. Finally,
coverslips were observed by fluorescence microscopy at ×400
magnification. The cells used for experiments were between passages
2 and 5.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis
Total RNA was extracted from rat lung tissue and
cultured rat PMVECs using TRIzol reagent (Sigma-Aldrich; Merck
KGaA). cDNA was synthesized from 2 µg total RNA using a PrimeScript
RT reagent kit with gDNA Eraser (Takara Bio, Inc., Otsu, Japan),
according to the manufacturer's protocol. qPCR was performed using
the SYBR Premix DimerEraser kit (Takara Bio, Inc.) in an ABI 7300
Real-Time PCR machine (Applied Biosystems; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). Reactions were performed under
the following thermocycling conditions: 95°C for 60 sec, followed
by 40 cycles at 95°C for 15 sec, 61°C for 15 sec and 72°C for 45
sec. The relative expression level was determined by the
2−ΔCq method and normalized against GAPDH (20). The primers used are as follows:
GAPDH, 5′-GTGCTGAGTATGTCGTGGAG-3′ (forward) and
5′-CGGAGATGATGACCCTTTT-3′ (reverse); ACE,
5′-CGGTTTTCATGAGGCTATTGGA-3′ (forward) and
5′-TCGTAGCCACTGCCCTCACT-3′ (reverse); ACE2,
5′-ACCCTTCTTACATCAGCCCTACTG-3′ (forward) and
5′-TGTCCAAAACCTACCCCACATAT-3′ (reverse); AT1R,
5′-GAAGCCAGAGGACCATTTGG-3′ (forward) and
5′-CACTGAGTGCTTTCTCTGCTTCA-3′ (reverse); renin,
5′-TTACGTTGTGAACTGTAGCCA-3′ (forward) and
5′-AGTATGCACAGGTCATCGTTC-3′ (reverse); and Ang II,
5-GTGGAGGTCCTCGTCTTCCA-3′ (forward) and
5′-GTTGTAGGATCCCCGAATTTCC-3′ (reverse); AT2R,
5-GCCAACATTTTATTTCCGAGATG-3′ (forward) and
5′-TTCTCAGGTGGGAAAGCCATA-3′ (reverse).
Renin activity and angiotensin II
assays
Renin activity and angiotensin II concentration in
the bronchoalveolar lavage fluid or culture medium of rat PMVECs
were determined using a Rat Renin ELISA kit (cat. no. CSB-E08702r;
Flarebio Biotech LLC, College Park, MD, USA) or a Rat Ang II ELISA
kit (cat. no. CSB-E04494r; Flarebio Biotech LLC), according to
manufacturer's protocol.
Western blot analysis
Rat lung tissue or cultured rat PMVECs were lysed in
ice-cold radioimmunoprecipitation assay cell lysis buffer (Beyotime
Institute of Biotechnology, Haimen, China) supplemented with 1 mM
phenylmethylsulfonyl fluoride (Beijing Transgen Biotech Co., Ltd.).
Protein concentration of samples were estimated by BCA. The
proteins (20 µg/lane) were separated with a SDS-PAGE (4–12% gel;
Bio-Rad Laboratories, Inc., Hercules, CA, USA) and
electrotransferred onto a polyvinylidene fluoride membrane. The
membrane was put in glass bottle containing 20 ml sealing liquid (1
g skimmed milk powder, 20 ml PBS-Tween 20) and then was placed on
the horizontal shaker and left to agitate at 25°C for 2 h. Probing
was performed with specific primary antibodies (4°C overnight) and
horseradish peroxidase-conjugated secondary antibodies (25°C for 2
h). The primary antibodies used were ACE (1:1,000; ab11734), ACE2
(1:500; ab108252), AT1R (1:500; ab124734) and AT2R (1:100; ab92445)
from Abcam (Cambridge, UK) and GAPDH (1:1,000; HC301) from Beijing
Transgen Biotech Co., Ltd. The secondary antibodies were
horseradish peroxidase-conjugated affinity-purified goat
anti-rabbit IgG (1:3,000; HS101-01) from Beijing Transgen Biotech
Co., Ltd. Signals were detected using SuperSignal West Femto Trial
kit (Thermo Fisher Scientific, Inc.; 34094; Lot#QE218149). The
relative amount of proteins was quantified using gel analysis
software UNSCAN-IT gel (version 5.3; Silk Scientific, Orem, UT,
USA) and normalized to GAPDH internal loading control.
Statistical analysis
Data are presented as the mean ± standard deviation
from at least three independent experiments. Statistical
comparisons were performed by one-way analysis of variance followed
by Tukey's test for multiple comparisons using SPSS (version 10.01;
SPSS, Inc., Chicago, IL, USA). P<0.05 was considered to indicate
a statistically significant difference.
Results
Calcitriol impairs the effect of LPS
on the expression of ACE and ACE2 in rat PMVECs
As previously described in our previous study
(19), PMVECs were isolated from
the outer periphery of the rat lung tissue and grew in monolayers
with morphology consistent with endothelial cells, examined by
phase-contrast microscopy. The cells grew initially as
capillary-like structures and assumed cobblestone morphology,
typical of endothelial cells at confluence. These cells were
characterized as endothelial cells by factor VIII-associated Ag
expression, and combined with FITC-PHA to demonstrate yellow green
fluorescence. (Fig. 1A). To
investigate the effects of calcitriol and LPS on the expression of
ACE and ACE2 in PMVECs, rat PMVECs were incubated with 100 µg/ml
LPS in the presence or absence of different concentrations of
calcitriol (5, 20 or 100 nM). As demonstrated in Fig. 1B, compared with the NT group, LPS
significantly induced ACE mRNA expression (8.1967±0.5749 vs.
4.7257±0.4767; P<0.01), and inhibited ACE2 mRNA expression
(0.7553±0.0723 vs. 1.3931±0.0714; P<0.01; Fig. 1B). ACE and ACE2 have opposing
functions in ALI; ACE2 counterbalances the deleterious effects of
ACE and prevents lung injury (21,22).
Therefore, the results demonstrated that LPS successfully induced
ALI in the present study.
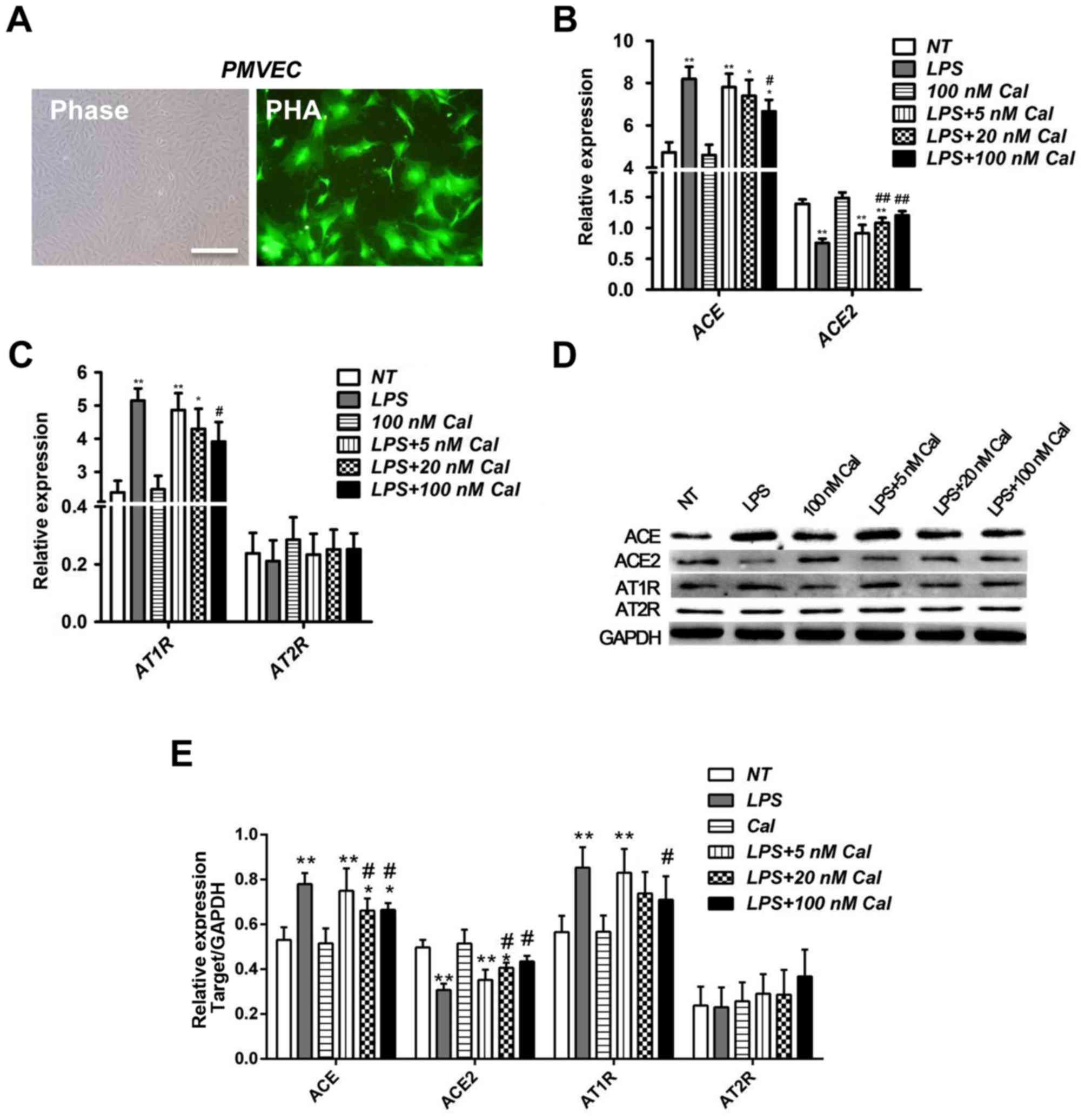 | Figure 1.Cal inhibits ACE and AT1R expression,
and induces ACE2 expression in LPS-treated rat PMVECs. (A)
Morphology of rat PMVECs and immunostaining for PMVEC marker
FITC-PHA. Phase: Normal PMVEC morphometrics under phase-contrast
microscopy (magnification, ×200). The cells grew initially as
capillary-like structures and assumed typical cobblestone
morphology of endothelial cells at confluence. PHA: PMVECs bound to
FITC-PHA under fluorescence microscopy to reveal yellow green
fluorescence (magnification, ×400). Reverse
transcription-quantitative polymerase chain reaction analysis of
(B) ACE and ACE2, and (C) AT1R and AT2R mRNA expression levels in
rat PMVECs in various treatment groups. ACE2 is a counter-regulator
of ACE and AT1R is a downstream effector of ACE. AT2R was employed
as a control. (D) Western blot analysis of the protein levels of
ACE, ACE2, AT1R and AT2R in rat PMVECs in various treatment groups.
(E) Densitometric analysis of the relative protein expression
levels of ACE, ACE2, AT1R and AT2R. Data are presented as the mean
+ standard deviation of three biological replicates. *P<0.05 and
**P<0.01 vs. NT; #P<0.05 and
##P<0.01 vs. LPS-only. Cal, calcitriol; Ang,
angiotensin; ACE, Ang I-converting enzyme; AT1R, Ang II type 1
receptor; LPS, lipopolysaccharide; PMVECs, pulmonary microvascular
endothelial cells; AT2R, Ang II type 2 receptor; NT, no treatment;
FITC-PHA, fluorescein isothiocyanate-endothelial marker protein phy
agglutinin. |
To determine whether calcitriol has an effect on
LPS-induced ALI, concentrations of 5, 20 and 100 nM calcitriol were
used in LPS-treated PMVECs. The results indicate that
calcitriol-only treatment exhibited no obvious effect on ACE and
ACE2 mRNA expression levels, compared with NT cells (Fig. 1B). However, the highest
concentration of calcitriol (100 nM) significantly reduced the
effects of LPS on ACE and ACE2 levels (Fig. 1B), which indicates an important
calcitriol-dependent pathway in attenuating ALI caused by LPS.
As AT1R is a downstream effector of ACE, the effects
of LPS and calcitriol on AT1R expression were subsequently
investigated. As demonstrated in Fig.
1C, consistent with results for the ACE expression pattern, LPS
significantly upregulated AT1R mRNA expression compared with NT
cells, while calcitriol reduced the effect of LPS on AT1R mRNA
levels in a dose-dependent manner. The mRNA expression of Ang II
type 2 receptor (AT2R), employed as a control (21,22),
was not significantly altered (Fig.
1C). Furthermore, alterations in the expression levels of ACE,
ACE2, AT1R and AT2R at the protein level were confirmed by western
blot analysis (Fig. 1D and E).
Therefore, these results indicate that calcitriol may prevent
LPS-induced ALI in rat PMVECs.
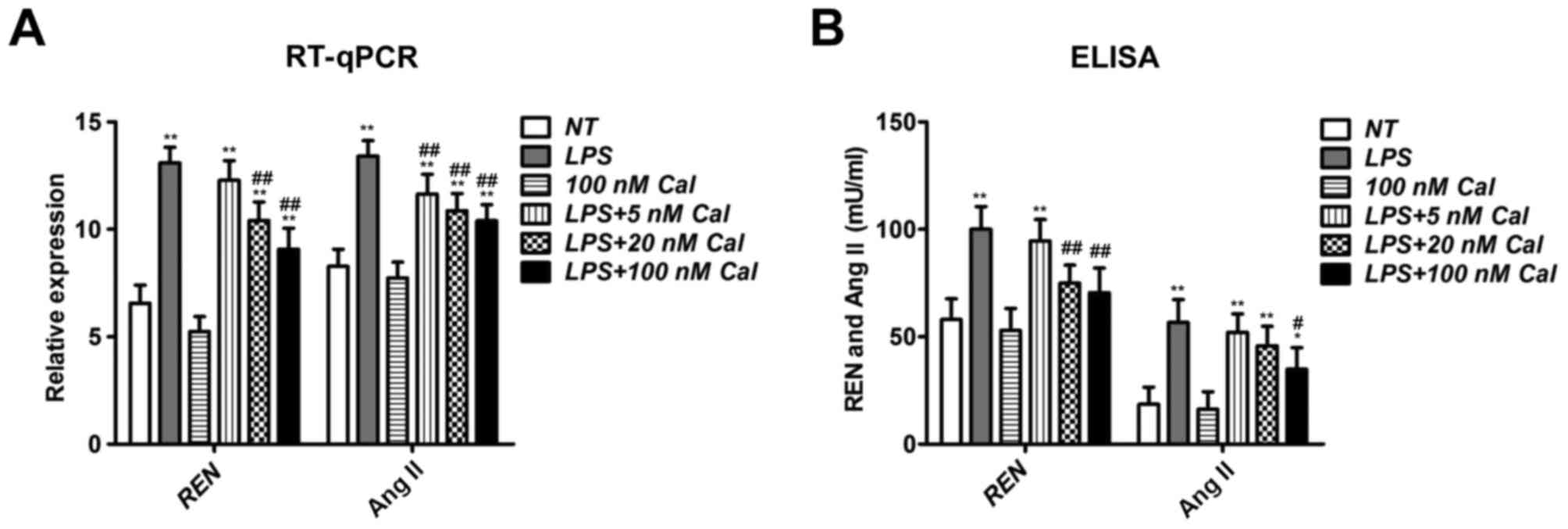
Calcitriol suppresses renin and Ang II
expression in LPS-treated rat PMVECs
Increased renin activity stimulates the conversion
of Ang I and ultimately Ang II (23), and Ang II is a key factor in the
ACE/AT1R axis. Therefore, the expression of renin and Ang II in
LPS-treated rat PMVECs was also investigated in the current study.
As presented in Fig. 2A, LPS
addition significantly increased renin and Ang II expression
compared with NT cells. Although calcitriol-only treatment
exhibited no effect on their expression compared with NT cells,
calcitriol inhibited renin and Ang II expression in LPS-treated
PMVECs in a concentration-dependent manner (Fig. 2A). ELISA was also performed to
determine renin and Ang II concentrations in the culture medium,
and the results were similar to RT-qPCR results (Fig. 2B). These results indicate that LPS
supplement may trigger the ACE/Ang II/AT1R axis, while calcitriol
exhibits an opposing function.
Calcitriol inhibits the effect of LPS
on the expression of ACE and ACE2 in rat lung tissue
To determine whether calcitriol may prevent
LPS-induced ALI in vivo, LPS was used to induce ALI in rats,
as previously reported (15), and
the total number of rat bronchoalveolar lavage cells were counted.
Compared with the NT group, a significant increase was observed in
the LPS group (Fig. 3A). However,
high concentrations of calcitriol (5 and 25 mg/kg) reduced the
number of bronchoalveolar lavage cells compared with the LPS-only
group (Fig. 3A). To assess the
severity of the lung vascular leakage, a leak index was calculated
using Evans blue dye. Compared with the NT group, the Evans blue
leakage significantly increased in the LPS group, and was reduced
in calcitriol groups in a dose-dependent manner compared with the
LPS-only group (Fig. 3B). These
results indicate that calcitriol reduces rat lung permeability
damage induced by LPS.
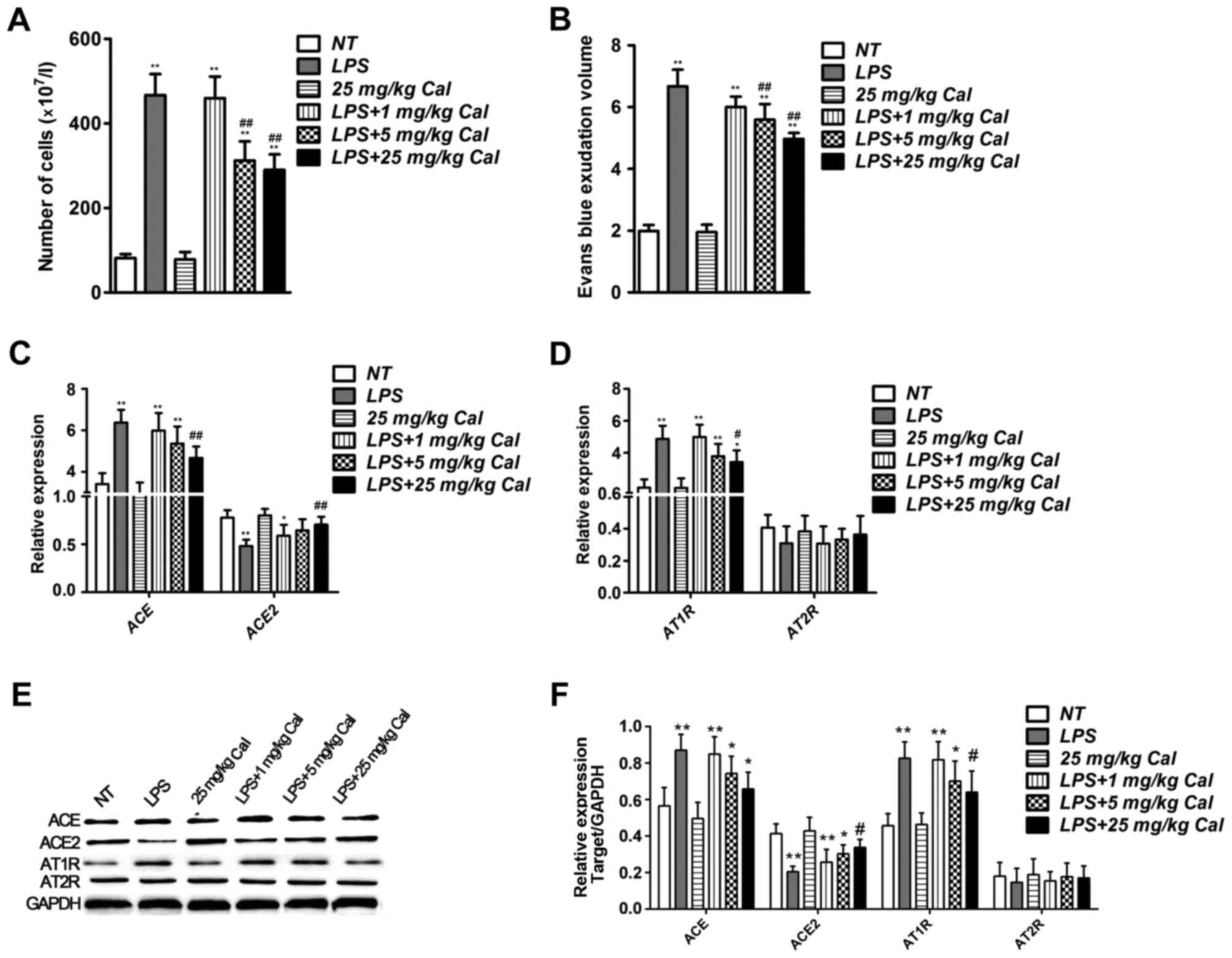 | Figure 3.Cal reduces ACE and AT1R expression,
and increases ACE2 expression in LPS-treated rat lung tissue. (A)
Number of cells in rat bronchoalveolar lavage fluid collected from
rat lung tissues in various treatment groups. (B) Results of Evans
blue permeability assays in rat lung tissues from various treatment
groups. Reverse transcription-quantitative polymerase chain
reaction analysis of (C) ACE and ACE2, and (D) AT1R and AT2R
expression in rat lung tissues from various treatment groups. (E)
Western blot analysis of the protein levels of ACE, ACE2, AT1R and
AT2R in rat lung tissues from various treatment groups. (F)
Densitometric analysis of the relative protein expression levels of
ACE, ACE2, AT1R and AT2R. Data are presented as the mean + standard
deviation of three biological replicates. *P<0.05 and
**P<0.01 vs. NT; #P<0.05 and
##P<0.01 vs. LPS-only. Cal, calcitriol; Ang,
angiotensin; ACE, Ang I-converting enzyme; AT1R, Ang II type 1
receptor; LPS, lipopolysaccharide; AT2R, Ang II type 2 receptor;
NT, no treatment. |
The mRNA expression levels of ACE and ACE2 in NT,
LPS and LPS + calcitriol-treated lung tissues were also determined,
and the results demonstrated that LPS increased ACE expression and
decreased ACE2 levels compared with the NT group. Calcitriol
treatment inhibited the effects of LPS on ACE and ACE2 mRNA
expression (Fig. 3C). Furthermore,
mRNA expression of the downstream factor AT1R was also induced by
LPS in rat lung tissue, which was suppressed when combined with
calcitriol treatment (Fig. 3D).
Western blot results confirmed alterations in expression levels at
the protein level (Fig. 3E and F).
These results indicate that calcitriol may attenuate LPS-induced
ALI in vivo.
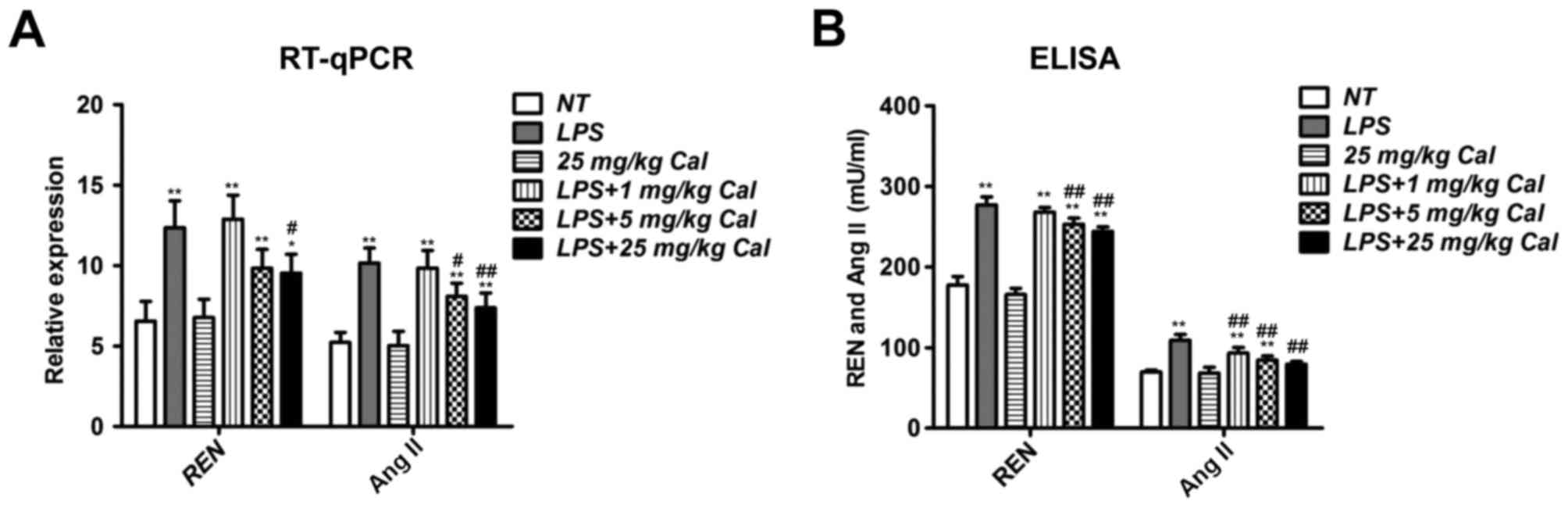
Calcitriol suppresses renin and Ang II
expression in LPS-treated rat lung tissue
As previously stated, renin and Ang II are two key
members of the RAS (23).
Therefore, the mRNA expression of renin and Ang II in rat lung
tissues was also determined. LPS treatment induced pulmonary renin
and Ang II mRNA expression in rat lung tissues significantly
compared with the NT group (Fig.
4A). However, this induction was suppressed when combined with
calcitriol treatment (Fig. 4A).
ELISA was also performed to confirm renin and Ang II concentrations
in the bronchoalveolar lavage fluid (Fig. 4B), and the results were similar to
RT-qPCR results (Fig. 4A).
Together, these results indicate that calcitriol may inhibit
LPS-induced renin and Ang II expression in vivo.
Discussion
ALI is a feature of LPS-induced sepsis, therefore,
the present study employed a LPS-induced sepsis model to
investigate the function of vitamin D in ALI. The results
demonstrated that LPS stimulation led to lung cell death and
increased vascular permeability, which may partially occur by
inducing the expression of renin, ACE, Ang II and AT1R, and
inhibiting ACE2 expression. However, in vivo and in
vitro results indicated that a vitamin D agonist, calcitriol,
significantly alleviated LPS-induced ALI to protect lungs.
ALI/ARDS is a major cause of morbidity and mortality
in seriously ill patients, and is characterized by endothelial
disruption and increased barrier permeability (24). Several previous studies have
reported that the vitamin D-mediated pathway has a potential
function in protecting against ALI. The VDR is highly expressed in
the lung, and by comparing the phenotype of lungs from LPS-treated
wild-type and VDR knockout mice, Kong et al (10) reported that VDR-knockout mice
exhibited ALI with increased severity and higher mortality compared
with wild-type mice following LPS treatment. In addition, Shi et
al (25) also demonstrated
that VDR-knockout mice experienced higher severity ALI induced by
LPS, which may primarily occur due to degeneration of the alveolar
epithelial tight junctions via reduced occludin and zonula
occludens-1 expression. However, a vitamin D analog, paricalcitol,
alleviated LPS-induced ALI and preserved alveolar barrier function.
Furthermore, calcitriol pretreatment was reported to reduce
seawater aspiration-induced ALI by inhibiting inflammatory
responses and reducing lung epithelial-endothelial barrier
permeability. The mechanism of these effects may be via inhibition
of NF-κB and the ras homolog family member A/Rho kinase signaling
pathways (26). The results of the
present study supported these previous studies by demonstrating
that vitamin D is capable of alleviating LPS-induced lung injury to
maintain the integrity of the lung.
Additionally, the present study revealed that
vitamin D may attenuate LPS-induced ALI by modulating the RAS
cascade. Previous studies have reported that 1,25(OH)2D3, the
hormonal form of vitamin D, is a negative endocrine regulator of
the RAS and inhibits renin biosynthesis (16,27).
Inhibition of the (pro)renin receptor reduced interstitial edema,
hemorrhage, neutrophil count and the amount of
non-proteolytically-activated prorenin in the lung tissues of rats
(28). In addition, dysregulation
of local and circulating RAS, with enhanced ACE/Ang II expression
levels and reduced ACE2/Ang-(1–7)
expression, was reported to contribute to
ischemia-reperfusion-induced ALI in mice (29). Furthermore, systemic infusion of
Ang II promoted ALI (30,31). However, ACE2 negatively regulated
the RAS by converting Ang II to Ang-(1–7)
(32), and preventing the
downregulation of ACE2 protected mice against LPS-induced ALI
(33). Similarly, Ang-(1–7) was
also reported to decrease the severity of ALI and inflammation
induced by a combination of acid aspiration and high-stretch
ventilation (34). The results of
the present study demonstrated that vitamin D inhibited renin, ACE
and Ang II expression, and induced ACE2 levels in LPS-induced ALI.
Therefore, vitamin D may attenuate LPS-Induced ALI by, at least
partially, inducing ACE2/Ang-(1–7) axis
activity and inhibiting renin and the ACE/Ang II/AT1R cascade.
In conclusion, the results of the current study, in
conjunction with previous findings, provide evidence for a
protective function of vitamin D in ALI and also provide insight
into the potential underlying molecular mechanism. These results
indicate that further research regarding vitamin D as a potential
therapy for ALI is required.
Acknowledgements
This research was funded by the Scientific Research
Fund of Anhui Medical University (grant no. 2011×kj083) and the
Scientific Research Fund of The First People's Hospital of Hefei
(grant no. 2016–42).
References
|
1
|
Rubenfeld GD, Caldwell E, Peabody E,
Weaver J, Martin DP, Neff M, Stern EJ and Hudson LD: Incidence and
outcomes of acute lung injury. N Engl J Med. 353:1685–1693. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Phua J, Badia JR, Adhikari NK, Friedrich
JO, Fowler RA, Singh JM, Scales DC, Stather DR, Li A, Jones A, et
al: Has mortality from acute respiratory distress syndrome
decreased over time? A systematic review. Am J Respir Crit Care
Med. 179:220–227. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Matute-Bello G, Frevert CW and Martin TR:
Animal models of acute lung injury. Am J Physiol Lung Cell Mol
Physiol. 295:L379–L399. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wheeler AP and Bernard GR: Acute lung
injury and the acute respiratory distress syndrome: A clinical
review. Lancet. 369:1553–1564. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Su X, Wang L, Song Y and Bai C: Inhibition
of inflammatory responses by ambroxol, a mucolytic agent, in a
murine model of acute lung injury induced by lipopolysaccharide.
Intensive Care Med. 30:133–140. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Haussler MR, Whitfield GK, Haussler CA,
Hsieh JC, Thompson PD, Selznick SH, Dominguez CE and Jurutka PW:
The nuclear vitamin D receptor: Biological and molecular regulatory
properties revealed. J Bone Miner Res. 13:325–349. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hansdottir S, Monick MM, Hinde SL, Lovan
N, Look DC and Hunninghake GW: Respiratory epithelial cells convert
inactive vitamin D to its active form: Potential effects on host
defense. J Immunol. 181:7090–7099. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jeffery LE, Burke F, Mura M, Zheng Y,
Qureshi OS, Hewison M, Walker LS, Lammas DA, Raza K and Sansom DM:
1,25-Dihydroxyvitamin D3 and IL-2 combine to inhibit T cell
production of inflammatory cytokines and promote development of
regulatory T cells expressing CTLA-4 and FoxP3. J Immunol.
183:5458–5467. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
D'Alessio FR, Tsushima K, Aggarwal NR,
West EE, Willett MH, Britos MF, Pipeling MR, Brower RG, Tuder RM,
McDyer JF and King LS: CD4+CD25+Foxp3+ Tregs resolve experimental
lung injury in mice and are present in humans with acute lung
injury. J Clin Invest. 119:2898–2913. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kong J, Zhu X, Shi Y, Liu T, Chen Y, Bhan
I, Zhao Q, Thadhani R and Li YC: VDR attenuates acute lung injury
by blocking Ang-2-Tie-2 pathway and renin-angiotensin system. Mol
Endocrinol. 27:2116–2125. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Tipnis SR, Hooper NM, Hyde R, Karran E,
Christie G and Turner AJ: A human homolog of angiotensin-converting
enzyme. Cloning and functional expression as a
captopril-insensitive carboxypeptidase. J Biol Chem.
275:33238–33243. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Santos RA, Ferreira AJ, Verano-Braga T and
Bader M: Angiotensin-converting enzyme 2, angiotensin-(1–7) and
Mas: New players of the renin-angiotensin system. J Endocrinol.
216:R1–R17. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Imai Y, Kuba K, Rao S, Huan Y, Guo F, Guan
B, Yang P, Sarao R, Wada T, Leong-Poi H, et al:
Angiotensin-converting enzyme 2 protects from severe acute lung
failure. Nature. 436:112–116. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Treml B, Neu N, Kleinsasser A, Gritsch C,
Finsterwalder T, Geiger R, Schuster M, Janzek E, Loibner H,
Penninger J and Loeckinger A: Recombinant angiotensin-converting
enzyme 2 improves pulmonary blood flow and oxygenation in
lipopolysaccharide-induced lung injury in piglets. Crit Care Med.
38:596–601. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Li Y, Zeng Z, Cao Y, Liu Y, Ping F, Liang
M, Xue Y, Xi C, Zhou M and Jiang W: Angiotensin-converting enzyme 2
prevents lipopolysaccharide-induced rat acute lung injury via
suppressing the ERK1/2 and NF-κB signaling pathways. Sci Rep.
6:279112016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Li YC, Qiao G, Uskokovic M, Xiang W, Zheng
W and Kong J: Vitamin D: A negative endocrine regulator of the
renin-angiotensin system and blood pressure. J Steroid Biochem Mol
Biol 89–90. 1–392. 2004.
|
|
17
|
Shi Y, Liu T, Li Y, Xing Y, Zhao X, Fu J
and Xue X: Chronic vitamin D deficiency induces lung fibrosis
through activation of the renin-angiotensin system. Sci Rep.
7:33122017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Zhou C, Lu F, Cao K, Xu D, Goltzman D and
Miao D: Calcium-independent and 1,25(OH)2D3-dependent regulation of
the renin-angiotensin system in 1alpha-hydroxylase knockout mice.
Kidney Int. 74:170–179. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhang H and Sun GY: Expression and
regulation of AT1 receptor in rat lung microvascular endothelial
cell. J Surg Res. 134:190–197. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) Method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Yang P, Gu H, Zhao Z, Wang W, Cao B, Lai
C, Yang X, Zhang L, Duan Y, Zhang S, et al: Angiotensin-converting
enzyme 2 (ACE2) mediates influenza H7N9 virus-induced acute lung
injury. Sci Rep. 4:70272014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Rey-Parra GJ, Vadivel A, Coltan L, Hall A,
Eaton F, Schuster M, Loibner H, Penninger JM, Kassiri Z, Oudit GY
and Thébaud B: Angiotensin converting enzyme 2 abrogates
bleomycin-induced lung injury. J Mol Med (Berl). 90:637–647. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Borghi C, Boschi S, Ambrosioni E, Melandr
G, Branzi A and Magnani B: Evidence of a partial escape of
renin-angiotensin-aldosterone blockade in patients with acute
myocardial infarction treated with ACE inhibitors. J Clin
Pharmacol. 33:40–45. 1993. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ware LB and Matthay MA: The acute
respiratory distress syndrome. N Engl J Med. 342:1334–1349. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Shi YY, Liu TJ, Fu JH, Xu W, Wu LL, Hou AN
and Xue XD: Vitamin D/VDR signaling attenuates
lipopolysaccharide-induced acute lung injury by maintaining the
integrity of the pulmonary epithelial barrier. Mol Med Rep.
13:1186–1194. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhang M, Dong M, Liu W, Wang L, Luo Y, Li
Z and Jin F: 1α,25-dihydroxyvitamin D3 ameliorates seawater
aspiration-induced acute lung injury via NF-κB and RhoA/Rho kinase
pathways. PLoS One. 9:e1045072014. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Li YC, Kong J, Wei M, Chen ZF, Liu SQ and
Cao LP: 1,25-Dihydroxyvitamin D(3) is a negative endocrine
regulator of the renin-angiotensin system. J Clin Invest.
110:229–238. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Ishii K, Takeuchi H, Fukunaga K, Hirano Y,
Suda K, Hagiwara T, Miyasho T, Yamada S, Nakamura R, Takahashi T,
et al: Attenuation of lipopolysaccharide-induced acute lung injury
after (pro)renin receptor blockade. Exp Lung Res. 41:199–207. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Chen LN, Yang XH, Nissen DH, Chen YY, Wang
LJ, Wang JH, Gao JL and Zhang LY: Dysregulated renin-angiotensin
system contributes to acute lung injury caused by hind-limb
ischemia-reperfusion in mice. Shock. 40:420–429. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Liu L, Qiu HB, Yang Y, Wang L, Ding HM and
Li HP: Losartan, an antagonist of AT1 receptor for angiotensin II,
attenuates lipopolysaccharide-induced acute lung injury in rat.
Arch Biochem Biophys. 481:131–136. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Wang F, Xia ZF, Chen XL, Jia YT, Wang YJ
and Ma B: Angiotensin II type-1 receptor antagonist attenuates
LPS-induced acute lung injury. Cytokine. 48:246–253. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Burrell LM, Johnston CI, Tikellis C and
Cooper ME: ACE2, a new regulator of the renin-angiotensin system.
Trends Endocrinol Metab. 15:166–169. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Shi Y, Zhang B, Chen XJ, Xu DQ, Wang YX,
Dong HY, Ma SR, Sun RH, Hui YP and Li ZC: Osthole protects
lipopolysaccharide-induced acute lung injury in mice by preventing
down-regulation of angiotensin-converting enzyme 2. Eur J Pharm
Sci. 48:819–824. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zambelli V, Bellani G, Borsa R, Pozzi F,
Grassi A, Scanziani M, Castiglioni V, Masson S, Decio A, Laffey JG,
et al: Angiotensin-(1–7) improves oxygenation, while reducing
cellular infiltrate and fibrosis in experimental acute respiratory
distress syndrome. Intensive Care Med Exp. 3:442015. View Article : Google Scholar : PubMed/NCBI
|