Introduction
Myocarditis is an inflammatory cardiac muscle
disease in association with cardiac dysfunction (1,2). The
natural history of acute myocarditis varies from complete recovery
to severe heart failure, including ventricular arrhythmias and
development of dilated cardiomyopathy (DCM) (3). Epidemiological studies of viral
myocarditis have been limited because the majority of patients have
a clinically unapparent course of myocarditis (4,5).
However, it is assumed that 2–5% of patients with viral infection
have myocardial involvement (5,6).
In general, myocarditis arises from infectious,
toxic, and immunologic causes. Infectious causes of myocarditis
include viruses, parasites, protozoa, and fungi, with viruses being
the most common cause (7,8). Coxsackieviruses are enteroviruses
that can be divided into two classes; class A is composed of 23
serotypes and class B is comprised of six serotypes.
CoxsackievirusB3 (CVB3) is believed to be the most common causative
agent of myocarditis, which can lead to DCM (5,9).
CVBs are single-stranded RNA viruses that have natural tropisms for
epithelial cells, immune cells, and cardiomyocytes (8).
Chemokines are low-molecular-mass cytokines that are
classified into the C, CC, CXC, and CX3C families based on the
spacing of their N-terminus cysteine residues (10,11).
Fractalkine (FKN; CX3CL1), the only member of the CX3C family, is
involved in several inflammatory diseases, including cardiovascular
diseases (12). FKN exists as
membrane-bound and soluble forms; the membrane-bound form is
synthesized as a transmembrane molecule with an extracellular
N-terminal domain that is attached by a mucin-like stalk to the
cell surface, whereas soluble FKN is generated via cleavage at the
base of the mucin-like stalk by two metalloproteinases, a
disintegrin and metalloproteinase (ADAM) 10 and ADAM17 (12–14).
The mitogen-activated protein kinase (MAPK)
signaling pathway links extracellular signals with intracellular
targets to control fundamental cellular processes such as
proliferation, growth, migration, differentiation, embryogenesis,
and death (15). MAPKs consist of
three subfamilies, whose members include extracellular
signal-regulated kinase 1/2 (ERK1/2), p38, and c-Jun N-terminal
kinase (JNK) (16). The
significance of MAPK in myocarditis has been highlighted in several
reviews. Ribosomal S6 kinases (RSK) comprise a family of
serine/threonine kinases that lie at the terminus of the
mitogen-regulated ERK-MAPK pathway (17). The stimulation of ERK initiates a
cascade of activating events including phosphorylation of RSK by
ERK and translocation to the nucleus where they phosphorylate
nuclear substrates. Many RSK (p90rsk contained)
substrates have been identified, implicating RSK in a myriad of
cellular processes (18,19). Previously reported a relationship
between myocarditis and the MAPK pathway (20).
Myocardial microvascular endothelial cells was one
of the role protected the myocardial, in the procedure of CVB3
infection. How did the myocardial microvascular endothelial cells
show its protecting function? And in protected procedure, how the
KFN and MAPK expression level changed. In this study, the role of
FKN in CVB3 infection of myocardial microvascular endothelial cells
were demonstrated, and the KFN and MAPK expression level also were
showed.
Materials and methods
Materials
All chemicals and reagents were purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany), unless otherwise
specified. Anti-ERK1/2 (cat. no. 16-284), anti-phospho-ERK1/2 (cat.
no. 05-797), anti-JNK (cat. no. 04-210), anti-phospho-JNK (cat. no.
46-613MAG), anti-p38 (cat. no. ABS29), anti-phospho-p38 (Cat.
MABS64), anti-p90rsk (cat. no. 04-417) and
anti-phospho-p90rsk antibodies (cat. no. ABS1849) were
purchased from EMD Millipore (Billerica, MA, USA). U0126 (cat. no.
CAS 109511-58-2), an ERK1/2 inhibitor, was purchased from Santa
Cruz Biotechnology, Inc. (Santa Cruz, CA, USA). CVB3 was provided
from China Center for Disease Control and Prevention (Beijing,
China).
Myocardial microvascular endothelial
cell culture
Human myocardial microvascular endothelial cells
(cat. no. 6000) were purchased from ScienCell Research Laboratories
(Carlsbad, CA, USA). Cells were cultured at 37°C in a humidified
atmosphere of 5% CO2 in air. Cells was grown as a
monolayer in RPMI-1640 (Hyclone; GE Healthcare Life Sciences,
Logan, UT, USA) containing 10% fetal bovine serum.
50% tissue culture infective dose
(TCID50) assay
Myocardial microvascular endothelial cells
(1×103) were seeded into each well of a 96-well plate
and incubated at 37°C overnight with 5% CO2, so that the
cells were 80% confluent at the time of infection. CVB3 viral
supernatants were collected after 6 h of cultivation and diluted
from 10−1 to 10−7. Each dilution was repeated
8 times. After incubation at 37°C with 5% CO2 for 2 h,
the diluted supernatant was replaced with 100 µl of DMEM
supplemented with 2% FBS. The plates were incubated for 24 to 96 h.
The viral titer (TCID50) was calculated according to the
method of Reed and Muench.
Western blot analysis
Myocardial microvascular endothelial cells were
homogenized. Proteins were separated by sodium dodecyl sulfate
polyacrylamide gel electrophoresis using 8–12% T-gels. The
separated proteins were transferred to a nitrocellulose membrane,
which was blocked in 5% fat-free milk for 60 min at room
temperature. The membrane was incubated with a primary antibody
(anti-ERK1/2 1:1,000, anti-phospho-ERK1/2 1:800, anti-JNK 1:1,500,
anti-phospho-JNK 1:1,000, anti-p38 1:500, anti-phospho-p38 1:800,
anti-p90rsk1:1,000, and anti-phospho-p90rsk
1:1,000), overnight at 4°C, after which it was washed in PBS and
incubated with a species-compatible secondary antibody.
Immunoreactive bands were developed using an enhanced
chemiluminescent reagent (Pierce; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) and quantified by densitometry using Quantity One
software (Bio-Rad Laboratories, Inc., Hercules, CA, USA).
RNA interference and cell
transfection
To confirm the role of FKN in AngII-induced
proliferation of myocardial microvascular endothelial cells,
Fkn siRNA was developed to silence Fkn gene expression, as
Fkn expression has been found in myocardial microvascular
endothelial cells under certain stimuli. Fkn siRNA (sense
strand, 5-GCU GUG GUA GUA AUU CAU AdT dT-3; antisense strand, 3-dTd
TCG ACA CCA UCA UUA AGU AU-5) was synthesized by RayBiotech
(Guangzhou, China) and transfected into myocardial microvascular
endothelial cells using Lipofectamine® 2000 (Invitrogen;
Thermo Fisher Scientific, Inc.) according to the manufacturer's
instructions. siRNA transfection was conducted according to the
protocol supplied by Invitrogen. Briefly, 1×105 cells
were seeded into 6-well plates containing an antibiotic-free medium
and incubated overnight. For each well, 5 µl siRNA were mixed with
125 µl OPTI-MEM I. The mixture was then combined with a solution of
5 µl lipofectamine in 125 µl OPTI-MEM I. After a 20-min incubation
period at RT, the mixture was applied to the cells in an
appropriate volume of Opti-MEM I so as to achieve a final
concentration of 100 nmol/l for each siRNA. After incubation for 6
h at 37°C, RPMI-1640 supplemented with serum was added to the
wells. Cells were cultured for an additional 24 h at 37°C before
analysis.
Statistical analysis
Differences in protein expression levels between
groups were detected using one-way analysis of variance (ANOVA)
with SPSS 13.0 and the figure was made by GraphPad Prism 5 software
(GraphPad Software, Inc., La Jolla, CA, USA). When the overall F
test result of ANOVA was significant, a multiple-comparison Tukey
test was applied. Student's t-test was used in two-mean
comparisons. Three independent replicates were performed for all
experiments. P<0.05 was considered to indicate a statistically
significant difference. Data are presented mean ± standard
deviation.
Results
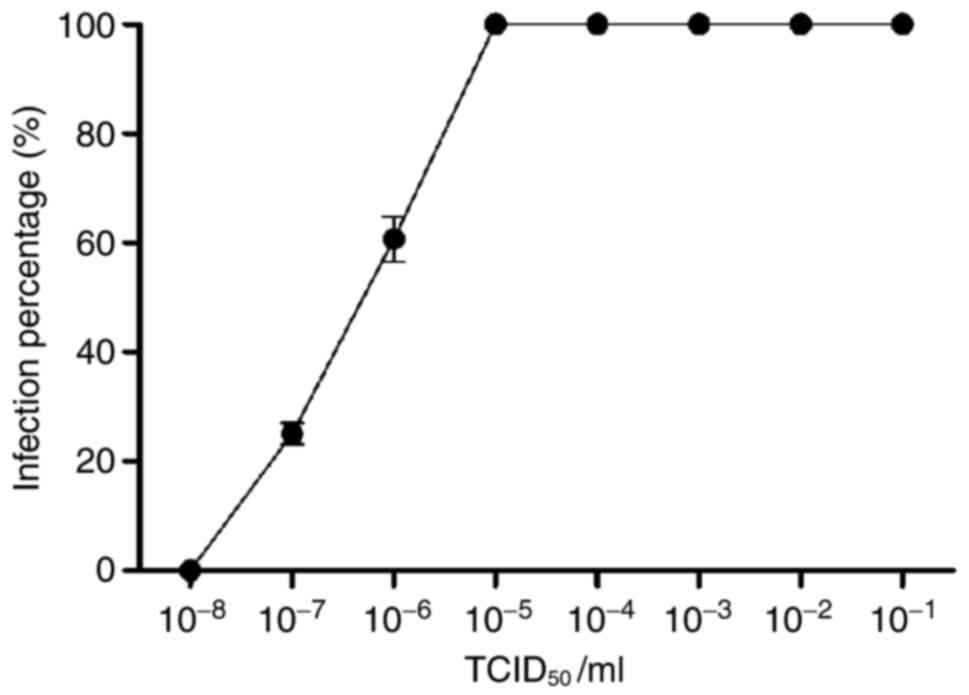
CVB3 infects myocardial microvascular
endothelial cells
The myocardial microvascular endothelial cells which
received CVB3 were randomly sacrificed everyday within 96 h after
CVB3 infection as described in the methods section, after which
TCID50 values were measured. At 10−6.825/ml
of CVB3, approximately 50% of the myocardial microvascular
endothelial cells were not viable; the TCID50 of CVB3
was 6. 825 (Fig. 1).
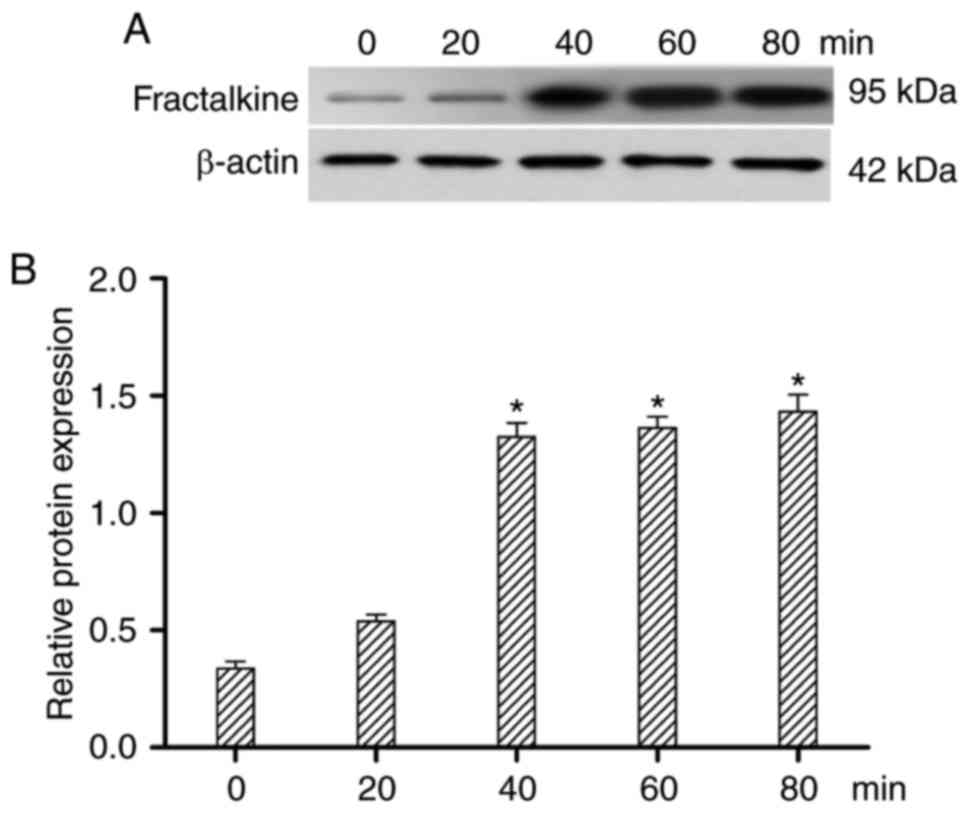
Influence of CVB3 on FKN
expression
To assess whether CVB3 treatment altered FKN
expression, FKN was detected by western blotting at 0, 20, 40, 60
and 80 min after CVB3 exposure. As shown in Fig. 2, FKN expression was increased at 0,
20, 40 min (P<0.05), but FKN expression was unchanged at 60 and
80 min. FKN protein expression peaked 40 min after CVB3
infection.
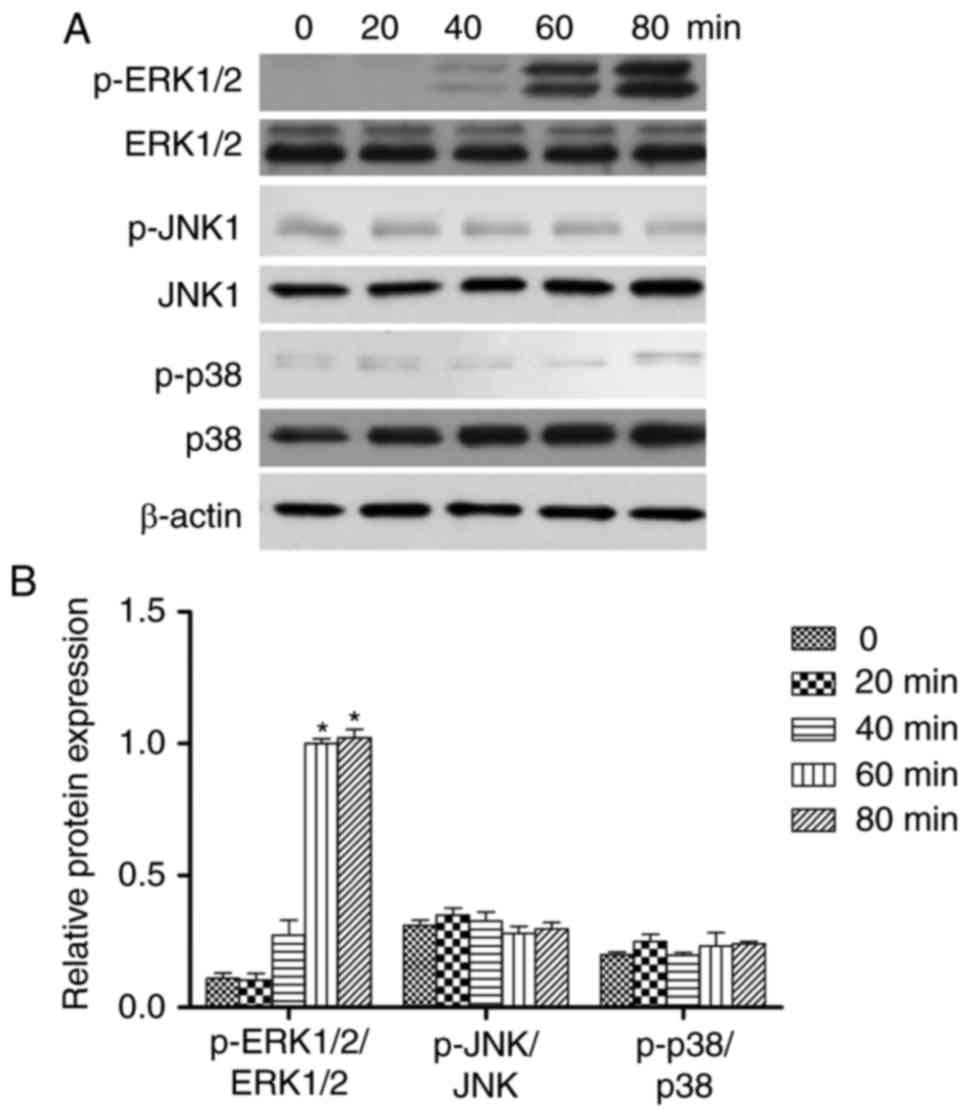
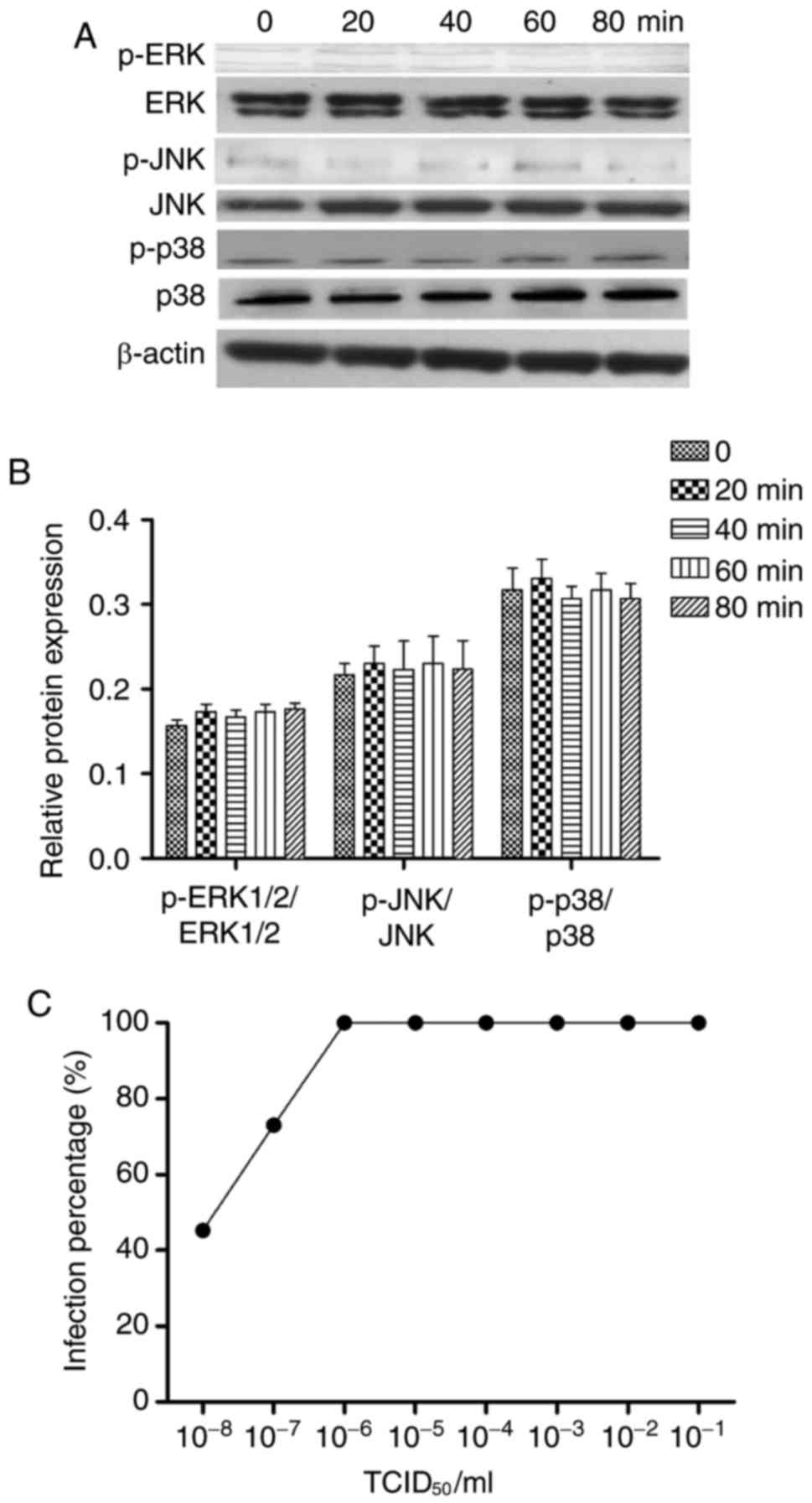
CVB3 activates the MAPK pathway
To assess MAPK signaling activity, levels of total
and phosphorylated ERK1/2, JNK, and p38 were measured 0, 20, 40, 60
and 80 min after CVB3 infection by western blot analysis. Following
CVB3 infection, the expression of phosphorylated ERK1/2 was
increased at 20, and 40 min, compared with 0 min (P<0.05), but
the expression of phosphorylated ERK1/2 was unchanged between the
60 and 80 min time points (Fig.
3). However, the expression of phosphorylated p-JNK and p-p38
were not changed in all the time. Therefore, ERK1/2 may be the
major protein regulating FKN in response to CVB3 infection.
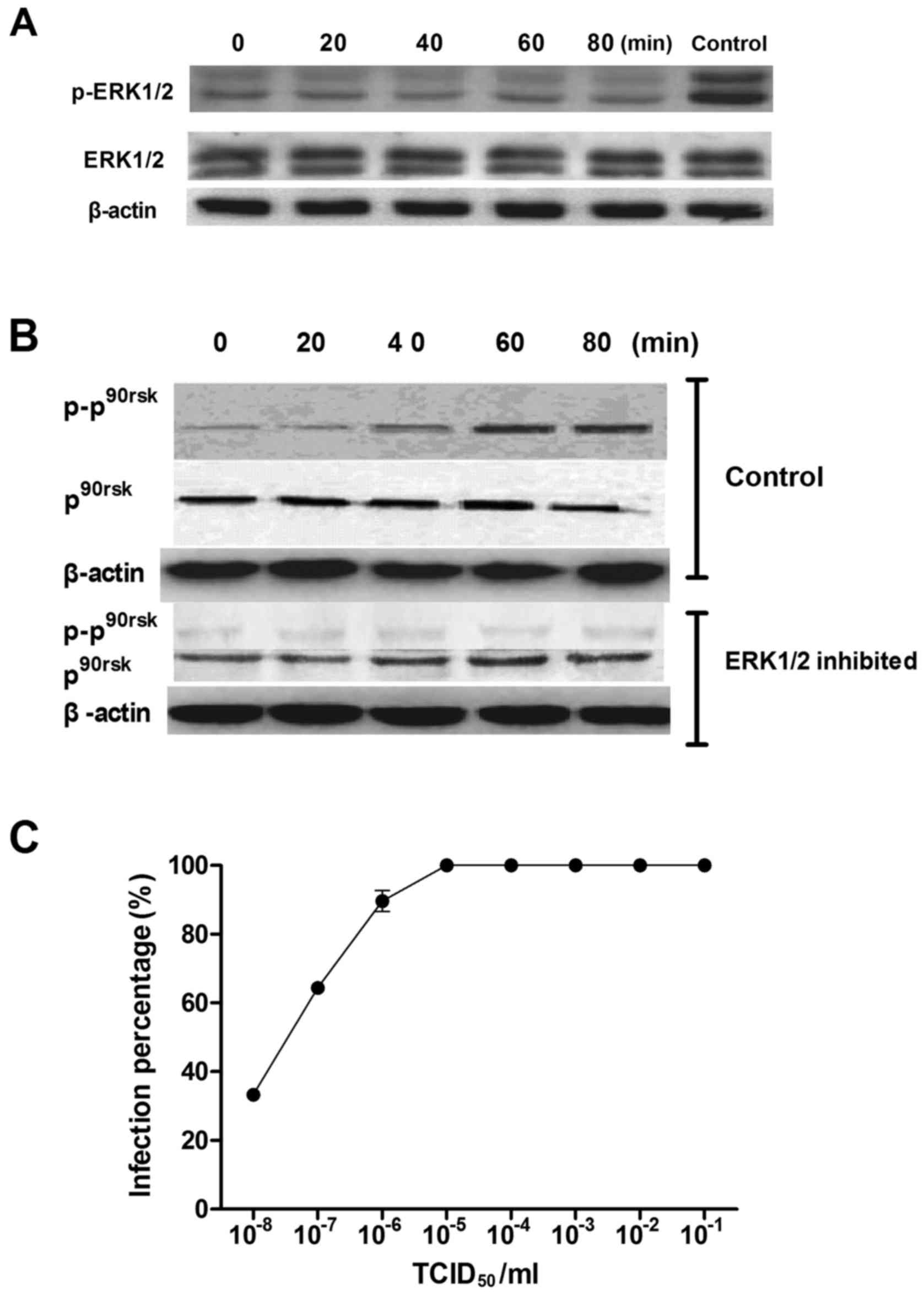
The expression of FKN was reduced following
inhibition of ERK1/2 by U0126 (Fig.
4); there was no change in the expression of phosphorylated
ERK1/2 and phosphorylated Rsk90 after CVB3 infection (Fig. 5A and B). Moreover, CVB3 showed
reduced invasiveness of myocardial microvascular endothelial cells
after they were exposed to U0126. At 10−7.48/ml of CVB3,
approximately 50% of the cells were not viable; the
TCID50 of CVB3 was 7.48 (Fig. 5C).
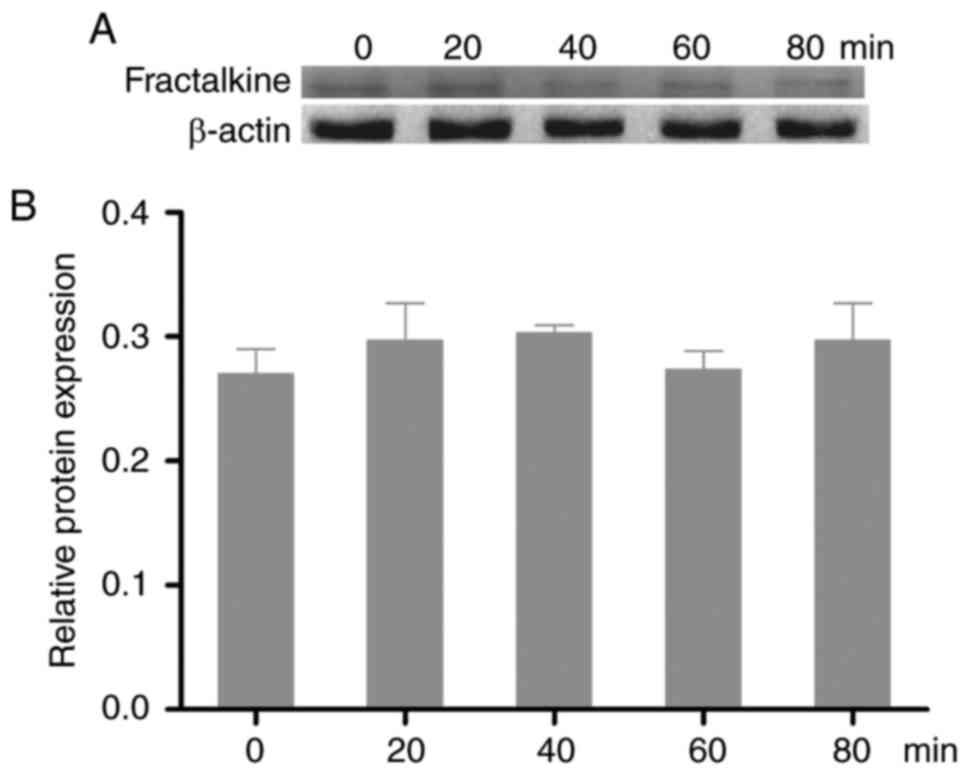
FKN influence the MAPK pathway and
CVB3 infectious
To explore the function of FKN in CVB3 infection,
FKN translation was inhibited by shRNA. As shown in Fig. 6, there was no difference in the
expression of p-ERK1/2, p-JNK, or p-p38 at any time point after FKN
was silenced (Fig. 6A and B). FKN
shRNA exposure reduced the TCID50 of CVB3 to 7.99
(Fig. 6C).
Discussion
Viral myocarditis has been recognized as a cause of
heart failure and premature death, but diagnosis is challenging;
therefore, epidemiological studies of this condition are limited
(9). During the course of
infection by CVB3, a causative agent of viral myocarditis, primary
replication occurs in the cells of the nasopharynx and the Peyer's
patches of the intestine. Next, viruses spread via the blood
circulation and interact inevitably with the vascular endothelium
to reach target organs like the myocardium (8). Only a few publications have assessed
infection of human endothelial cells by CVB3 (21,22).
Viruses interact with cell surface proteins that play integral
roles in the biology of the target cell. Endothelial cells are
exquisitely capable of responding to changes in their environment
such as tissue damage and infection (23,24).
Few studies of coxsackieviral infection of the
endothelium have been published. Recently, it was shown that CVs
infect polarized brain microvasculature and aortic endothelial
cells (8); CV infection was
associated with altered calcium signaling, which may be involved in
mechanisms enabling the virus to cross endothelial barriers.
Moreover, RNAi screening methods have shown that CV and poliovirus
infections induce numerous alterations in brain endothelial cells
(25). CVB3 replication in rat
cardiac microvascular endothelial cells alters expression of
numerous cellular factors in a manner that may contribute to the
progression of cardiac fibrosis (2).
In this study, CVB3 increased the expression of
phosphorylated ERK1/2 in infected myocardial microvascular
endothelial cells, while expression of FKN, a CX3C chemokine first
identified in endothelial cells, was increased. FKN acts as a
chemoattractant and adhesion molecule (10). Subsequent research identified
several additional cellular sources of FKN, including neurons,
microglial cells, osteoblasts, smooth muscle cells, dendritic
cells, lymphocytes, and macrophages (11). The membrane-bound form of FKN
functions as an adhesion protein, whereas soluble FKN acts as a
chemokine (26). MAPKs ERK1/2,
p38, and JNK play significant roles in regulating the synthesis of
inflammatory mediators such as NF-κB and activator protein-1.
Conversely, the production of inflammatory mediators is completely
blocked by the suppression of multiple MAPK family members
(16,27). In this study, the total ERK1/2
protein level was increased.
To better understand the role of MAPK signaling in
CVB3 infection, ERK1/2 was inhibited with U1206. The
TCID50 of CVB3 in ERK1/2-inhibited cells was higher than
that of ERK1/2 uninhibited cells. In addition, there was no change
in the expression of phosphorylated Rsk90, while FKN expression was
inhibited.
CVB3 infection of myocardial microvascular
endothelial cells activates FKN expression (against infetious) via
the ERK1/2 signaling pathway. These findings may lead to the
development of new treatments for patients with CVB3-induced
myocarditis.
References
|
1
|
Zhou X, Xin Q, Wang Y, Zhao Y, Chai H,
Huang X, Tao X and Zhao M: Total flavonoids of astragalus plays a
cardioprotective role in viral myocarditis. Acta Cardiol Sin.
32:81–88. 2016.PubMed/NCBI
|
|
2
|
Yu Y, Yu Y, Liu M, Yu P, Liu G, Liu Y, Su
Y, Jiang H and Chen R: Ethyl pyruvate attenuated coxsackievirus
B3-induced acute viral myocarditis by suppression of
HMGB1/RAGE/NF-KB pathway. Springerplus. 5:2152016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rose NR: Viral myocarditis. Curr Opin
Rheumatol. 28:383–389. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Sun XH, Fu J and Sun DQ: Halofuginone
alleviates acute viral myocarditis in suckling BALB/c mice by
inhibiting TGF-β1. Biochem Biophys Res Commun. 473:558–564. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huber SA: Viral myocarditis and dilated
cardiomyopathy: Etiology and pathogenesis. Curr Pharm Des.
22:408–426. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Werner L, Deutsch V, Barshack I, Miller H,
Keren G and George J: Transfer of endothelial progenitor cells
improves myocardial performance in rats with dilated cardiomyopathy
induced following experimental myocarditis. J Mol Cell Cardiol.
39:691–697. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Wen C, Xie G, Zeng P, Huang LF and Chen
CY: Tranilast inhibits myocardial fibrosis in mice with viral
myocarditis. Zhongguo Dang Dai Er Ke Za Zhi. 18:446–454. 2016.(In
Chinese). PubMed/NCBI
|
|
8
|
Li-Sha G, Jing-Lin Z, Guang-Yi C, Li L,
De-Pu Z and Yue-Chun L: Erratum: Dose-dependent protective effect
of nicotine in a murine model of viral myocarditis induced by
coxsackievirus B3. Sci Rep. 5:172472015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu H, Lou C and Liu P: Interleukin-27
ameliorates coxsackievirus-B3-induced viral myocarditis by
inhibiting Th17 cells. Virol J. 12:1892015. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Xu MX, Yu R, Shao LF, Zhang YX, Ge CX, Liu
XM, Wu WY, Li JM and Kong LD: Up-regulated fractalkine (FKN) and
its receptor CX3CR1 are involved in fructose-induced
neuroinflammation: Suppression by curcumin. Brain Behav Immun.
58:69–81. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Poniatowski ŁA, Wojdasiewicz P, Krawczyk
M, Szukiewicz D, Gasik R, Kubaszewski Ł and Kurkowska-Jastrzębska
I: Analysis of the role of CX3CL1 (Fractalkine) and its receptor
CX3CR1 in traumatic brain and spinal cord injury: Insight into
recent advances in actions of neurochemokine agents. Mol Neurobiol.
54:2167–2188. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Julia V, Staumont-Salle D and Dombrowicz
D: Role of fractalkine/CX3CL1 and its receptor CX3CR1 in allergic
diseases. Med Sci (Paris). 32:260–266. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Bagci B, Bagci G, Huzmeli C, Sezgin I and
Ozdemir O: Associations of fractalkine receptor (CX3CR1) and CCR5
gene variants with hypertension, diabetes and atherosclerosis in
chronic renal failure patients undergoing hemodialysis. Int Urol
Nephrol. 48:1163–1170. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zanier ER, Marchesi F, Ortolano F, Perego
C, Arabian M, Zoerle T, Sammali E, Pischiutta F and De Simoni MG:
Fractalkine receptor deficiency is associated with early protection
but late worsening of outcome following brain trauma in mice. J
Neurotrauma. 33:1060–1072. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Kim SM, Kim H, Jang KW, Kim MH, Sohn J,
Yun MR, Kang HN, Kang CW, Kim HR, Lim SM, et al: EGFR-mediated
reactivation of MAPK signaling induces acquired resistance to
GSK2118436 in BRAF V600E mutant NSCLC cell lines. Mol Cancer Ther.
15:1627–1636. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Chandaka GK, Wang L, Senogles S and
Armstrong WE: Late pregnancy is a critical period for changes in
phosphorylated mitogen-activated protein kinase/extracellular
signal-regulated kinase 1/2 in oxytocin neurons. J Neuroendocrinol.
doi: 10.1111/jne.12398. PubMed/NCBI
|
|
17
|
Itoh S, Ding B, Bains CP, Wang N, Takeishi
Y, Jalili T, King GL, Walsh RA, Yan C and Abe J: Role of p90
ribosomal S6 kinase (p90RSK) in reactive oxygen species and protein
kinase C beta (PKC-beta)-mediated cardiac troponin I
phosphorylation. J Biol Chem. 280:24135–24142. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Roux PP, Richards SA and Blenis J:
Phosphorylation of p90 ribosomal S6 kinase (RSK) regulates
extracellular signal-regulated kinase docking and RSK activity. Mol
Cell Biol. 23:4796–4804. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Neise D, Sohn D, Stefanski A, Goto H,
Inagaki M, Wesselborg S, Budach W, Stühler K and Jänicke RU: The
p90 ribosomal S6 kinase (RSK) inhibitor BI-D1870 prevents gamma
irradiation-induced apoptosis and mediates senescence via RSK- and
p53-independent accumulation of p21WAF1/CIP1. Cell Death Dis.
4:e8592013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Dou Zhongxia ZX: Role of NF-kappa B
pathway in viral myocarditis in mice. J Nanchang Univ (Med Sci).
2016.
|
|
21
|
Ju Y, Wang T, Li Y, Xin W, Wang S and Li
J: Coxsackievirus B3 affects endothelial tight junctions: possible
relationship to ZO-1 and F-actin, as well as p38 MAPK activity.
Cell Biol Int. 31:1207–1213. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dai Q, Zhang D, Yu H, Xie W, Xin R, Wang
L, Xu X, He X, Xiong J, Sheng H, et al: Berberine restricts
coxsackievirus B type 3 replication via inhibition of c-Jun
N-Terminal Kinase (JNK) and p38 MAPK Activation in vitro. Med Sci
Monit. 23:1448–1455. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Liu J, Zhao J, Petrochenko P, Zheng J and
Hewlett I: Sensitive detection of influenza viruses with Europium
nanoparticles on an epoxy silica sol-gel functionalized
polycarbonate-polydimethylsiloxane hybrid microchip. Biosens
Bioelectron. 86:150–155. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Cusinato R, Pacenti M, Martello T, Fattori
P, Morroni M and Palù G: Effectiveness of hydrogen peroxide and
electron-beam irradiation treatment for removal and inactivation of
viruses in equine-derived xenografts. J Virol Methods. 232:39–46.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Elmastour F, Jaidane H, Aguech-Oueslati L,
Benkahla MA, Aouni M, Gharbi J, Sane F and Hober D: Immunoglobulin
G-dependent enhancement of the infection with Coxsackievirus B4 in
a murine system. Virulence. 7:527–535. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Merino JJ, Muñetón-Gómez V, Alvárez MI and
Toledano-Díaz A: Effects of CX3CR1 and fractalkine chemokines in
amyloid beta clearance and p-Tau Accumulation in Alzheimer's
disease (AD) rodent models: Is fractalkine a systemic biomarker for
AD? Curr Alzheimer Res. 13:403–412. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Rodriguez-Carballo E, Gámez B and Ventura
F: p38 MAPK signaling in osteoblast differentiation. Front Cell Dev
Biol. 4:402016. View Article : Google Scholar : PubMed/NCBI
|