Introduction
Diabetic nephropathy (DN) is a leading cause of
end-stage renal failure worldwide. The pathogenetic mechanisms for
DN may start with reactive oxygen species (ROS) as the common link
connecting to various signaling pathways that lead to kidney
impairment (1–4). The ROS are a family of molecules
including superoxide anion (O2−), hydroxyl racial
(HO−), hydrogen peroxide (H2O2),
peroxynitrite (ONOO−), nitric oxide (NO) and lipid
radicals. In mammalian cells, potential origins of ROS generation
include mitochondrial respiratory chain, xanthine oxidase, NOXs,
and nitric oxide synthase (NOS), with increasing evidence that NOXs
appear to be the major sources or more specifically initial trigger
of ROS generation in the diabetic complications including kidney
(5–7).
Various subtypes of NOXs (e.g., NOX1, NOX2, NOX4)
are discovered abundantly in the mesangial cells (8). Lee et al (9)showed that mitochondrial derived ROS
was elevated in DN subsequent to NOX4 activation. Although NOX4 is
likely implicated in the mediation of kidney fibrosis in diabetes
including mesangial impairment, other subtypes such as NOX1, NOX2
and their cytosolic component NADPH oxidase organizer 1 (NOXO1) are
also possible candidates involved in this pathogenesis (10). Additionally, other researches
reported that NOS uncoupling instead of mitochondria dysfunction in
the mesangial cells is responsible for the early glomerular
pathology in diabetes and is associated with inflammation and
oxidative stress of DN (11).
Thus, the downstream of NOXs activation or the main source of
oxidative stress is still in debate and should be under extensive
investigation. Recently, both in vivo and in vitro
studies have demonstrated that nitroxidative/nitrosative stress
(ONOO−) has been actively involved in the development of
DN or the hyperglycemia induced damage in mesangial cells (12). Therefore, NOS uncoupling as the
most possible cause of ONOO− production should be
examined. It is well known that NOS coupling status is governed by
its cofactor tetrahydrobiopterin (H4B) abundance, for instance:
when H4B is deficient, NOS becomes dysfunctional to produce
superoxide instead of NO which changes into ONOO− on
site instantly and vastly. Moreover, H4B synethesis is controlled
by dihydrofolate reductase (DHFR), the key enzyme responsible for
H4B salvage pathway, whose expression level would determine the
abundance of H4B. Notably, DHFR suppression could play an important
role in the development of DN via dysfunctional NOS and restoration
of DHFR may well be the target for antioxidant treatment.
As an antioxidant, epithelium-derived factor (PEDF)
is a glycoprotein that belongs to the superfamily of serine
protease inhibitors, which is assumed to have beneficial effects on
retinopathy. It was first retrieved from the culture media of human
retinal pigment epithelial cells which possessed potential neuronal
differentiating activity. PEDF is expressed broadly in human
tissues, including the kidney. Previous studies have shown that
decreased PEDF levels in the kidney are implicated in DN,
especially in the early stage (13). It is reported that the injection of
adenovirus expressing PEDF recombinant gene significantly reduced
the albuminuria and the production of extracellular matrix (ECM)
protein, and ameliorated glomerular hypertrophy in the diabetic
kidney (14). Although, according
to previous studies, it is assumed that PEDF protects the renal
function from diabetic injury, via its anti-oxidative and
anti-fibrogenic activities, the particular mechanism has not been
fully illustrated (15–18). Therefore, the purpose of the
present study is to investigate how PEDF protects the kidney from
oxidative stress and determine whether this beneficiary effect is
through rectifying dysfunctional iNOS and de-activation of NOXs
including NOXO1.
Materials and methods
Cell culture
The use of human glomerular mesangial cells (HMCs;
Xiangya School of Medicine, Central South University, Changsha,
China) in the present study was approved by the Ethics Committee of
Renmin Hospital of Wuhan University (Wuhan, China) which was in
adherence with the Declaration of Helsinky. Dulbecco Modified
Eagle's Medium (DMEM) were used to culture HMCs. Cells of passages
3–6 were used and after reaching nearly confluence, they were
quiescenced with serum starvation for 48 h, and then exposed to
different conditions without elevated glucose or advanced glycation
end products (AGEs) in the presence or absence of PEDF for 48 h. In
some experiments, the cells were also treated with 20 µM SB203580
for p38MAPK inhibition. Bovine serum albumin (BSA; 200 mg/l) was
used as a control for AGE treatment. The PEDF was purchased from
Peprotech (Princeton, USA); AGEs were purchased from Jiamay Biotech
(Beijing, China); the rest mediums and reagents were purchased from
Sigma-Aldrich (Merck KGaA; Darmstadt, Germany) unless stated
otherwise. PEDF at 100 nmol/l was used in this study since our
previous dose-dependent experiment showed that this was a proper
dose for the antioxidant protection of mesangial cells (18).
Western blotting analysis
The protein levels of NOX1, NOXO1, NOX2, NOX4, DHFR,
iNOS and p-p38MAPK were detected by immunoblotting analysis. After
the treatment, cells were lysed in 50 mM Tris-HCl buffer. Equal
amounts of lysates (20–40 µg) were loaded, separated by 10%
SDS-PAGE and transferred to PVDF membranes. Membranes were then
blocked with 5% milk overnight, and blotted with primary antibody
against TGF-β (Rabbit; 1:500); iNOS (mouse; 1:1,000); NOX1 (Goat;
1:1,000), NOXO1 (Rabbit; 1:1,000); NOX2 (Rabbit; 1:1,000) and NOX4
(Rabbit; 1:1,000), DHFR (Rabbit; 1:1,000; all Abcam, Cambridge, UK)
and p-p38MAPK (Rabbit; 1:1,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA) separately following standard procedure. The
protein band density was measured by Image J software and the
quantification was reflective of the relative amounts as a ratio of
each protein band over loading control (β-actin).
Measurements of TGF-β1 by
enzyme-linked immunosorbent assay (ELISA)
Spectrophotometre was used to detect the protein
levels of TGF-β 1 and FN which was prepared with the FN ELISA kit
(USCN Life Sciences, Inc., Wuhan, China) and the TGF-β 1 ELISA kit
(Boster, Wuhan, China) according to the manufacturers'
protocol.
In vitro silence of NOXO1
The RNAi against NOXO1 (5′-CCTCGCCCATTTCAGGAAT-3′)
was obtained from Invitrogen (Thermo Fisher Scientific Inc.,
Waltham, MA, USA). HMCs were transfected with siRNA/oligofectamine
(Invitrogen; Thermo Fisher Scientific Inc.) mixtures according to
the manufacturer's protocol.
HPLC-based H4B
measurement
HMCs were lysed by trichloroacetic acid containing
10 mmol/l DTT. Lysates were subjected to differential oxidation in
acidic or alkalytic solutions (19). In summary, total biopterin (H4B,
dihydropterin [H2B], and oxidized biopterin) were determined by
acid oxidation, whereas H2B and oxidized biopterin were measured by
alkali oxidation. The ratio of H4B over H2B was calculated and used
as an index for DHFR activity.
Intracellular ONOO-measurement
ONOO− contents were detected using
hydroxyphenyl fluorescein (HPF) (20). Briefly, cell suspension (200 µl,
107/ml) was incubated with 10 µM HPF in 96 well plates
for 30 min at 37°C in the dark. The fluorescence generated was
measured at excitation 485 nm and emission 585 nm.
Statistical analysis
One-way ANOVA was used to test for statistical
significance, and P<0.05 was considered to indicate a
statistically significant difference. All data shown in the
figures were presented as means ± SD.
Results
PEDF abolishes iNOS upregulation
induced by diabetic environment via p38MAPK inhibition
Both p38MAPK and iNOS protein levels were
upregulated in diabetic environment (Fig. 1). However, p38MAPK and iNOS
upregulations were annihilated by PEDF treatment (Fig. 1A-C). Moreover, p38MAPK inhibitor,
SB203580 abolished iNOS induction completely but only canceled
ONOO− and TGF-β overproduction partially (Fig. 1E-H). In comparison, iNOS induction,
peroxynitite and TGF-β overproduction were resumed back to the
normal level by PEDF (Fig.
1A-H).
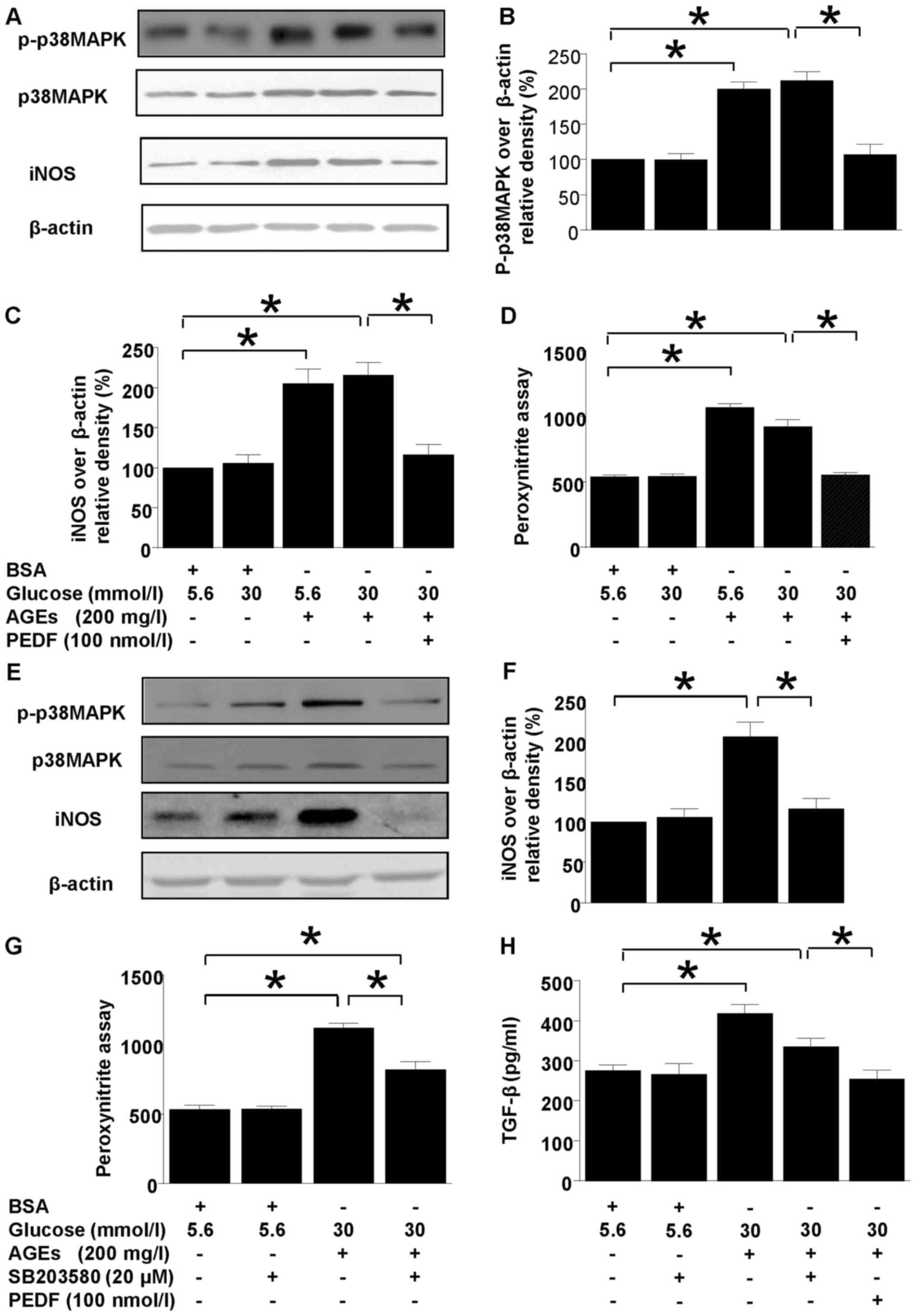 | Figure 1.Effects of PEDF or p38MAPK inhibition
on iNOS and peroxynitrite production. Cells were cultured in 5.6
mmol/l glucose, 30 mmol/l glucose, 5.6 mmol/l glucose + 200 mg/l
AGEs, 30 mmol/l glucose + 200 mg/l AGEs and 30 mmol/l glucose + 200
mg/l AGEs + 100 nmol/l PEDF for 48 h (A-D) or cells were cultured
in 5.6 mmol/l glucose, 5.6 mmol/l glucose + 20 µmol/l SB203580, 30
mmol/l glucose +200 mg/l AGEs, and 30 mmol/l glucose + 200 mg/l
AGEs + 20 µmol/l SB203580 for 48 h (E-H). (A) p-p38MAPK, iNOS and
β-actin protein levels were assessed by immunoblotting analysis;
(B) Relative p-p38 MAPK blot density was analyzed and plotted; (C
and F) The density analysis of iNOS/β-actin from immunoblots; (D
and G) The peroxynitrite production was analyzed by HPF
fluorescence assay; (E) p-p38MAPK, iNOS and β-actin protein levels
were assessed by immunoblotting analysis in the presence or absence
of SB 203580. (H) The TGF-β expression tested by ELISA. Data are
expressed as the means ± standard error of the mean, n=3–5 and
*P<0.05 as indicated. PEDF, pigment epithelium-derived factor;
iNOS, nitric oxide synthase; AGEs, advanced glycation end products;
TGF, transforming growth factor; BSA, bovine serum albumin; MAPK,
mitogen activated protein kinase; p, phosphorylated. |
PEDF reverses iNOS uncoupling induced
by diabetic environment
H4B is the cofactor for iNOS and its abundance
decides the coupling status of iNOS. DHFR is a key enzyme of H4B
salvage pathway which could modulate iNOS coupling status via
regulation of H4B synthesis (Fig.
2). Interestingly, DHFR protein level was downregulated in
diabetic environment and PEDF retrieved DHFR protein level back to
basal level (Fig. 2A and C). As a
result of DHFR reduction in diabetes environment, the H4B contents
and H4B/H2B ratio were diminished (Fig. 2E-H), but PEDF restored H4B level as
well as H4B/H2B ratio in accordance with upregulation of DHFR
protein level (Fig. 2E, G).
However, SB203580 had no effect on DHFR protein expression, H4B
contents as well as H4B/H2B ratio (Fig. 2B, D, F and H).
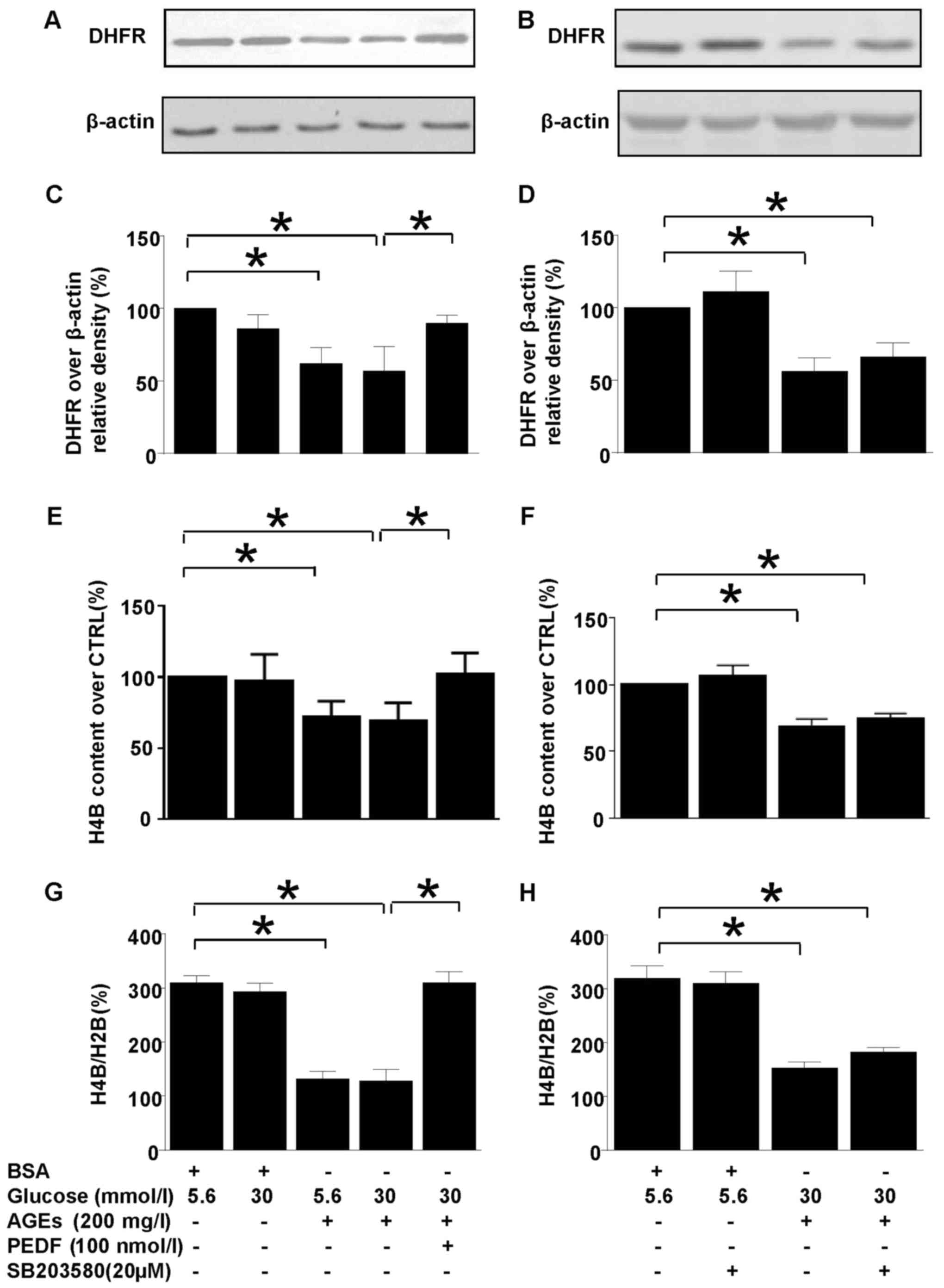 | Figure 2.The effects of PEDF or SB203580 on
DHFR protein expression and intracellular H4B contents. Cells were
cultured in 5.6 mmol/l glucose, 30 mmol/l glucose, 5.6 mmol/l
glucose + 200 mg/l AGEs, 30 mmol/l glucose +200 mg/l AGEs and 30
mmol/l glucose + 200 mg/l AGEs + 100 nmol/l PEDF for 48 h (A, C,
E), or cells were cultured in 5.6 mmol/l glucose, 5.6 mmol/l
glucose +20 µmol/l SB203580, 30 mmol/l glucose +200 mg/l AGEs and
30 mmol/l glucose + 200 mg/l AGEs + 20 µmol/l SB203580 for 48 h (B,
D, F). (A and B) DHFR and β-actin protein levels were assessed by
immunoblotting analysis; (C and D) The density analysis of
DHFR/β-actin from immunoblot; (E and F) the intracellular H4B
contents were measured with high-performance liquid chromatography;
(G and H) the ratio of H4B/H2B was calculated and analyzed. Data
are expressed as the means ± standard error of the mean, n=3–5 and
*P<0.05 as indicated. PEDF, pigment epithelium-derived factor;
AGEs, advanced glycation end products; H4B, tetrahydrobiopterin;
H2B, dihydropterin; CTRL, control; DHFR, dihydrofolate reductase;
BSA, bovine serum albumin. |
PEDF acts independent of NOXs
Our previous study has shown that NOX1-iNOS pathway
played a major role in diabetes induced oxidative stress of
mesangial cells. However, this study showed that although NOX1,
NOX2 and NOX4 were all upregulated in diabetes environment, PEDF
had no effect on their protein levels (Fig. 3), suggesting that PEDF may protect
kidney at probably the downstream of NOX1 pathway. However, NOXO1,
the adaptor protein for NOX1 was downregulated by PEDF suggesting
that NOXO1 suppression could be responsible for the protective
mechanism of PEDF.
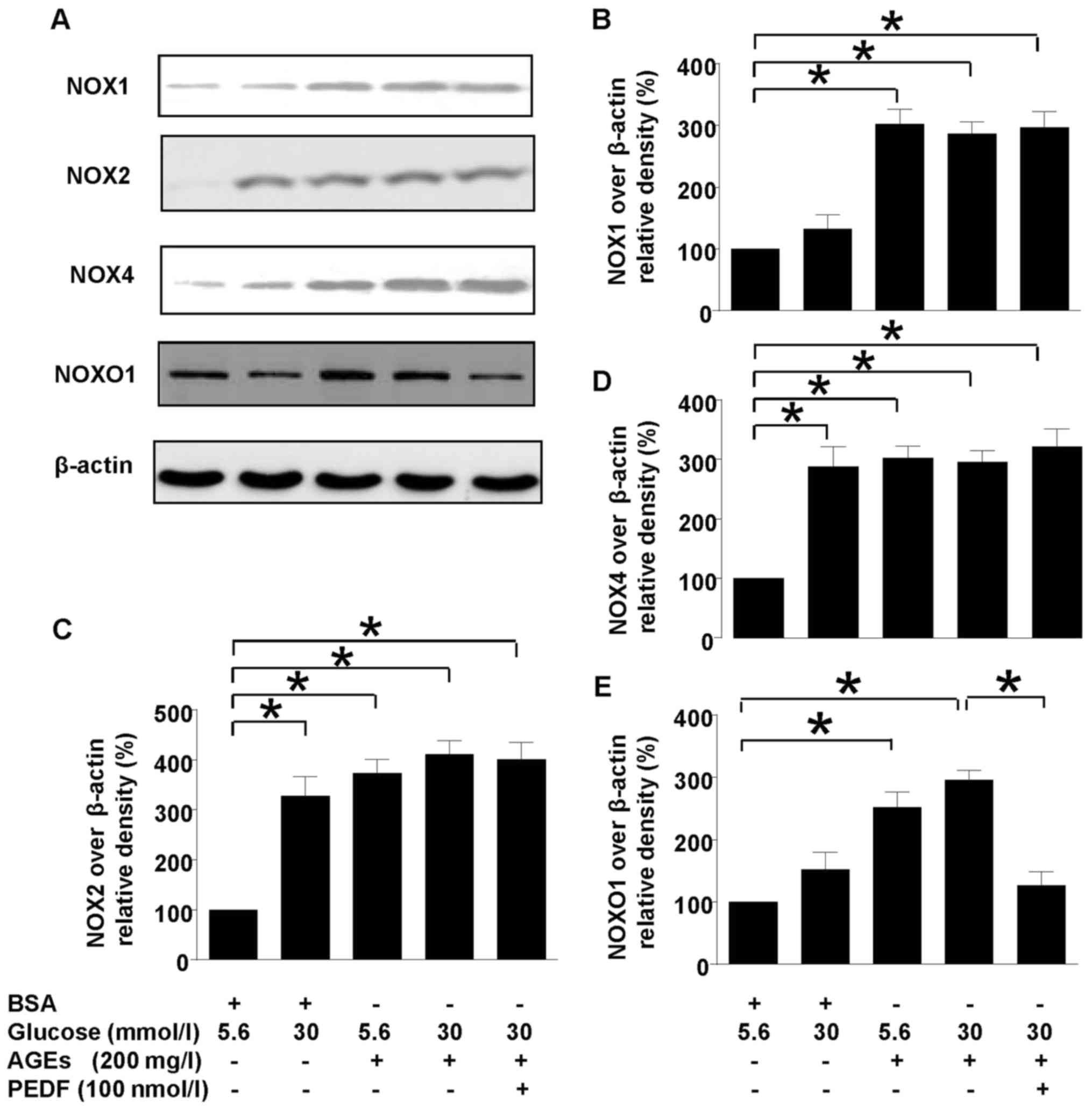 | Figure 3.Effects of PEDF on NOXs activation.
Cells were cultured in 5.6 mmol/l glucose, 30 mmol/l glucose, 5.6
mmol/l glucose + 200 mg/l AGEs, 30 mmol/l glucose + 200 mg/l AGEs
and 30 mmol/l glucose + 200 mg/l AGEs+100 nmol/l PEDF for 48 h. (A)
NOX1, NOX2, NOX4, NOXO1 and β-actin protein levels were assessed by
immunoblotting analysis; (B) The density analysis of NOX2/β-actin
from immunoblot; (C) The density analysis of NOX4/β-actin from
immunoblot; (D) The density analysis of NOX1/β-actin from
immunoblot; (E) The density analysis of NOXO1/β-actin from
immunoblot; Data are expressed as the means ± standard error of the
mean, n=3–5 and *P<0.05 as indicated. PEDF, pigment
epithelium-derived factor; NOXs, nicotinamide adenine dinucleotide
phosphate (NAPDH) oxidases; BSA, bovine serum albumin. |
PEDF confers renal protection via
NOXO1 inhibition
Silencing NOXO1 with RNAi downregulated NOXO1
protein expression (Fig. 4) and
reproduced the effect of PEDF on iNOS expression and coupling
status. NOXO1 silencing abolished the iNOS suppression (Fig. 4A and C). Meanwhile, uncoupling of
iNOS was also annihilated by NOXO1 silencing as demonstrated by
measurement of DHFR expression (Fig.
4D), H4B contents, and H4B/H2B ratio (Fig. 4D and E). Consequently, in mesangial
cells the protective effect of PEDF on oxidative stress
(ONOO− overproduction, Fig.
4G) and kidney fibrosis (TFGF-β production 4H) were also
duplicated by NOXO1 silencing.
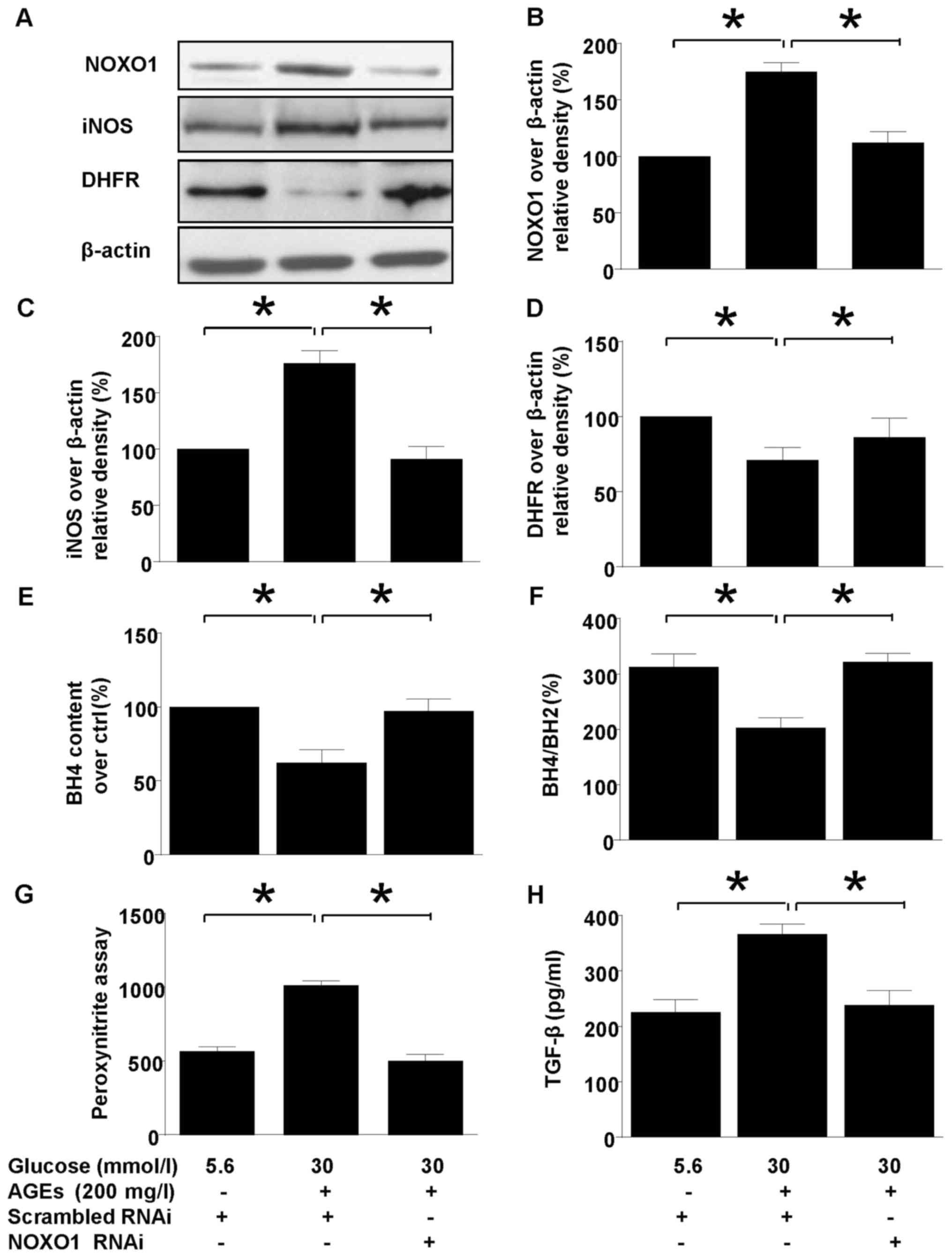 | Figure 4.The effect of NOXO1 silencing on
diabetes induced iNOS upregulation and uncoupling. Cells were
cultured in 5.6 mmol/l glucose, 30 mmol/l glucose + 200 mg/l AGEs
in the presence or absence of scrambled RNAi or NOXO1 RNAi for 48
h. (A) NOXO1, DHFR, iNOS and β-actin protein levels were assessed
by immunoblotting; (B) The density analysis of NOXO1/β-actin from
immunoblot; (C) The density analysis of iNOS/β-actin from
immunoblot; (D) The density analysis of DHFR/β-actin from
immunoblot. (E) Intracellular H4B content was measured by
high-performance liquid chromatography; (F) The ratio of H4B/H2B
was calculated and analyzed; (G) Peroxynitrite (ONOO−)
production was examined by hydroxyphenyl fluorescein staining; (H)
TGF-β protein levels were measured using ELISA. Data are expressed
as the means ± standard error of the mean, n=3–5 and *P<0.05 as
indicated. iNOS, inducible nitric oxide synthase; NOXO1,
nicotinamide adenine dinucleotide phosphate oxidase 1; AGEs,
advanced glycation end products; H4B, tetrahydrobiopterin; H2B,
dihydropterin; DHFR, dihydrofolate reductase; RNAi, RNA
inteference; TGF, transforming growth factor. |
Discussion
The present study explored the possible mechanism or
the molecular pathway of PEDF action on mesangial cells in
diabetes. We found that PEDF was protective for mesangial cells in
diabetic environment by reversing TGF-β overexpression,
ONOO− overproduction, iNOS induction and uncoupling. The
ONOO− derived from iNOS induction and uncoupling was
completely abolished consequent to PEDF treatment via p38MAPK
inactivation and restoration of DHFR protein level. However, those
salutary effects of PEDF were mediated by NOXO1 but not individual
NOXs downregulation. Taken together, our findings provided initial
evidence to reveal novel mechanisms that PEDF prevented oxidative
stress and protected mesangial cells from fibrogenesis in diabetic
environment via dual effects converging at NOXO1 suppression:
Retrivation of iNOS overexpression through p38MAPK inactivation and
restoration of iNOS coupling through DHFR restoration.
Sheikpranbabu et al (21) have shown that PEDF significantly
decreased NADPH oxidase and ROS generation in pericytes following
AGE-BSA treatment. In the present study, we observed an attenuation
of ROS production by PEDF in mesangial cells treated with high
glucose and AGEs (diabetes environment). As NOX1, NOX2 and NOX4 are
the three major NADPH oxidases existed in the mesangial cells and
our previous study showed that the activation of NOX1 and its
derived superoxide led to direct cell injury and initiated a chain
of deleterious stress signaling such as iNOS induction and
uncoupling. The effect of PEDF on NOXs protein level was examined
(22). However, NOXs protein level
was not altered by PEDF. Interestingly, NOXO1, the cytosolic
regulation protein for NOXs (23),
was suppressed by PEDF and downregulation of NOXO1 by RNA silencing
reproduced the antioxidant effect of PEDF and its restoration
effect on iNOS induction and uncoupling. All of these suggested
that NOXO1 mediated the PEDF protective effect.
It has been demonstrated that p38MAPK activation is
involved in the development of DN (24). However, the exact role of p38MAPK
is not clear since it is traditionally considered as an
anti-apoptotic signaling molecule. The p38MAPK activation is not
self explanatory for the mesangial cells proliferation in the early
stage of DN. Our data, among a few other researches suggested that
p38MAPK is a pro-fibrogenetic rather than a pro-apoptotic molecule
in high glucose induced mesangial damage (25,26).
Previously, it was suggested that p38MAPK was a
possible upstream for iNOS and it has been reported that p38MAPK is
activated in glomeruli isolated from streptozotocin-induced
diabetic rats and glomerular mesangial cells cultured under high
glucose conditions (27). Yuan
et al (28) showed that
high glucose (33 mM) caused activation of p38MAPK and inhibition
for p38MAPK abrogated the high glucose induced iNOS expression,
cell injury and levels of NO and nitrotyrosine in cultured human
retinal pigmented epithelial cells. Therefore, activation of
p38MAPK plays an important role in the iNOS induction and oxidative
stress of DN. Recently, ATF-2 and NF-KB are suggested to be the
possible downstreams or targets of p38MAPK whose activation may
contribute to iNOS unregulation (29). In this study, we established that
iNOS was induced in diabetes environment consequent to p38MAPK
upregulation, but PEDF reversed the process. However, the p38MAPK
inhibitor, SB203580 completely abolished iNOS induction but only
partially canceled ONOO− and TGF-β overproduction in
diabetic enviroment. Therefore, we speculated that PEDF may protect
kidney from diabetes via multiple pathways including p38MAPK and
iNOS induction.
In the endothelial cells, PEDF has been shown to
inhibit the cellular permeability via p38MAPK de-activation
(30). While, PEDF also protected
against retinal neovascularization by suppression of p38MAPK
activation and dampening iNOS induction in the macrophage (31,32).
Likewise, our data showed that PEDF, by targeting NOXO1, protected
mesangial cells from fibrogenesis and nitroxidative/nitrosative
stress, at least partly via abrogation of p38MAPK mediated iNOS
induction.
It is well established that oxidative stress has a
major role in the development of diabetic complications including
DN. ONOO−, as a notorious radical form created from the
interaction between superoxide and NO, attacks various biomolecules
in the tissues and induces damages in the microcirculations
(33,34). Prabhakar et al (12) reported that renal ONOO−
formation, which acts as a main contributor to oxidative stress of
DN and accounts partly for decreased NO bioavailability of kidney,
was elevated in the kidney homogenates of obese ZSF1 rats (~150%
up) vs. lean ZSF rats. However, there is a lack of quantitative
information of ONOO− about the formation and biological
relevance because of its difficult and indirect measurements.
Indeed, serum level of oxidative stress indicated by
ONOO− is associated with the severity of diseases such
as Parkinson's (35).
Since iNOS is the main source for ONOO−
overproduction, iNOS uncoupling could be another contributing
factor for the nitrosative stress. Basically, when iNOS becomes
uncoupled, it will fail to produce NO, and begin to produce
superoxide which eventually culminates in ONOO−
formation. H4B is the key co-factor for iNOS whose deficiency
results in iNOS uncoupling. Indeed, DHFR (the enzyme responsible
for H4B replenishment) is diminished in diabetes environment which
consequently leads to H4B consumption (36). As we expected, PEDF relieved the
suppression of DHFR and retrieved H4B contents (H4B/H2B ratio) in
diabetes which eventually lead to iNOS recoupling. This effect is
independent of p38MAPK pathway because SB203508, the p38MAPK
inhibitor has no effect on DHFR suppression and H4B deficiency.
The deleterious effects of diabetes are attributed
mainly to the formation of sugar-derived substances called AGEs.
The AGEs formation is markedly accelerated in diabetes because of
the increased availability of glucose. We always believe that the
detrimental effects of diabetes are the combination of AGEs and
elevated glucose, although ONOO− formation is majorly
due to AGEs, not elevated glucose. Likewise, the AGE effect is more
detrimental and advanced damage. On the other hand, the effect of
high glucose is more of an acute damage and represents an early
stage of oxidative stress such as NOX activation (37,38).
Overall, AGEs represented the chronic effect of diabetic condition
on renal impairment, and elevated glucose acted as an acute insult
(within min). In our study, the cells were examined after 48 h
during which the acute effect of elevated glucose may have faded
away (so as the glucose concentration) and its extended effect may
act via protein glycation which is AGEs-related. Nevertheless, it
is important to expose the mesangial cells with elevated glucose
and AGE together in order to mimic the in vivo diabetic
condition.
NOXO1 is originally believed to be the cytosolic or
inner component/subunit that solely regulates NOX1
activity/mediated superoxide generation, but is also recently found
to interact with other NOXs such as NOX2, NOX4 and be required for
their assembly and activation. Collectively, it is still unclear
about NOXO1 regarding its function in the mesangial cells. The
current data suggests that although NOX1, NOX2, NOX4 and NOXO1 are
involved in the high glucose induced oxidative stress in the
mesangial cells, only NOXO1 has a key role in the regulation of
detrimental events. Moreover, since NOX1, NOX2 and NOX4 all
possibly interact with NOXO1 (10,39),
they could be linked to the effect of PEDF as a complex with
NOXO1.
In summary, iNOS is the main source of ROS in
diabetic kidney and PEDF protects against diabetic damage via iNOS
suppression mediated by p38MAPK inactivation and iNOS recoupling
via H4B restoration consequent to DHFR upregulation (Fig. 5). Therefore, we have verified the
antioxidant effects of PEDF, which is consistent with previous
studies. This is the first time we clarify the anti-oxidation
mechanism of PEDF on DN which could lead to the discovery of a
potential target for PEDF and provide a theoretical basis for the
clinical treatment of DN. It is noticed in this study that PEDF
curbed the origination and progression of oxidative stress by NOXO1
suppression, but the mechanism of PEDF on the initial link of
oxidative stress is still puzzled.
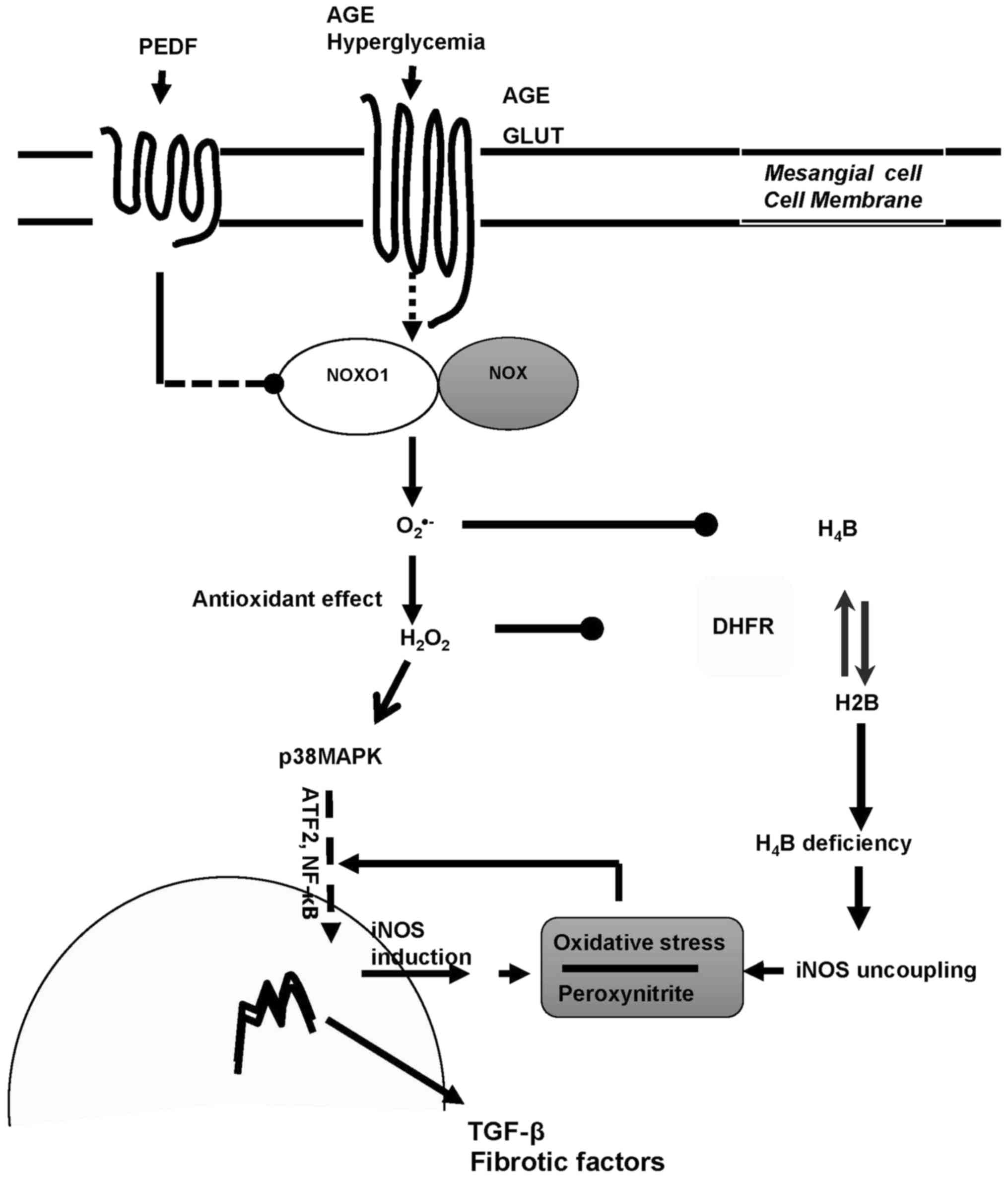 | Figure 5.The dual mechanisms of PEDF on
diabetic kidney protection. PEDF prevented oxidative stress and
protected against fibrogenesis of mesangial cells in diabetic
environment via dual effects mediated by NOXO1 inhibition:
Annhilation of iNOS induction through p38MAPK inactivation and
retrivation of iNOS coupling through DHFR restoration. PEDF,
pigment epithelium-derived factor; iNOS, inducible nitric oxide
synthase; NOXO1, nicotinamide adenine dinucleotide phosphate
oxidase 1; AGE, advanced glycation end product; H4B,
tetrahydrobiopterin; H2B, dihydropterin; DHFR, dihydrofolate
reductase; TGF, transforming growth factor; NF, nuclear factor;
GLUT, glucose transporter; MAPK, mitogen activated protein kinase;
ATF. Activating transcription factor. |
Acknowledgements
The present study is supported by National Natural
Science Foundation of China (grant nos. 81170767 and 81571376 to Dr
Gao) and Diabetes Study Fund from Chinese Medical Association
(grant no. 13060906481 to Dr Gao) and Young Investigator Grant for
diabetes study from Novo Nordisk (grant no. 2012 to Dr Gao).
References
|
1
|
Lee HB, Yu MR, Yang Y, Jiang Z and Ha H:
Reactive oxygen species-regulated signaling pathways in diabetic
nephropathy. J Am Soc Nephrol. 14 8 Suppl 3:S241–S245. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nishikawa T and Araki E: Impact of
mitochondrial ROS production in the pathogenesis of diabetes
mellitus and its complications. Antioxid Redox Signal. 9:343–353.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Forbes JM, Coughlan MT and Cooper ME:
Oxidative stress as a major culprit in kidney disease in diabetes.
Diabetes. 57:1446–1454. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Giacco F and Brownlee M: Oxidative stress
and diabetic complications. Circ Res. 107:1058–1070. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sedeek M, Callera G, Montezano A, Gutsol
A, Heitz F, Szyndralewiez C, Page P, Kennedy CR, Burns KD, Touyz RM
and Hébert RL: Critical role of Nox4-based NADPH oxidase in
glucose-induced oxidative stress in the kidney: Implications in
type 2 diabetic nephropathy. Am J Physiol Renal Physiol.
299:F1348–F1358. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Hancock JT, Desikan R and Neill SJ: Role
of reactive oxygen species in cell signalling pathways. Biochem Soc
Trans. 29:345–350. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Youn JY, Gao L and Cai H: The p47phox- and
NADPH oxidase organiser 1 (NOXO1)-dependent activation of NADPH
oxidase 1 (NOX1) mediates endothelial nitric oxide synthase (eNOS)
uncoupling and endothelial dysfunction in a streptozotocin-induced
murine model of diabetes. Diabetologia. 55:2069–2079. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Jha JC, Thallas-Bonke V, Banal C, Gray SP,
Chow BS, Ramm G, Quaggin SE, Cooper ME, Schmidt HH and
Jandeleit-Dahm KA: Podocyte-specific Nox4 deletion affords
renoprotection in a mouse model of diabetic nephropathy.
Diabetologia. 59:379–389. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Lee DY, Wauquier F, Eid AA, Roman LJ,
Ghosh-Choudhury G, Khazim K, Block K and Gorin Y: Nox4 NADPH
oxidase mediates peroxynitrite-dependent uncoupling of endothelial
nitric-oxide synthase and fibronectin expression in response to
angiotensin II: Role of mitochondrial reactive oxygen species. J
Biol Chem. 288:28668–28686. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gorin Y, Cavaglieri RC, Khazim K, Lee DY,
Bruno F, Thakur S, Fanti P, Szyndralewiez C, Barnes JL, Block K and
Abboud HE: Targeting NADPH oxidase with a novel dual Nox1/Nox4
inhibitor attenuates renal pathology in type 1 diabetes. Am J
Physiol Renal Physiol. 308:F1276–F1287. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Eid AA, Lee DY, Roman LJ, Khazim K and
Gorin Y: Sestrin 2 and AMPK connect hyperglycemia to Nox4-dependent
endothelial nitric oxide synthase uncoupling and matrix protein
expression. Mol Cell Biol. 33:3439–3460. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Prabhakar S, Starnes J, Shi S, Lonis B and
Tran R: Diabetic nephropathy is associated with oxidative stress
and decreased renal nitric oxide production. J Am Soc Nephrol.
18:2945–2952. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Chen H, Zheng Z, Li R, Lu J, Bao Y, Ying
X, Zeng R and Jia W: Urinary pigment epithelium-derived factor as a
marker of diabetic nephropathy. Am J Nephrol. 32:47–56. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang JJ, Zhang SX, Mott R, Knapp RR, Cao
W, Lau K and Ma JX: Salutary effect of pigment epithelium-derived
factor in diabetic nephropathy: Evidence for antifibrogenic
activities. Diabetes. 55:1678–1685. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tombran-Tink J, Chader GG and Johnson LV:
PEDF: A pigment epithelium-derived factor with potent neuronal
differentiative activity. Exp Eye Res. 53:411–414. 1991. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Tombran-Tink J and Barnstable CJ: PEDF: A
multifaceted neurotrophic factor. Nat Rev Neurosci. 4:628–636.
2003. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Becerra SP: Focus on molecules: Pigment
epithelium-derived factor (PEDF). Exp Eye Res. 82:739–740. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Mao T, Gao L, Li H and Li J: Pigment
epithelium-derived factor inhibits high glucose induced oxidative
stress and fibrosis of cultured human glomerular mesangial cells.
Saudi Med J. 32:769–777. 2011.PubMed/NCBI
|
|
19
|
Gao L, Pung YF, Zhang J, Chen P, Wang T,
Li M, Meza M, Toro L and Cai H: Sepiapterin reductase regulation of
endothelial tetrahydrobiopterin and nitric oxide bioavailability.
Am J Physiol Heart Circ Physiol. 297:H331–H339. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Setsukinai K, Urano Y, Kakinuma K, Majima
HJ and Nagano T: Development of novel fluorescence probes that can
reliably detect reactive oxygen species and distinguish specific
species. J Biol Chem. 278:3170–3175. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sheikpranbabu S, Haribalaganesh R and
Gurunathan S: Pigment epithelium-derived factor inhibits advanced
glycation end-products-induced cytotoxicity in retinal pericytes.
Diabetes Metab. 37:505–511. 2011.PubMed/NCBI
|
|
22
|
Gao L, Huang W and Li J: NOX1 abet
mesangial fibrogenesis via iNOS induction in diabetes. Mol Cell
Biochem. 382:185–191. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Cheng G and Lambeth JD: NOXO1, regulation
of lipid binding, localization, and activation of Nox1 by the Phox
homology (PX) domain. J Biol Chem. 279:4737–4742. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Li X, Liu W, Wang Q, Liu P, Deng Y, Lan T,
Zhang X, Qiu B, Ning H and Huang H: Emodin suppresses cell
proliferation and fibronectin expression via p38MAPK pathway in rat
mesangial cells cultured under high glucose. Mol Cell Endocrinol.
307:157–162. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Ho C, Lee PH, Huang WJ, Hsu YC, Lin CL and
Wang JY: Methylglyoxal-induced fibronectin gene expression through
Ras-mediated NADPH oxidase activation in renal mesangial cells.
Nephrology (Carlton). 12:348–356. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Liu CM, Qi XL, Yang YF and Zhang XD:
Betulinic acid inhibits cell proliferation and fibronectin
accumulation in rat glomerular mesangial cells cultured under high
glucose condition. Biomed Pharmacother. 80:338–342. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Fang S, Jin Y, Zheng H, Yan J, Cui Y, Bi
H, Jia H, Zhang H, Wang Y, Na L, et al: High glucose condition
upregulated Txnip expression level in rat mesangial cells through
ROS/MEK/MAPK pathway. Mol Cell Biochem. 347:175–182. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yuan Z, Feng W, Hong J, Zheng Q, Shuai J
and Ge Y: p38MAPK and ERK promote nitric oxide production in
cultured human retinal pigmented epithelial cells induced by high
concentration glucose. Nitric Oxide. 20:9–15. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Shatanawi A, Lemtalsi T, Yao L, Patel C,
Caldwell RB and Caldwell RW: Angiotensin II limits NO production by
upregulating arginase through a p38 MAPK-ATF-2 pathway. Eur J
Pharmacol. 746:106–114. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Yang J, Duh EJ, Caldwell RB and Behzadian
MA: Antipermeability function of PEDF involves blockade of the MAP
kinase/GSK/beta-catenin signaling pathway and uPAR expression.
Invest Ophthalmol Vis Sci. 51:3273–3280. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Gao S, Li C, Zhu Y, Wang Y, Sui A, Zhong
Y, Xie B and Shen X: PEDF mediates pathological neovascularization
by regulating macrophage recruitment and polarization in the mouse
model of oxygen-induced retinopathy. Sci Rep. 7:428462017.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wen H, Liu M, Liu Z, Yang X, Liu X, Ni M,
Dong M, Luan X, Yuan Y, Xu X and Lu H: PEDF improves
atherosclerotic plaque stability by inhibiting macrophage
inflammation response. Int J Cardiol. 235:37–41. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Pacher P and Szabo C: Role of the
peroxynitrite-poly(ADP-ribose) polymerase pathway in human disease.
Am J Pathol. 173:2–13. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kar S and Kavdia M: Endothelial NO and
O2− production rates differentially regulate
oxidative, nitroxidative, and nitrosative stress in the
microcirculation. Free Radic Biol Med. 63:161–174. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Kouti L, Noroozian M, Akhondzadeh S,
Abdollahi M, Javadi MR, Faramarzi MA, Mousavi S and Ghaeli P:
Nitric oxide and peroxynitrite serum levels in Parkinson's disease:
Correlation of oxidative stress and the severity of the disease.
Eur Rev Med Pharmacol Sci. 17:964–970. 2013.PubMed/NCBI
|
|
36
|
Harrison DG, Chen W, Dikalov S and Li L:
Regulation of endothelial cell tetrahydrobiopterin
pathophysiological and therapeutic implications. Adv Pharmacol.
60:107–132. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Stern D, Yan SD, Yan SF and Schmidt AM:
Receptor for advanced glycation endproducts: A multiligand receptor
magnifying cell stress in diverse pathologic settings. Adv Drug
Deliv Rev. 54:1615–1625. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Yamagishi S, Nakamura K, Matsui T, Noda Y
and Imaizumi T: Receptor for advanced glycation end products
(RAGE): A novel therapeutic target for diabetic vascular
complication. Curr Pharm Des. 14:487–495. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Schröder K, Weissmann N and Brandes RP:
Organizers and activators: Cytosolic Nox proteins impacting on
vascular function. Free Radic Biol Med. 109:22–32. 2017. View Article : Google Scholar : PubMed/NCBI
|














