Introduction
Sepsis is a systemic and overwhelming lethal
syndrome following local infection (1); however, therapeutic choices are
limited for sepsis. Therefore, it is important to elucidate the
mechanisms that mediate the development of sepsis. The pathogenesis
of sepsis is attributed to pro-inflammatory cytokines that cause
inflammation in tissues. Thus, the targeting of late
proinflammatory mediators may be an ideal therapy for sepsis.
High-mobility group box 1 (HMGB1) is a late mediator of lethal
endotoxemia and circulating HMGB1 level is elevated in a delayed
fashion in septic mice (2). HMGB1
has previously been reported to disturb the barrier integrity of
endothelial cells and induce sepsis by promoting proinflammatory
cytokines secretion (3,4). Therefore, HMGB1 may provide an
opportunity for clinical intervention for sepsis, which suggested a
need to investigate the mechanisms by which HMGB1 is modulated
during sepsis.
Post-transcriptional regulation is a crucial event,
which includes regulating mRNA stability in inflammatory processes
(5). Human antigen R (HuR) is an
important RNA-binding protein; its levels of expression have been
reported to be increased in various tumors (6,7),
which indicated that HuR may exert a promotive role in tumor
development. However, whether HuR is involved in sepsis and the
related mechanisms remain unclear.
The present study used bioinformatics analyses to
demonstrate that HuR may bind to HMGB1. Reverse
transcription-quantitative polymerase chain reaction (RT-qPCR) and
western blot analyses revealed that ectopic expression of HuR led
to increased HMGB1 expression. Luciferase reporter and
co-immunoprecipitation assays demonstrated that HuR was able to
bind to HMGB1 directly and thus enhanced the stability of HMGB1
mRNA. In addition, treatment with HuR small interfering (si)RNA
suppressed the lipopolysaccharide (LPS)-mediated HMGB1 release and
attenuated HMGB1-mediated hyperpermeability and leukocyte migration
in vitro and in vivo. Injection of HuR-siRNA also
downregulated cecal ligation and puncture (CLP)-induced HMGB1
release, reduced the production of interleukin (IL)-6 and lowered
mortality rates. Finally, the promotive effects of HuR
overexpression on the inflammatory response were attenuated when
human umbilical vein endothelial cells (HUVECs) were co-treated
with HMGB1 short hairpin (sh)RNA. These results suggested that HuR
may be able to directly bind to and regulate HMGB1 expression
during sepsis.
Materials and methods
Cell culture
HUVECs were obtained from American Type Culture
Collection (Manassas, VA, USA) and cultured in Dulbecco's Modified
Eagle's Medium (Gibco; Thermo Fisher Scientific, Inc., Waltham, MA,
USA) supplemented with 10% heat-inactivated fetal bovine serum
(Gibco; Thermo Fisher Scientific, Inc.), 80 U/ml penicillin, 0.08
mg/ml streptomycin and 2 mM glutamine at 37°C under humidified
atmosphere with 5% CO2.
Animal model of sepsis
All animal experiments were performed with the
approval of the Cangzhou Central Hospital Ethics Committee for
Animal Experimentation (Cangzhou, China). A total of 60 male
C57BL/6 mice (age, 8 weeks; weight, 20–25 g) were purchased from
the Medical Center of Yangzhou University (Yangzhou, China). Mice
were housed and fed at 27°C, and 12-h light/dark cycle and had
access to food/water. Sepsis was induced by CLP, following a
previously published protocol (8).
HuR-siRNA (10 nM; Biomics Biotechnology Inc., Nantong, China;
Table I) was injected in the tail
vein 3 days post-sepsis to detect the effects of HuR on CLP-induced
HMGB1 release, IL-6 production and mortality, a scrambled-siRNA was
used as a control, following the manufacture's siRNA injection
procedure.
 | Table I.Primer sequences used for reverse
transcription-quantitative polymerase chain reaction, siRNA and
shRNA. |
Table I.
Primer sequences used for reverse
transcription-quantitative polymerase chain reaction, siRNA and
shRNA.
| Gene | Sequences
(5′→3′) |
|---|
| HMGB1 | F:
CTTCCTCTTCTGCTCTGAGTATCG |
|
| R:
CTTCCTTCTTTTTCTTGCTTTTTT |
| E-selectin | F:
AACTGCGAGAAGAACGGATAGAGA |
|
| R:
AGCGAGGAGAACAAAAACAAGAGC |
| HuR | F:
CTGGTGTCCAAAAGTCAACAATAA |
|
| R:
AAAAAAAAAAATAAAAAGGCAATG |
| β-actin | F:
ATCCACGAAACTACCTTCAACTCC |
|
| R:
GATCTTGATCTTCATTGTGCTGGG |
| ICAM-1 | F:
GATTGTCATCATCACTGTGGTAGC |
|
| R:
GGCCTGTTGTAGTCTGTATTTCTT |
| VCAM-1 | F:
ATACAACCGTCTTGGTCAGCCCTT |
|
| R:
CATTCATATACTCCCGCATCCTTC |
| HuR siRNA |
GAGGCAAUUACCAGUUUCA |
|
| F:
CCGGTGAGGCAATTACCAGTTTCACTC |
|
|
GAGTGAAACTGGTAATTGCCTCTTTTTG |
|
| R:
AATTCAAAAAAGGCAATTACCAGTTT |
|
|
CACTCGAGTGAAACTGGTAATTGCCTCA |
ELISA
ELISA kits of HMGB1 (cat. no. H6-NBP1-42888),
interleukin (IL)-6 (cat. no. NBP1-92668), tumor necrosis factor
(TNF)-α (cat. no. NBP2-31085), IL-1β (cat. no. KA0357) and IL-10
(cat. no. KA1038) were purchased from Novus Biologicals, LLC,
Littleton, CO, USA). Toll-like receptor (TLR)2 (cat. no. EK1130)
ELISA kit was purchased from Wuhan Boster Biological Technology,
Ltd., (Wuhan, China) TLR4 (cat. no. kt56334) and nuclear factor
(NF)-κB (cat. no. kt21004) ELISA kits were purchased from MSK
(Wuhan, China), which were used to detect the cytokines level in
septic mice serum according to the manufacture's protocols.
Adenovirus (Ad) vector
construction
OBiO Technology Corp., Ltd. (Shanghai, China)
constructed the HuR overexpression (Ad-HuR), knockdown
(Ad-HuR-shRNA; Table I) and HMGB1
overexpression (Ad-HMGB1) adenovirus vectors, which were verified
by DNA sequencing by SprinGenBioTech (Nanjing, China).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from 1×106 HUVECs
with TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.),
following the manufacturer's protocol. First-strand cDNA was
synthesized from the total RNA using a QuantiTect Rev.
Transcription kit (cat. no. 205311; Qiagen China Co., Ltd.,
Shanghai, China). qRT-PCR was performed on the StepOnePlus PCR
system with SYBR Green kit (Biomics Biotechnologies Co., Ltd.,
Nantong, China). β-actin was used as an internal control and gene
expression levels were normalized to β-actin, and qRT-PCR reactions
were performed at 58°C for 35 cycles in triplicate for each
experiment. Primer sequences are listed in Table I. The relative gene expression
levels were analyzed via 2−ΔΔCq method (9).
mRNA stability assays
HuR expression level was upregulated or
downregulated by infecting with Ad-HuR or Ad-HuR-shRNA at
1×106 multiplicity of infection and 70–80% of cell
density, respectively, for at 37°C with DMEM medium for 48 h.
Subsequently, de novo RNA synthesis was blocked with the
addition of 5 µg/ml actinomycin D (ActD; Apexbio, Houston, TX USA)
to the medium. Total RNA was harvested at the 0, 2, 4, 6, 8 and 10
h post-transfection, and HMGB1 mRNA expression levels were measured
by RT-qPCR assay. mRNA half-life was examined by comparing with the
mRNA expression level prior to the addition of ActD.
RNA immunoprecipitation (RIP)
assays
RIP assays were conducted as previously described
(10). Briefly, HUVECs
(1×107) were lysed with 25 mM Tris-HCl buffer (pH 7.5)
and 100 U/ml RNase inhibitor (Sigma-Aldrich; Merck KGaA, Darmstadt,
Germany), and subsequently incubated with protein-A sepharose beads
(P001, 7Sea Biotech, Shanghai, China) precoated with 3 µg anti-HuR
antibody or control rabbit immunoglobulin G for 1.5 h at 4°C. The
RNA-protein complexes were pulled-down by protein A/G agarose
beads, RNA was extracted with TRIzol and HMGB1 expression levels
were detected with RT-qPCR.
Luciferase reporter assays
The HMGB1 promoter sequence was inserted into the
pGL3 vector (pGL3-HMGB1; Promega Corporation, Madison, WI, USA),
which was transfected in HUVECs with Ad-HuR or Ad-HuR-shRNA
co-infection. Transfection efficiency was normalized with β-gal
activity, and the luciferase activity of pGL3-HMGB1 was measured
for HMGB1 promoter transcriptional activity assays. Cells at a
density of 80% were used for transfection with
Lipofectamine® 2000 (Thermo Fisher Scientific, Inc) at
37°C with DMEM medium for 72 h. The luciferase activity was
measured by a Glomax 96 luminometer (Promega Corporation).
Cell viability assays
Cell Counting Kit-8 (CCK-8, Vazyme Biotech Co.,
Ltd., Nanjing, China) was used to examine cell viability. Cells
infected with Ad-HuR-shRNA or Ad-empty vector were seeded (4,000
cells/well) in 96-well plates and incubated for 24, 48 and 72 h at
37°C; growth rates were determined by measuring optical density
with a microplate reader. Each experiment was performed in
triplicate.
In vitro permeability assay
HUVECs were seeded (8×104 cells/well)
onto Transwell inserts (Millipore; Merck KGaA) for 6 h and
subsequently treated with HMGB1 cytokine (1 µg/ml; cat. no.
34-8401-82; Thermo Fisher Scientific, Inc.) for 16 h followed by
infection with or without Ad-HuR-shRNA for 24 h. Transwell inserts
were then washed with PBS, followed by addition of 0.5 ml of Evans
blue (0.67 mg/ml) diluted in growth medium containing 5% bovine
serum albumin (BSA). Fresh growth medium was then added to the
lower chamber, and the medium in the upper chamber was replaced
with Evans blue/BSA. Following 10 min incubation, optical density
was measured at 650 nm in the lower chamber.
In vivo permeability and leukocyte
migration assays
CLP-operated 12 mice or control 12 mice were
pretreated with HMGB1 (2 µg) intravenously for 16 h, after which
Ad-HuR-siRNA was injected intravenously. A total of 12 mice treated
with a scrambled siRNA were used as a control group. After 48 h, 1%
Evans blue dye solution in normal saline was injected intravenously
into each mouse, and the mice were sacrificed 30 min
post-injection. The peritoneal exudates were collected following
washes with 5 ml normal saline and centrifuged at 200 × g for 10
min at 4°C. The absorbance of the supernatant was read at 650 nm
via a 96-well plate by GloMax® Discover System (GM3000;
Promega Corporation). Vascular permeability was expressed in terms
of dye (µg)/mouse. To assess leukocyte and neutrophil migration, 20
µl of peritoneal fluid was mixed with 0.38 ml of Turk's solution
(0.01% crystal violet in 3% acetic acid), and the leukocyte number
was counted under a light microscope. The results were expressed as
neutrophil ×106 per peritoneal cavity.
Statistical analysis
All data are presented as mean ± standard deviation.
The differences between the groups were analyzed with the Student's
t-test or one-way analysis of variance of three independent
experiments. The differences between the groups were analyzed using
a one-way analysis of variance with the Tukey-Kramer post-hoc test.
P<0.05 was considered to indicate a statistically significant
difference.
Results
Overexpression of HuR increases HMGB1
expression in HUVECs
The present study identified that HuR has 21
potential binding sites on HMGB1 in 11 different cancers and the
binding ability ranked first by the starBase v2.0 (http://starbase.sysu.edu.cn/browseMrnaCeRNA.php)
analysis (Table II).
Subsequently, the ability of HuR to increase HMGB1 expression in
HUVECs was examined. Transfection with Ad-HuR or Ad-HuR-shRNA
resulted in the subsequent increase or decrease, respectively, of
HuR mRNA and protein expression levels in HUVECs (Fig. 1A-D). In addition, overexpression of
HuR significantly upregulated HMGB1 mRNA and protein expression
levels (Fig. 1E and F,
respectively). By contrast, HuR-knockdown led to a significant
decrease in HMGB1 mRNA and protein expression (Fig. 1G and H, respectively). These
results indicated that HuR was able to positively regulate HMGB1
expression.
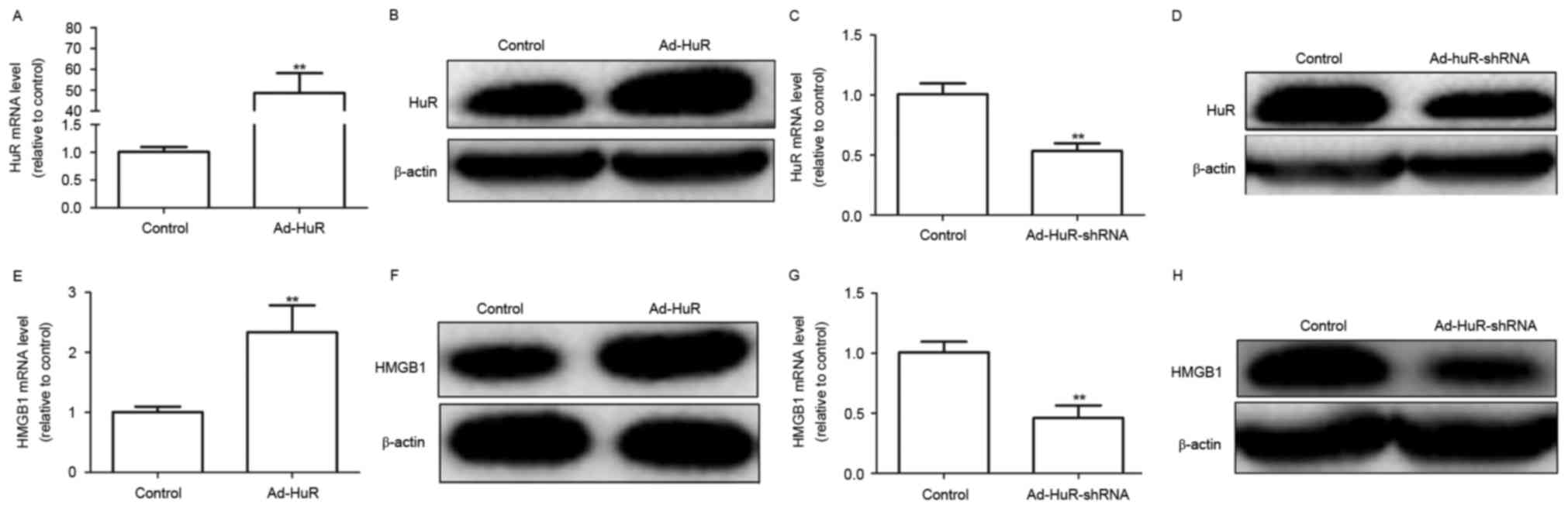 | Figure 1.Ectopic expression of HuR promotes
HMGB1 expression in HUVECs. (A) HuR mRNA and (B) HuR protein levels
were detected in HUVECs infected with Ad-HuR by RT-qPCR and western
blot analysis, respectively. (C) RT-qPCR and (D) western blot
analyses were applied to examine the mRNA and protein levels,
respectively, of HuR in HUVECS transfected with Ad-HuR-shRNA. (E)
HMGB1 mRNA and (F) HMGB1 protein levels were detected in HUVECs
infected with Ad-HuR by RT-qPCR and western blot assays,
respectively. (G) RT-qPCR and (H) western blot analyses were
applied to examine the mRNA and protein levels, respectively, of
HMGB1 in HUVECs transfected with Ad-HuR-shRNA. Data are presented
as the mean ± standard deviation; **P<0.01 vs. empty vector
control. Ad, adenovirus; HMGB1, high-mobility group box 1; HuR,
human antigen R; HUVECs, human umbilical vein endothelial cells;
RT-qPCR, reverse transcription-quantitative polymerase chain
reaction; shRNA, short hairpin RNA. |
| Table II.Human RNA-binding protein-mRNA
interactions. |
Table II.
Human RNA-binding protein-mRNA
interactions.
| Protein | Gene | Target sites | Biocomplex | ClipReadNum | cancerNum |
|---|
| HuR | HMGB1 | 21 | 3 | 21 | 11 |
| PTB | HMGB1 | 3 | 1 | 28 | 9 |
| IGF2BP1 | HMGB1 | 7 | 1 | 141 | 5 |
| IGF2BP2 | HMGB1 | 8 | 1 | 278 | 5 |
| IGF2BP3 | HMGB1 | 12 | 1 | 310 | 6 |
| TNRC6 | HMGB1 | 1 | 1 | 14 | 9 |
| eIF4AIII | HMGB1 | 18 | 2 | 86 | 11 |
| FMRPH | HMGB1 | 4 | 2 | 4000 | 7 |
| FXR2 | HMGB1 | 2 | 1 | 2000 | 7 |
| FUS | HMGB1 | 17 | 4 | 1052 | 9 |
| LIN28A | HMGB1 | 4 | 1 | 25 | 5 |
| LIN28B | HMGB1 | 3 | 1 | 28 | 3 |
| MOV10 | HMGB1 | 7 | 1 | 7000 | 11 |
| CAPRIN1 | HMGB1 | 2 | 1 | 1350 | 8 |
| EWSR1 | HMGB1 | 1 | 1 | 1000 | 10 |
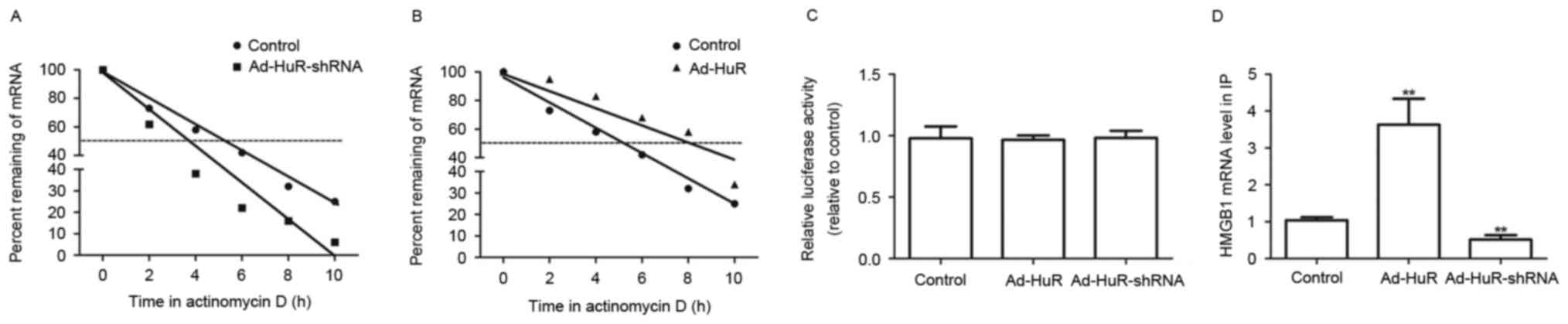
HuR may physically interact with HMGB1
in HUVECs
Whether HuR was able to regulate HMGB1 mRNA
stability in HUVECs was examined. HuR expression was induced or
knocked down followed by the inhibition of de novo mRNA
synthesis by ActD treatment. The decay rate of HMGB1 mRNA was
faster in Ad-HuR-shRNA infected cells (t1/2=3.8±0.4 h)
compared with control cells (t1/2=5.4±0.3 h; Fig. 2A), and slower in the Ad-HuR-treated
group (t1/2=7.9± 0.2 h) compared with the control cells
(t1/2=5.3± 0.4 h; Fig.
2B). However, the luciferase reporter assay demonstrated that
the promoter transcriptional activity of HMGB1 was not affected by
Ad-HuR or Ad-HuR-shRNA infection in HUVECs (Fig. 2C), which indicate that HuR could
not regulate the transcriptional activity of HMGB1. Furthermore,
RIP assays demonstrated that HuR interacted with HMGB1 more
specifically than the control group (Fig. 2D). Our results demonstrate that HuR
may interact with HMGB1 and enhances HMGB1 mRNA stability in
HUVECs.
Effects of HuR on LPS- and
CLP-mediated release of HMGB1
Previous studies have shown that LPS induced HMGB1
release in human endothelial cells (11,12).
The release of HMGB1 was stimulated with LPS (100 ng/ml) treatment
in HUVECs (Fig. 3A). HUVECs were
transfected with Ad-HuR-shRNA and subsequently stimulated with LPS
for 24 h. Knockdown of HuR with Ad-HuR-shRNA attenuated the
inductive effects of LPS on HMGB1 release (Fig. 3B). However, HUVECs treated with
Ad-HuR-shRNA alone did not affect the release of HMGB1 at 24 h, but
decreased HMGB1 release at 48 and 72 h (Fig. 3C). As CLP-induced sepsis in mice
closely resembles human sepsis (13), mice were subjected to sepsis by CLP
treatment. Notably, injection of HuR-siRNA inhibited the
CLP-mediated release of HMGB1 (Fig.
3D).
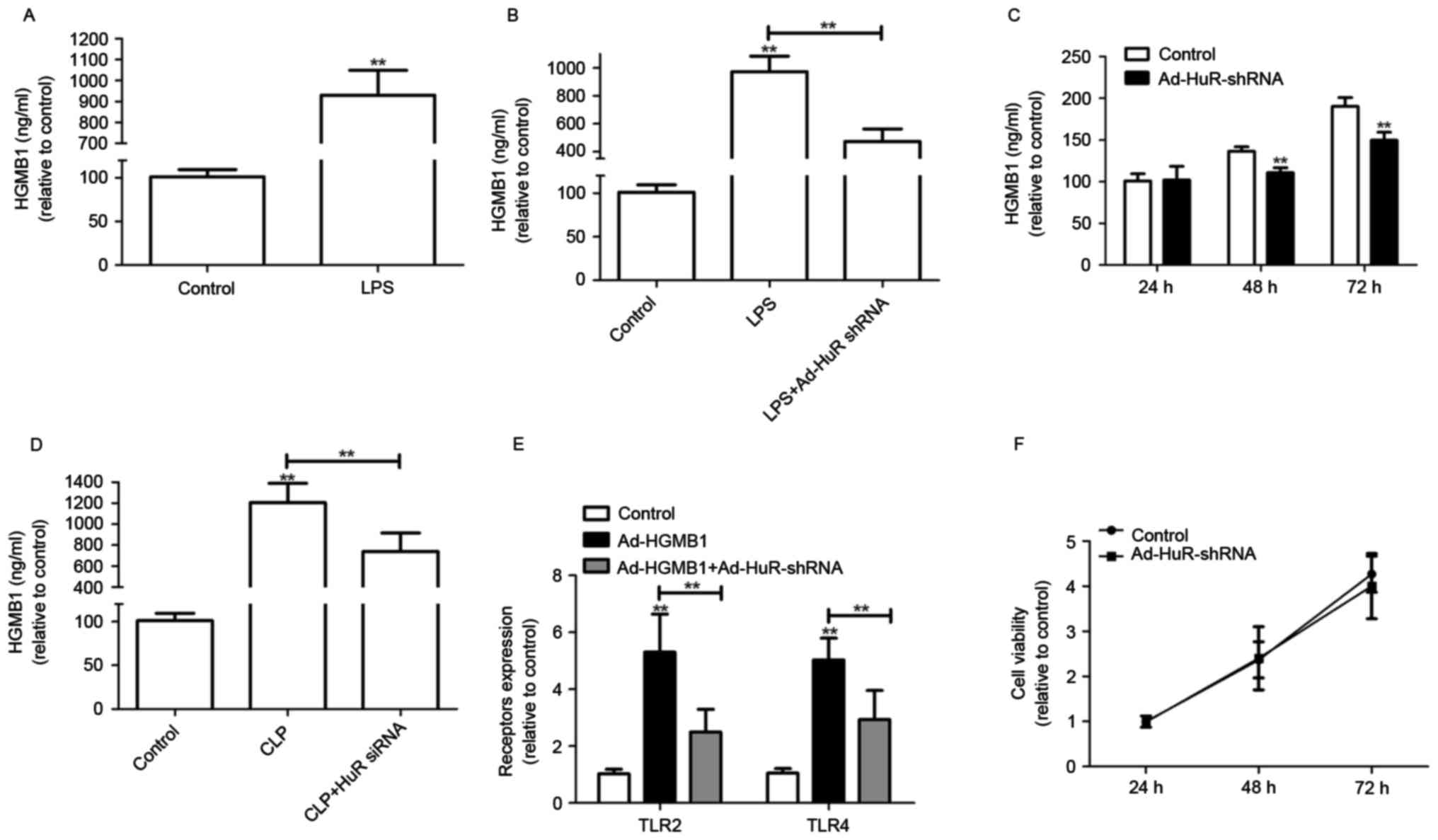 | Figure 3.HuR knockdown impairs HMGB1 release
and receptor expression. (A) HUVECs were treated with 100 ng/ml LPS
for 16 h, and HMGB1 release was measured by ELISA. (B) HUVECs were
transfected with Ad-HuR-shRNA for 24 h following stimulation with
LPS (100 ng/ml) for 16 h; HMGB1 release was examined by ELISA. (C)
HMGB1 release was tested by ELISA in Ad-HuR-shRNA-transfected
HUVECs for 24, 48 and 72 h. (D) Male C57BL/6 mice underwent CLP
surgery and subsequently injected with HuR-siRNA intravenously at
12 h post-CLP (n=5). Mice were euthanized 24 h following CLP. Serum
HMGB1 levels were measured by ELISA. (E) Confluent HUVECs were
incubated with Ad-HMGB1 (1 µg/ml) for 16 h; cells were subsequently
infected with or without Ad-HuR-shRNA for 24 h. Expression levels
of TLR2 and TLR4 in HUVECs were measured by cell-based ELISA. (F)
The effects of Ad-HuR-shRNA on cellular viability were measured
using the Cell Counting Kit-8 assay. Values are presented as the
mean ± standard deviation; n=3; **P<0.01. Empty vectors and
Scrambled-siRNA were used as controls. Ad, adenovirus; CLP, cecal
ligation and puncture; HMGB1, high-mobility group box 1; HuR, human
antigen R; HUVECs, human umbilical vein endothelial cells; LPS,
lipopolysaccharide; shRNA, short hairpin RNA; siRNA, small
interfering RNA; TLR, toll-like receptor. |
In addition, the effects of HuR on HMGB1 receptors
TLR2 and TLR4 were detected in HUVECs. The results indicated that
infection with Ad-HMGB1 significantly induced TLR2 and TLR4 protein
expression, which was attenuated by co-infection with Ad-HuR-shRNA
(Fig. 3E). To exclude the
possibility that the decline of HMGB1 release was due to
cytotoxicity caused by Ad-HuR-shRNA infection, cell viability was
examined following Ad-HuR-shRNA infection by CCK-8 assay.
Ad-HuR-shRNA did not affect cell viability at 24, 48 or 72 h
post-transfection (Fig. 3F).
Therefore, these results suggested that HuR-knockdown may attenuate
the LPS- and CLP-mediated HMGB1 release.
Effects of HuR-siRNA transfection on
LPS or HMGB1-mediated barrier disruption
The effects of HuR on the barrier integrity of
HUVECs were also examined. No significant difference was identified
in barrier integrity in cells transfected with Ad-HuR-shRNA alone
compared with control transfected cells (Fig. 4A). As endothelial membrane barriers
were previously demonstrated to be disrupted by LPS induction
(14), HUVECs were transfected
with Ad-HuR-shRNA for 24 h following LPS treatment. Knockdown of
HuR attenuated the LPS-mediated membrane disruption (Fig. 4B). In addition, the barrier
integrity has been reported to be perturbed by HMGB1 induction
(15). Therefore, HUVECs were
transfected with HMGB1 followed by Ad-HuR-shRNA co-transfection.
Cells co-transfected with Ad-HuR-shRNA exhibited an decrease in
HMGB1-mediated membrane disruption (Fig. 4C). In addition, these effects were
investigated in vivo by examining HMGB1- or CLP-induced
vascular permeability in mice. Treatment with HuR-shRNA suppressed
HMGB1- or CLP-induced peritoneal leakage of dye (Fig. 4D and E, respectively). These
results demonstrated that the knockdown of HuR expression may
reduce HMGB1-mediated endothelial disruption and maintain the human
endothelial cell barrier integrity.
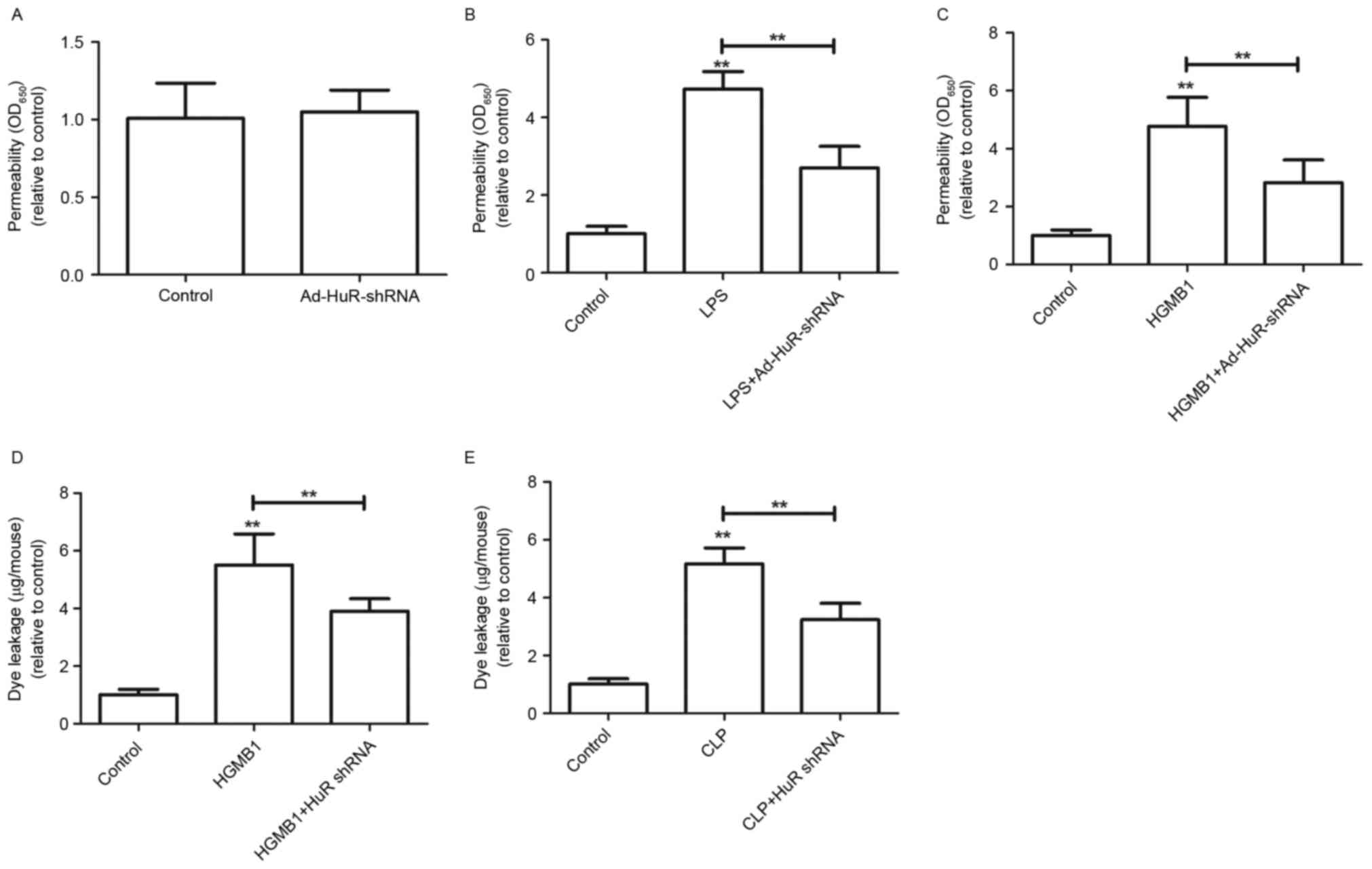 | Figure 4.Ad-HuR-shRNA impairs HMGB1-mediated
permeability in vitro and in vivo. (A) HUVECs were
infected with Ad-HuR-shRNA for 24 h, and permeability was monitored
by measuring the flux of Evans blue-bound albumin across HUVECs
(n=3). HUVECs were stimulated with (B) LPS (100 ng/ml for 4 h) or
(C) HMGB1 (1 µg/ml, 16 h) and subsequently infected with
Ad-HuR-shRNA for 24 h. Permeability was monitored by measuring the
flux of Evans blue-bound albumin across HUVECs (n=3). The effects
of HuR-siRNA injection on (D) HMGB1-induced (2 µg/mouse) or (E)
CLP-induced (24 h post-CLP) vascular permeability in mice were
examined by measuring the amount of Evans blue in peritoneal
washings (n=5). Values are presented as the mean ± standard
deviation; **P<0.01 vs. control. Empty vector and
Scrambled-siRNA were used as controls. Ad, adenovirus; CLP, cecal
ligation and puncture; HMGB1, high-mobility group box 1; HuR, human
antigen R; HUVECs, human umbilical vein endothelial cells; LPS,
lipopolysaccharide; shRNA, short hairpin RNA; siRNA, small
interfering RNA. |
HuR-knockdown inhibits the expression
of cell adhesion molecules (CAMs) and proinflammatory
responses
A previous study reported that the inflammatory
effects mediated by HMGB1 were caused by increasing the cell
surface expression of intracellular adhesion molecule (ICAM)-1,
vascular (V)CAM-1 and E-selectin, which promote leukocyte adhesion
and migration from the endothelium to inflammation sites and hence
of the proinflammatory response (16). Transfection with Ad-HuR-shRNA
suppressed the mRNA expression levels of ICAM-1, VCAM-1 and
E-selectin (Fig. 5A). The binding
ability of leukocytes to HMGB1-stimulated HUVECs was significantly
inhibited by HuR knockdown (Fig.
5B). In addition, HuR-knockdown inhibited the association of
leukocytes to HUVEC binding with the subsequent transendothelial
migration of leukocytes (Fig. 5C).
Notably, mice injected with HuR-siRNA exhibited an inhibition of
HMGB1- or CLP-induced migration of neutrophils (Fig. 5D and E). The results demonstrated
that knockdown of HuR inhibits HMGB1-induced proinflammatory
signaling and thereby downregulates the activation of inflammatory
pathways, such as the upregulation of CAMs, leukocyte adhesion and
migration induced by HMGB1.
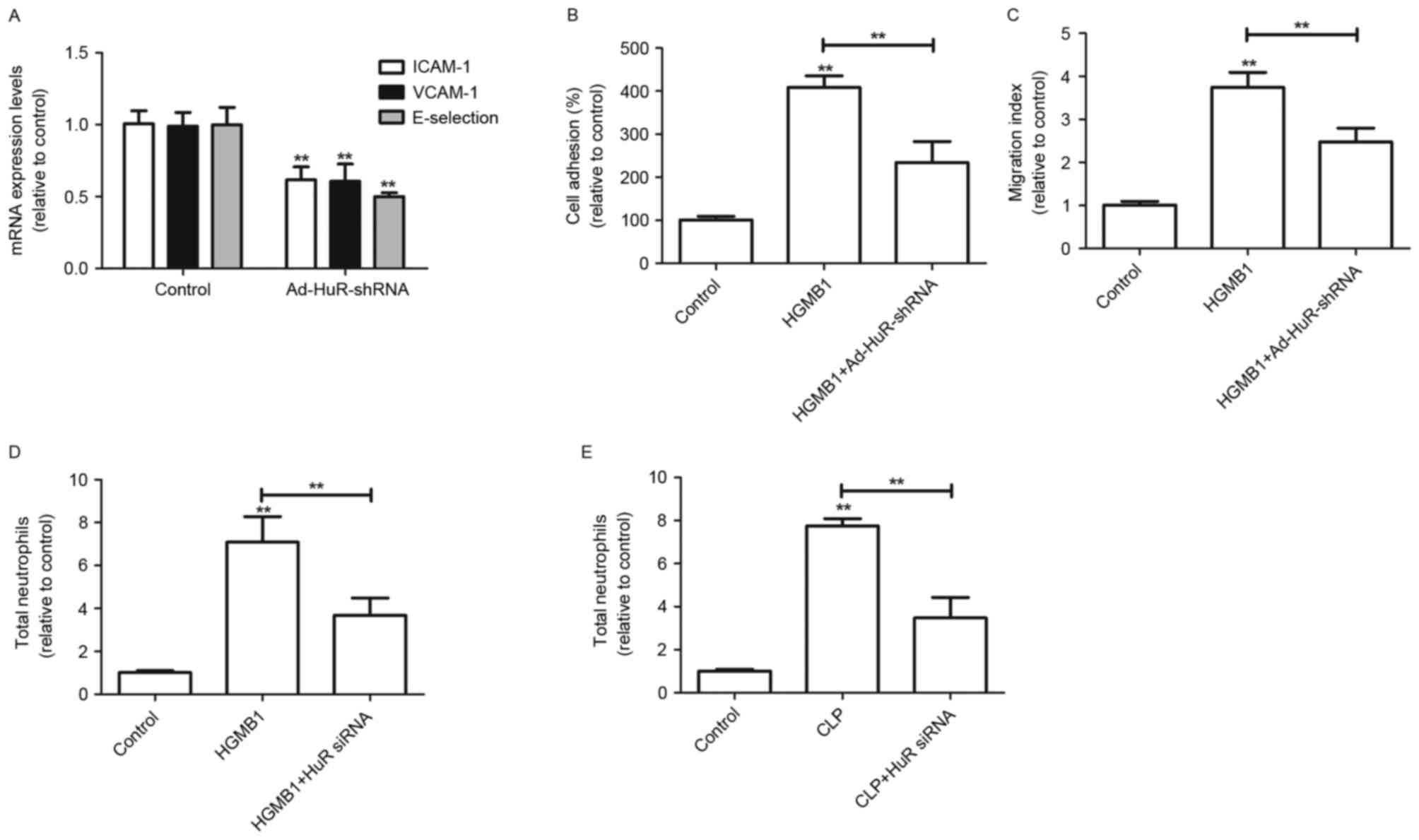 | Figure 5.Effects of HuR knockdown on
HMGB1-mediated pro-inflammatory responses. (A) HUVECs were
stimulated with HMGB1 (1 µg/ml) for 16 h followed by transfection
with Ad-HuR-shRNA. HMGB1-mediated expression levels of VCAM-1,
ICAM-1 and E-selectin in HUVECs were detected by reverse
transcription-quantitative polymerase chain reaction. (B) Adherence
of neutrophils to HUVECs and (C) migration of monocytes through
HUVECs were analyzed. Male C57BL/6 mice (D) were stimulated with
HMGB1 or (E) received CLP surgery and were subsequently co-treated
with HuR-siRNA, and HMGB1- or CLP-mediated migration of leukocytes
into the peritoneal cavity of mice was analyzed. Values are
presented as the mean ± standard deviation; n=3; **P<0.01 vs.
control. Empty vectors and Scramble-siRNA were used as a controls.
Ad, adenovirus; CLP, cecal ligation and puncture; HMGB1,
high-mobility group box 1; HuR, human antigen R; HUVECs, human
umbilical vein endothelial cells; ICAM, intercellular adhesion
molecule; shRNA, short hairpin RNA; siRNA, small interfering RNA;
VCAM, vascular cell adhesion molecule. |
HuR-knockdown inhibits
HMGB1-stimulated production of TNF-α and IL-1β
To investigate the potential effects of
HuR-knockdown on the induction of inflammatory signaling molecules,
the production levels of TNF-α and IL-1β were detected in
HMGB1-activated HUVECs. The production of TNF-α and IL-1β was
increased by HMGB1 treatment (Fig. 6A
and B, respectively), which was attenuated by HuR knockdown
with Ad-HuR-shRNA. In addition, the effects of HuR knockdown on the
NF-κB pathway were evaluated. Knockdown of HuR inhibited the
promotive effects of HMGB1-mediated NF-κB activity (Fig. 6C). IL-10 is an anti-inflammatory
cytokine (17); the production of
IL-10 was detected in HMGB1-activated HUVECs. The production of
IL-10 was decreased by HMGB1 treatment (Fig. 6D), and this effect was reversed by
HuR knockdown. These results indicated that HuR may regulate the
NF-κB pathway that is involved in the induction of proinflammatory
responses in human endothelial cells.
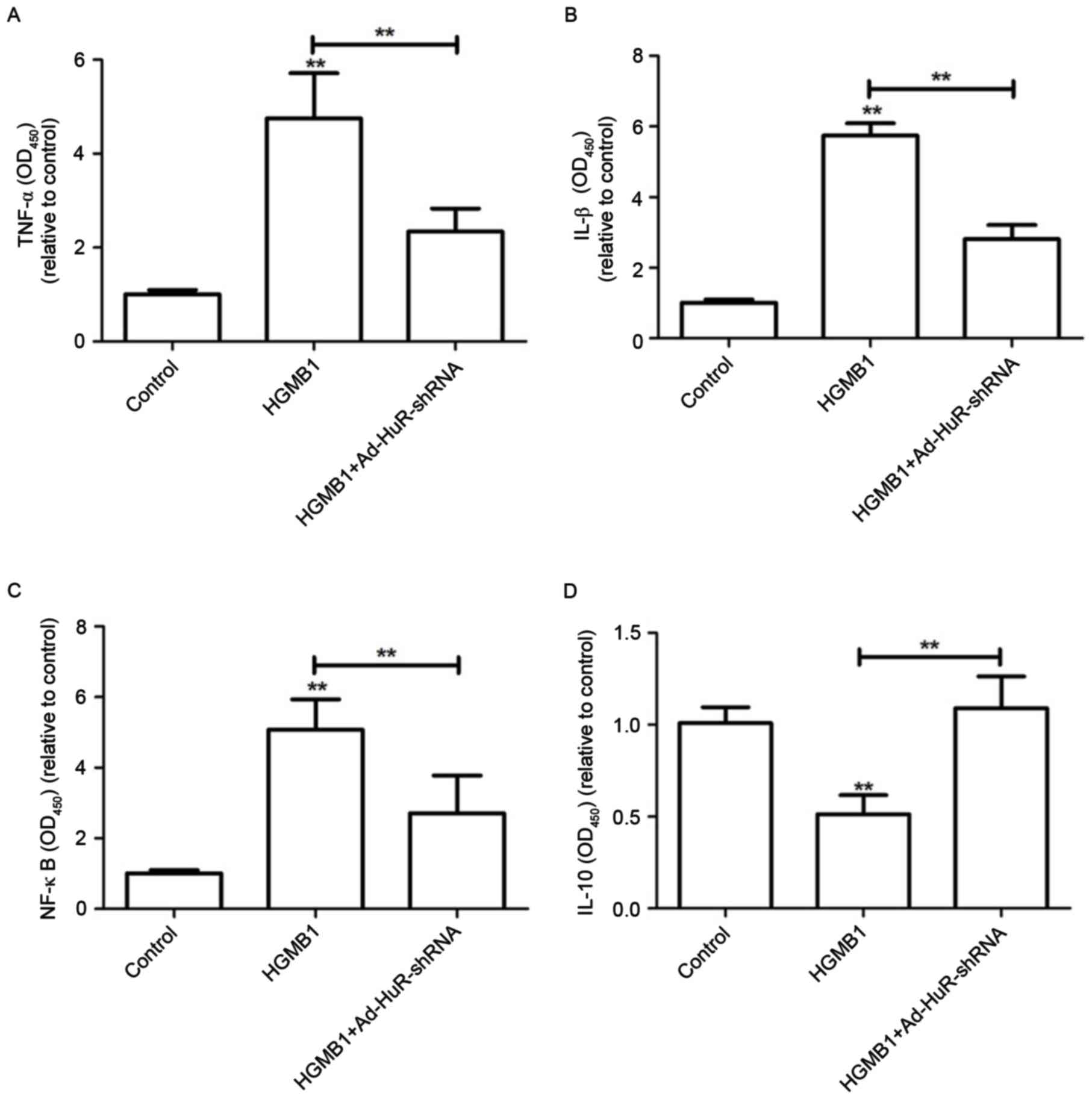 | Figure 6.Effects of HuR knockdown on
HMGB1-stimulated activation of NF-κB and production of TNF-α and
IL-1β in HUVECs. HUVECs were stimulated with HMGB1 (1 µg/ml) for 16
h, followed by transfection with or without Ad-HuR-shRNA for 24 h.
HMGB1-mediated production of (A) TNF-α, (B) IL-1β (C) total NF-κB
(D) IL-10 in HUVECs were analyzed. Values are presented as the mean
± standard deviation; n=3; **P 0.01 vs. control. Empty vectors were
used as control. Ad, adenovirus; HMGB1, high-mobility group box 1;
HuR, human antigen R; HUVECs, human umbilical vein endothelial
cells; IL, interleukin; NF-κB, nuclear factor κB; shRNA, short
hairpin RNA; TNF, tumor necrosis factor. |
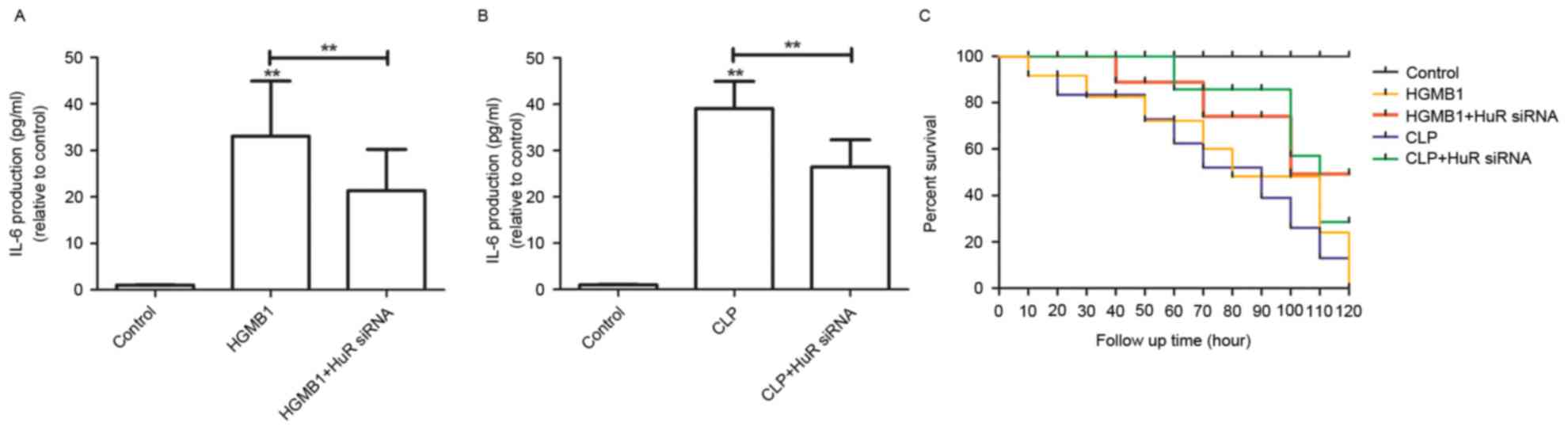
Protective effects of HuR-siRNA in
CLP-induced production of IL-6 and septic mortality
Sepsis has a poor prognosis, which is often
characterized as the production of IL-6 (18,19).
Therefore, the present study hypothesized that treatment with
HuR-siRNA would be able to reduce the production of IL-6 and
mortality rates in HMGB1- or CLP-induced sepsis mouse model.
Production of IL-6 in HMGB1- or CLP-induced mice was reduced by
HuR-siRNA injection (Fig. 7A and
B, respectively). In addition, HuR-siRNA injection resulted in
an increase in the survival rate, analyzed by Kaplan-Meier survival
plot (Fig. 7C), which suggests
that HuR might be a potential therapeutic target for sepsis.
Discussion
Survivors of sepsis may exhibit significant
cognitive and physical impairments (20). Therefore, it is important to
investigate the mechanisms underlying sepsis. Experimentally,
sepsis is routinely induced by a number of techniques, such as the
infusion of exogenous bacterial toxins and CLP-induced disruption
of host epithelial barrier (3).
Bacterial toxin-induced models have been previously used to
investigate the roles of cytokines in systemic inflammation;
however, the CLP-induced model is considered the most clinically
relevant experimental model for human sepsis (21). In the present study, CLP-induced
model rats were used. The results demonstrated that the knockdown
of HuR may attenuate LPS- and CLP-mediated HMGB1 release, and
reduce HMGB1-mediated endothelial disruption and maintain the human
endothelial cell barrier integrity. Notably, data from the present
study indicated that HuR knockdown provided protective effects on
CLP-induced production of IL-6 and mortality due to sepsis. A
previous study demonstrated that high plasma concentrations of
HMGB1 during sepsis may be related to poor prognosis and high
mortality (22); the prevention of
LPS- or CLP-induced release of HMGB1 by knockdown of HuR in the
present study suggested that HuR may be a potential therapeutic
target for the treatment of vascular inflammatory diseases.
In mouse models of endotoxemia and CLP-mediated
sepsis, HMGB1 was first detected in the circulation 8 h following
onset of the disease, and subsequently increased to plateau levels
from 16 to 32 h (21). However,
whether HMGB1 is regulated and the detailed mechanism was unknown.
Post-transcriptional regulation, particularly mRNA stability
alterations, may lead to the ectopic expression of
inflammation-related genes (5).
The present study demonstrated that HMGB1 may be a target of HuR in
HUVECs. To the best of the authors' knowledge, the present study
was the first to reveal a novel mechanism of HuR-mediated
regulation HMGB1 in HUVECs.
In conclusion, HuR/HMGB1 may be a potential
therapeutic target and provides a strong rationale for the
development of HMGB1-based therapeutic strategies for sepsis.
Acknowledgements
The authors thank Professor Hu for critical reading
of this study.
References
|
1
|
Riedemann NC, Guo RF and Ward PA: Novel
strategies for the treatment of sepsis. Nat Med. 9:517–524. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Wang H, Bloom O, Zhang M, Vishnubhakat JM,
Ombrellino M, Che J, Frazier A, Yang H, Ivanova S, Borovikova L, et
al: HMG-1 as a late mediator of endotoxin lethality in mice.
Science. 285:248–251. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Wang H, Ward MF and Sama AE: Targeting
HMGB1 in the treatment of sepsis. Expert Opin Ther Targets.
18:257–268. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Huang W, Tang Y and Li L: HMGB1, a potent
proinflammatory cytokine in sepsis. Cytokine. 51:119–126. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gerstberger S, Hafner M and Tuschl T: A
census of human RNA-binding proteins. Nat Rev Genet. 15:829–845.
2014. View
Article : Google Scholar : PubMed/NCBI
|
|
6
|
Zhu H, Berkova Z, Mathur R, Sehgal L,
Khashab T, Tao RH, Ao X, Feng L, Sabichi AL, Blechacz B, et al: HuR
suppresses fas expression and correlates with patient outcome in
liver cancer. Mol Cancer Res. 13:809–818. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Poria DK, Guha A, Nandi I and Ray PS:
RNA-binding protein HuR sequesters microRNA-21 to prevent
translation repression of proinflammatory tumor suppressor gene
programmed cell death 4. Oncogene. 35:1703–1715. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yang M, Cao L, Xie M, Yu Y, Kang R, Yang
L, Zhao M and Tang D: Chloroquine inhibits HMGB1 inflammatory
signaling and protects mice from lethal sepsis. Biochem Pharmacol.
86:410–418. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nabors LB, Gillespie GY, Harkins L and
King PH: HuR, a RNA stability factor, is expressed in malignant
brain tumors and binds to adenine- and uridine-rich elements within
the 3′ untranslated regions of cytokine and angiogenic factor
mRNAs. Cancer Res. 61:2154–2161. 2001.PubMed/NCBI
|
|
11
|
Bae JS and Rezaie AR: Activated protein C
inhibits high mobility group box 1 signaling in endothelial cells.
Blood. 118:3952–3959. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Bae JS and Rezaie AR: Thrombin inhibits
HMGB1-mediated proinflammatory signaling responses when endothelial
protein C receptor is occupied by its natural ligand. BMB Rep.
46:544–559. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Buras JA, Holzmann B and Sitkovsky M:
Animal models of sepsis: Setting the stage. Nat Rev Drug Discov.
4:854–865. 2005. View
Article : Google Scholar : PubMed/NCBI
|
|
14
|
Berman RS, Frew JD and Martin W:
Endotoxin-induced arterial endothelial barrier dysfunction assessed
by an in vitro model. Br J Pharmacol. 110:1282–1284. 1993.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Wolfson RK, Chiang ET and Garcia JG: HMGB1
induces human lung endothelial cell cytoskeletal rearrangement and
barrier disruption. Microvasc Res. 81:189–197. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hansson GK and Libby P: The immune
response in atherosclerosis: A double-edged sword. Nat Rev Immunol.
6:508–519. 2006. View
Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dagdeviren S, Jung DY and Friedline RH:
IL-10 prevents aging-associated inflammation and insulin resistance
in skeletal muscle. FASEB J. 31:701–710. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Cohen J: The immunopathogenesis of sepsis.
Nature. 420:885–891. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Teiten MH, Eifes S, Dicato M and Diederich
M: Curcumin-the paradigm of a multi-target natural compound with
applications in cancer prevention and treatment. Toxins. 2:128–162.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Perl TM, Dvorak L, Hwang T and Wenzel RP:
Long-term survival and function after suspected Gram-negative
sepsis. JAMA. 274:338–345. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Li W, Zhu S, Zhang Y, Li J, Sama AE, Wang
P and Wang H: Use of animal model of sepsis to evaluate novel
herbal therapies. J Vis Exp pii. 39262012.
|
|
22
|
Yoo H, Ku SK, Lee T and Bae JS: Orientin
inhibits HMGB1-induced inflammatory responses in HUVECs and in
murine polymicrobial sepsis. Inflammation. 37:1705–1717. 2014.
View Article : Google Scholar : PubMed/NCBI
|