Introduction
Pancreatic ductal adenocarcinoma (PDAC) with its
highly lethal malignancy affected >79,400 people in China in
2015 (1). While it is a rapidly
invasive, metastatic tumor, nearly 80% of PDAC patients are
unresectable at diagnosis due to advanced stages or distant
metastasis. The prognosis of PDAC has not been significantly
improved, despite advances in comprehensive treatment (2). The overall 5-year survival rate for
PDAC is still ~5% (1).
Comprehensive treatment, particularly chemotherapy, is the main
strategy for most PDAC patients. However, the effect of
chemotherapy is still limited, for both monotherapy and
polytherapy.
A variety of molecular and genetic changes exist in
the development of PDAC (3,4).
Aberrant DNA hypermethylation is considered to be associated with
tumorigenesis in human pancreatic tumors (5). DNA methyltransferases (DNMTs), the
key cellular enzymes in epigenetic modifications, serve a major
role in transferring the methyl group to cytosine in CpG islands,
and are comprised of the three families of DNMT1, DNMT2, DNMT3a and
DNMT3b.
DNMT1 is responsible for the maintenance of the DNA
methylation pattern during DNA replication, whereas DNMT3 functions
as a de novo methyltransferase acting on unmethylated and
hemimethylated DNA. DNMTs restrain tumor suppressor gene
transcription by promoting methylation of CpG islands in the
promoter, thus contributing to the occurrence and development of
the tumor. Previous studies have demonstrated that DNMT1 and DNMT3a
are overexpressed in a variety of human tumors, including gastric
cancer and pancreatic cancer (6,7), and
overexpression of DNMT1 and DNMT3a is inversely associated with the
prognosis of PDAC (8,9). Methylation-mediated tumor suppressor
gene silencing, which does not involve altering the DNA base
sequence, can be reversed by pharmacological or chemical
intervention. Therefore, DNMTs have been considered as potential
anti-cancer therapeutic targets (10). Inhibition of DNMT1 had synergic
effects on the cytotoxicity induced by chemotherapeutic drugs in
multiple tumor models, including pancreatic cancer (11,12).
However, the role of DNMT3a in chemosensitivity remained elusive in
PDAC.
Gemcitabine (GEM) and oxaliplatin (OXA) are DNA
damage agents, which have been applied in the treatment of PDAC
extensively. GEM and 5-fluorouracil (5-FU) have been used as the
main chemotherapeutic regimens for PDAC in the last two decades.
Recently, clinical studies demonstrated that combined GEM and
erlotinib or albumin-bound paclitaxel therapy improved overall
survival by <2 months in metastatic PDAC (13,14).
Furthermore, another combined chemotherapeutic regimen of
FOLFIRINOX, including OXA, irinotecan, 5-FU and leucovorin,
improved the median overall survival for 4.3 months compared with
GEM monotherapy as a first-line therapy for patients with
metastatic PDAC (15). Therefore,
the objective response of GEM and OXA still remains limited
(16). Thus, there is an urgent
need to improve chemotherapeutic efficacy in PDAC.
DNA damage in cancer cells caused by DNA damage
agents raises the activation of cellular responses, including p53
and serine-protein kinase ATM-cell cycle checkpoint kinase (CHK)2
and serine/threonine-protein kinase ATR (ATR)-CHK1 pathways, which
cause the DNA damage response. It induces cell cycle arrest to
repair DNA damage, evading the cytotoxicity of chemotherapeutic
agents. P53-deficient cancer cells, unlike normal cells, rely
mainly on phosphorylation of S or G2 CHK1, which induces S phase
arrest in response to DNA damage, instead of p53 (17). An accumulation of phosphorylated
CHK1 induced by GEM treatment leads to S-phase arrest in Panc-1
cells, which prevents premature mitotic entry, and CHK1 depletion
enhances GEM-mediated cytotoxicity and radiosensitization (18). Inhibition of CHK1 potentiates the
cytotoxicity of irinotecan in triple-negative breast cancer
(19), and overcomes the cisplatin
resistance in head and neck cancer cells with loss of functional
p53 (20). Therefore, CHK1 is
regarded as a potential target in p53-deficient cancer, such as
PDAC, with nearly 50% patients being p53-deficient. A previous
study demonstrated DNMT3a mediates the cell cycle progression in
PDAC cells. However, whether DNMTs affect the activation of CHK1 is
unknown.
Previous research demonstrated DNMT1 and DNMT3a are
widely expressed in PDAC, mediating the proliferation and cell
cycle progression in PDAC cells. However, in our previous work, it
was found that downregulation of DNMT3a had synergic effects with
GEM or OXA in p53-deficient PDAC cells, which was not detected in
DNMT1 inhibition (data not published). The present study
investigated the regulation of DNMT3a on chemosensitivity to GEM
and OXA, and the potential mechanisms in p53-deficient PDAC cells.
Additionally, the role of DNMT3a on CHK1 activity, which
contributes to GEM and OXA sensitivity, was assessed.
Materials and methods
Cell culture and reagents
The Panc-1 p53-deficient pancreatic cancer cells
were obtained from the Type Culture Collection of the Chinese
Academy of Sciences (Shanghai, China). Panc-1 cells were cultured
in RPMI1640 medium (Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) supplemented with 10% fetal bovine serum (FBS), and 100
U/ml penicillin-streptomycin at 37°C and 5% CO2. GEM was
obtained from Eli Lilly, Inc. (Indianapolis, IN, USA). Oxaliplatin
was purchased from Sanofi-Aventis, Inc. (Paris, France). Antibodies
against CHK1 (cat. no. 2360S), phosphorylated (p)-CHK1 (Ser345;
cat. no. 2348S), poly[ADP-ribose] polymerase (PARP; cat. no.
9542L), protein kinase B (AKT; cat. no. 9272S), p-AKT (Ser473; cat.
no. 9271L), p-extracellular signal-regulated kinase (ERK)1/2
(Thr202/Tyr204; cat. no. 4370S), Caspase-3 (cat. no. 9663S) and
γ-histone H2AX (γ-H2AX; cat. no. 9718S) were obtained from Cell
Signaling Technology, Inc. (Danvers, MA, USA). Anti-DNMT3a (cat.
no. sc-20703), anti-GAPDH (cat. no. sc-25778), anti-ERK (cat. no.
sc-514302), and secondary goat anti-rabbit (cat. no. sc-2007) and
goat anti-mouse antibodies (cat. no. sc-2039) were obtained from
Santa Cruz Biotechnology, Inc. (Dallas, TX, USA).
Transient transfection
Small interfering RNA (siRNA) of DNMT3a from
Shanghai Gemma Pharmaceutical Technology, Co., Ltd. (Shanghai,
China) was used: 5′-GCGUCACACAGAAGCAUAUTTAUAUGCUUCUGUGUGACGCTT-3′.
The negative-control siRNA sequence was
5′-AATTCTCCGAACGTGTCACGT-3′. Panc-1 cells were transfected with
DNMT3a siRNA, negative-control siRNA and Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol, and the transfection medium was replaced
4–6 h after transfection.
MTT assay
Panc-1 cells were seeded at 6×103 cells
per well into 96-well plates with three replicate wells for each
condition. Cells treated with GEM and OXA were cultured in 96-well
plates for 48 h. A total of 24 h after transfection, GEM and OXA
were added to each corresponding well to continue culturing for 48
h, with final concentration of 1 and 5 µM, respectively. Cells were
then harvested for MTT assay. MTT (20 µl; 5 mg/ml) reagent was
added to each well, and the incubation continued for 4 h at 37°C.
After removal of the supernatant, dimethyl sulfoxide (DMSO; 200
µl/well) was added to dissolve the formazan crystals. The optical
density was measured at 570 nm with a microplate reader (Model 550,
Bio-Rad Laboratories, Inc., Hercules, CA, USA). All experiments
were repeated three times.
Western blot analysis
The cells were washed with ice-cold PBS twice, lysed
in lysis buffer (1% Triton X-100, 50 mM Tris-HCl Ph 7.4, 150 mM
NaCl, 10 mM EDTA, 100 mM NaF, 1 mM Na3VO4, 1
mM PMSF and 2 µg/ml aprotinin) and quantified with the Coomassie
brilliant blue G-250 method (Shanghai Maikun Chemical Co., Ltd,
Shanghai, China). The supernatant was diluted in 3X SDS loading
buffer and then boiled for 5 min. Proteins (20 µg) were separated
on 8–12% gels by SDS-PAGE, then transferred onto a polyvinylidene
difluoride membrane. The membranes were blocked with 5% skimmed
milk in TBS with Tween 20 (TBST) buffer (10 mM Tris-HCl pH 7.4, 150
mM NaCl, 0.1% Tween 20) at room temperature for 1 h. The membranes
were incubated overnight with the antibodies CHK1 (1:1,000), p-CHK1
(1:500), PARP (1:1,000), AKT (1:1,000), p-AKT (1:500), p-ERK1/2
(1:500), Caspase-3 (1:1,000), γ-H2AX (1:1,000), DNMT3a (1:1,000),
GAPDH (1:1,000) and ERK1/2 (1:500) at 4°C. Following washing with
TTBS buffer three times, the membrane was incubated with secondary
goat anti-rabbit (1:1,000) and goat anti-mouse (1:500) antibodies
for 30 min at room temperature. Finally, the protein bands were
ultimately visualized with a MicroChemi 4.2 Gel Capture version
6.12 (DNR Bio-Imaging Systems, Ltd., Neve Yamin, Israel).
Cell cycle analysis
Panc-1 cells were treated with GEM (5 µM) and OXA (5
µM) for 12 and 24 h, respectively. In the meantime, the cells were
transfected with negative-control siRNA or DNMT3a siRNA for 48 h.
After transfection, GEM was added for 12 h with a final
concentration of 5 µM, and OXA was added for 24 h with a final
concentration of 5 µM. Cells were washed with cold PBS twice, and
fixed with ice-cold 70% ethanol overnight at 4°C and then incubated
with 100 µg/ml RNase A in PBS for 30 min at 37°C. Subsequently,
cells were stained with propidium iodide (PI; 5 mg/ml) for another
30 min at 37°C away from light. A BD Accuri C6 FACScanflow
cytometer (BD Biosciences, Franklin Lakes, NJ, USA) and ModFit LT
software version 3.3 (modfit-lt.software.informer.com/download; Verity
Software House, Inc., Topsham, ME, USA) were used to analyze cell
cycle distribution. All experiments were repeated three times.
Apoptosis analysis
Panc-1 cells were seeded at 3×105 cells
per well into 6-well plates. Cells were transfected with
negative-control siRNA, DNMT3a siRNA using Lipofectamine 2000
(Invitrogen; Thermo Fisher Scientific, Inc.) according to the
manufacturer's protocol. A total of 24 h after transfection, GEM
and OXA were added for 48 h with a final concentration of 5 µM.
Subsequently, cells were harvested and resuspended in binding
buffer containing Annexin V-FITC and PI according to the
instructions of the Annexin V-FITC/PI Apoptosis Detection kit
(Invitrogen; Thermo Fisher Scientific, Inc.). The percentage of
apoptosis was analyzed by flow cytometry. For each group, the
process was repeated three times.
Statistical analysis
All experiments were repeated at least three times.
All values are expressed as the mean ± standard error. Differences
between the multiple groups were evaluated by one-way analysis of
variance with a post-hoc LSD test, and differences between the two
groups were evaluated by Student's t-test (two-tailed). SPSS
software version 17.0 (SPSS Inc., Chicago, IL, USA) was used for
statistical analysis. P<0.05 was considered to indicate a
statistically significant difference.
Results
DNMT3a downregulation increases GEM
and OXA sensitivity of p53-deficient PDAC cells
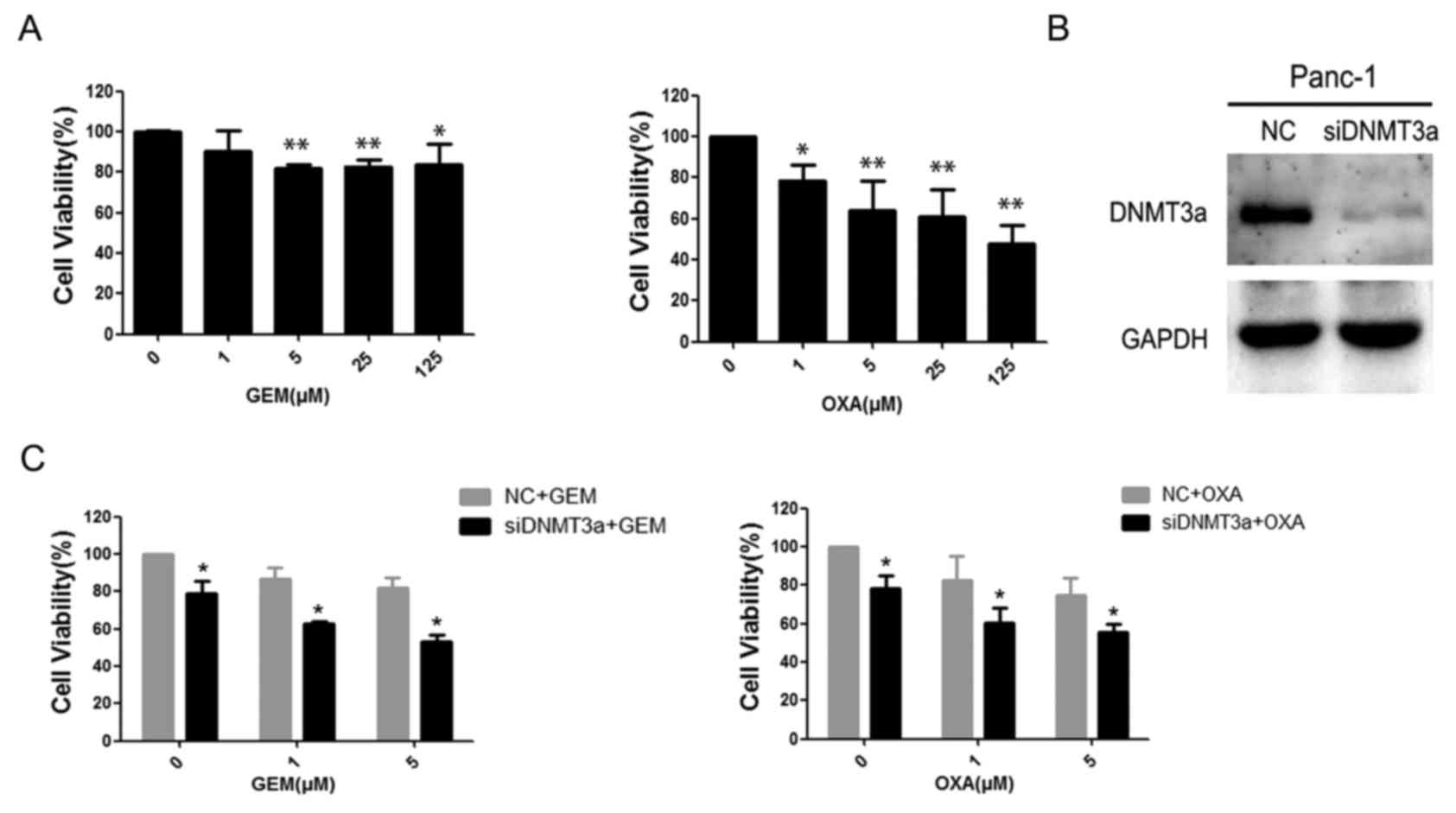
First, GEM and OXA sensitivity was examined in
Panc-1 cells. The results demonstrated that cell viability was
inhibited by GEM and OXA in a dose-dependent manner in 48 h. Panc-1
cells treated with GEM had significantly greater viability than
cells treated with OXA (Fig. 1A).
The cell viability rates of Panc-1 cells treated with 5 µM GEM for
48 h was 82.13±1.93%, (P=0.007), and for cells treated with 5 µM
OXA was 63.84±14.49% (P=0.001). To identify the role of DNMT3a in
regulating chemosensitivity, siRNA was used to knockdown the
expression of DNMT3a in Panc-1 cells, and the efficiency of DNMT3a
suppression by siRNA was confirmed by western blotting (Fig. 1B). Subsequently, the effects of
DNMT3a downregulation on the sensitivity of pancreatic cancer cells
to GEM and OXA was examined by MTT assay. The results demonstrated
that the downregulation of DNMT3a increased the drug-sensitivity of
Panc-1 to both GEM and OXA (Fig.
1C; Table I). These data
suggested that DNMT3a downregulation increased sensitivity of
Panc-1 cells to GEM and OXA.
 | Table I.Effect of DNMT3a downregulation on
sensitivity to GEM and OXA in Panc-1 cells. |
Table I.
Effect of DNMT3a downregulation on
sensitivity to GEM and OXA in Panc-1 cells.
|
| Cell viability
(%) |
|---|
|
|
|
|---|
| Concentration
(µM) | NC | siDNMT3a | P-value |
|---|
| GEM |
| 0 |
100.0±0.00 |
79.26±6.14 | 0.028 |
| 1 |
86.54±6.21 |
62.54±1.31 | 0.016 |
| 5 |
81.96±5.29 |
52.98±3.85 | 0.012 |
| OXA |
| 0 |
100.0±0.00 |
78.32±6.65 | 0.029 |
| 1 |
82.64±12.42 |
60.67±7.73 | 0.028 |
| 5 |
74.59±9.39 |
55.85±4.23 | 0.025 |
GEM and OXA induce the activation of
AKT and CHK1 in Panc-1 cells
To investigate the effect of GEM and OXA on cell
proliferation and DNA damage, western blot analysis and flow
cytometry were performed to identify the expression of associated
proteins and the cell cycle. p-AKT and p-ERK were gradually
elevated by GEM and OXA in Panc-1 cells, which reached a peak at 6
h following GEM or OXA treatment (Fig.
2A). Flow cytometry revealed that the accumulation in S phase
cells was increased with following treatment of GEM or OXA for 12
and 24 h, respectively (Fig. 2B).
The percentage of S phase cells increased from 39.19±5.43 to
49.27±3.99% following GEM treatment, and 41.48±2.43 to 51.81±3.28%
following OXA treatment (Fig. 2C).
Furthermore, γ-H2AX was activated in a time-dependent manner in
Panc-1 cells treated with GEM and OXA. In addition, a significant
phosphorylation of CHK1 was observed after drug treatment for 6 h
(Fig. 2D). Taken together, these
data suggested that GEM and OXA, as DNA damage agents, not only
induced cell cycle arrest, but also stimulated the cell
proliferation signal pathway, which may cause limited inhibition of
GEM and OXA in Panc-1 cells.
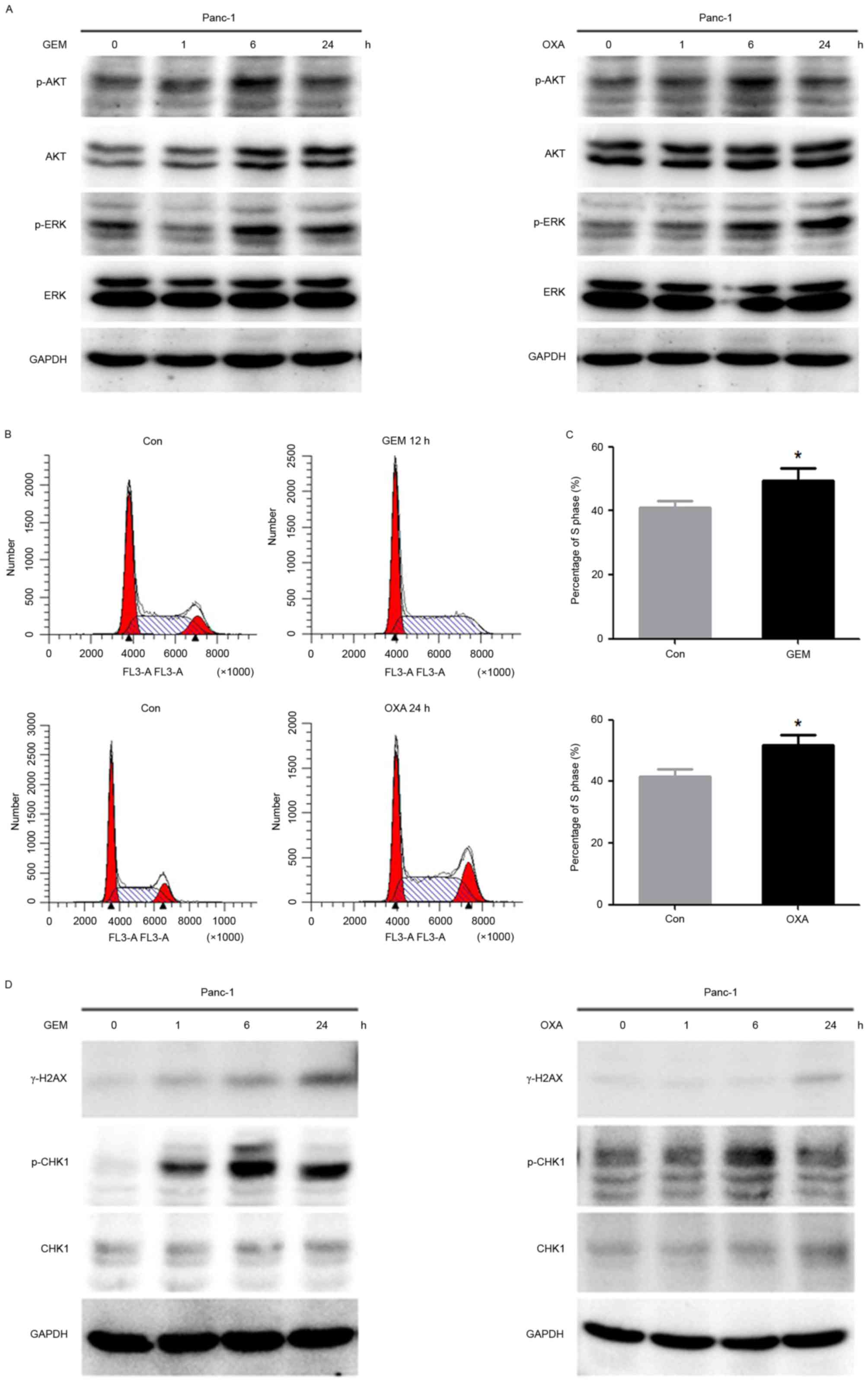 | Figure 2.GEM and OXA induces activation of AKT
and CHK1 in Panc-1 cells. (A) The expression of p-AKT, AKT, p-ERK
and ERK in Panc-1 cells treated with GEM (5 µM) and OXA (5 µM) for
0, 1, 6, 24 h was analyzed by western blot. (B) The cell cycle was
detected by flow cytometry in Panc-1 cells after GEM (5 µM) and OXA
(5 µM) treatment for 12 and 24 h and (C) percentage of cells in S
phase. (D) Cells were treated with GEM (5 µM) and OXA (5 µM) for 0,
1, 6 and 24 h. Expression of γ-H2AX, p-CHK1 and CHK1 were
determined by western blot analysis. Data are presented as the mean
± standard deviation. *P<0.05. Con, control; GEM, gemcitabine;
OXA, oxaliplatin; p, phosphorylated; AKT, protein kinase B; γ-H2AX,
γ-histone H2AX; ERK, extracellular signal-regulated kinase; CHK1,
cell cycle checkpoint kinase 1. |
DNMT3a downregulation promotes cell
apoptosis induced by GEM and OXA
The cytotoxicity of GEM and OXA not only inhibited
proliferative signal and cell cycle arrest, but also induced
apoptosis in cancer cells. Annexin V-FITC/PI and western blot
analysis were performed to elucidate cell apoptosis. Annexin
V-FITC/PI demonstrated that DNMT3a downregulation combined with GEM
or OXA increased cell apoptosis (GEM + NC vs. GEM + siDNMT3a:
7.97±2.11% vs. 19.87±3.23%, P<0.001; OXA + NC vs. OXA +
siDNMT3a: 4.4±1.65% vs. 16.57±2.44%, P<0.001; Fig. 3A and B). Similarly, dual inhibition
with DNMT3a siRNA and drug treatment distinctly induced cleavage of
PARP and caspase-3 compared to single agent treatment (Fig. 3C). Thus, DNMT3a downregulation
increased apoptosis in GEM and OXA-treated Panc-1 cells.
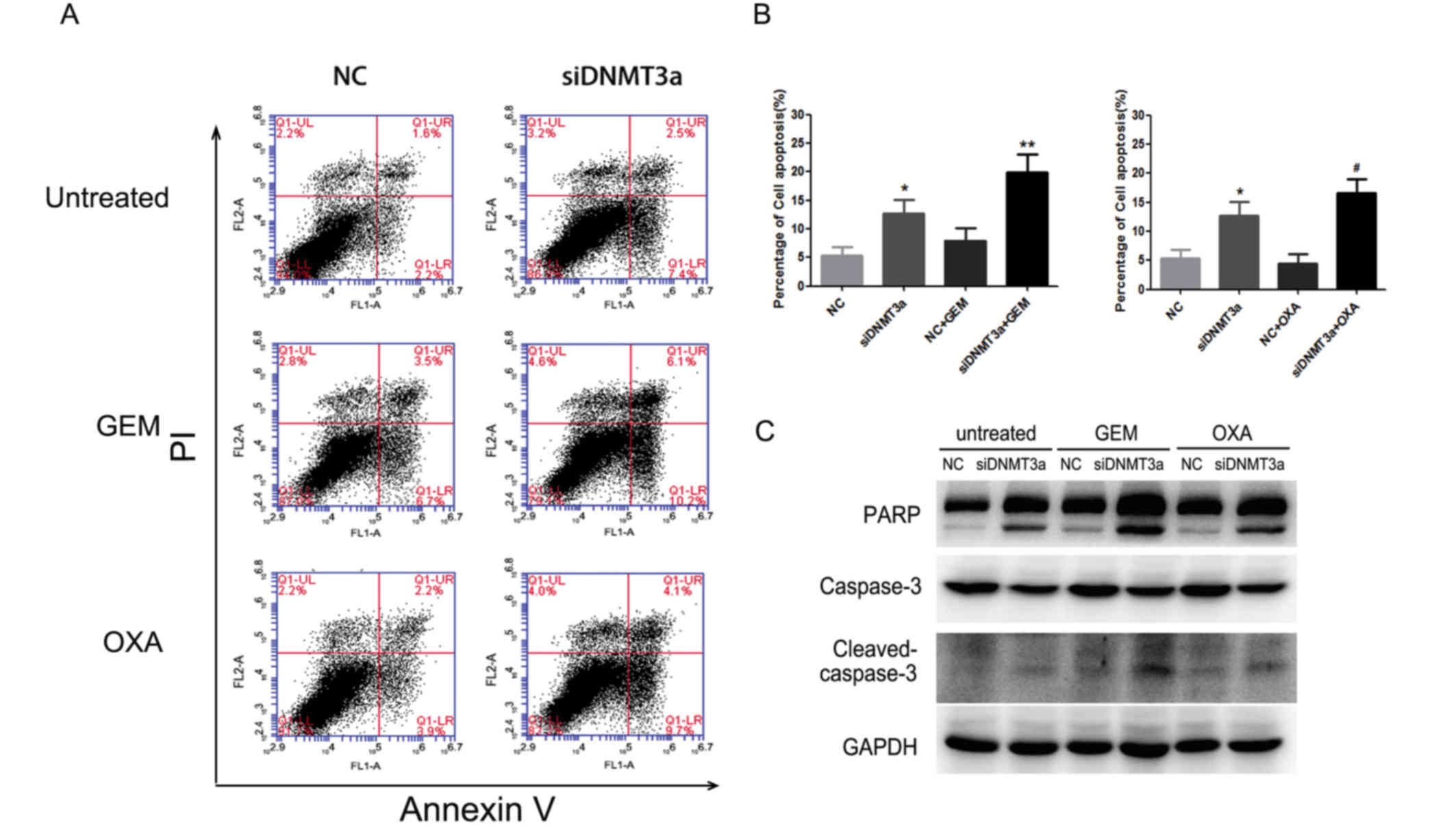 | Figure 3.Effect of a combination of DNMT3a
downregulation with GEM and OXA treatment on apoptosis. (A) After
transfection with DNMT3a siRNA for 72 h and treatment with GEM (5
µM) and OXA (5 µM) for 48 h, Annexin V-FITC/PI was applied to
detect cell apoptosis. (B) The percentage of apoptotic cells
(Annexin V-positive cells). (C) Panc-1 cells were transfected with
DNMT3a siRNA, followed by treatment of GEM (5 µM) or OXA (5 µM) for
48 h, and expression of the apoptosis-associated proteins PARP and
caspase-3 were determined by western blot analysis. Data are
presented as the mean ± standard deviation. *P<0.01 vs. NC,
**P<0.001 vs. NC + GEM, #P<0.001 vs. NC + OXA.
GEM, gemcitabine; OXA, oxaliplatin; NC, negative control; si, small
interfering; DNMT3a, DNA methyltransferase 3a; FITC, fluorescein
isothiocyanate; PI, propidium iodide; PARP, poly[ADP-ribose]
polymerase. |
DNMT3a downregulation abrogates the
activation of AKT and CHK1 and cell cycle arrest induced by GEM and
OXA
Next, the change of AKT and ERK signaling and cell
cycle when DNMT3a was downregulated was investigated. The results
demonstrated that downregulation of DNMT3a could significantly
inhibit the AKT activation induced by GEM and OXA at 6 h, while no
obvious change of p-ERK was observed in Panc-1 cells (Fig. 4A). Additionally, downregulation of
DNMT3a distinctly abolished the blockage of S phase arrest induced
by GEM and OXA at 12 and 24 h, and enhanced
G0/G1 phase arrest (Fig. 4B). Downregulation of DNMT3a
restored the S phase arrest response to GEM and OXA in Panc-1 cells
(GEM + NC vs. GEM + siDNMT3a: 35.49±6.37% vs. 17.88±4.25%, P=0.001;
OXA + NC vs. OXA + siDNMT3a: 45.35±6.15% vs. 24.18±4.93%,
P<0.001), and raised G0/G1 percentage (GEM
+ NC vs. GEM + siDNMT3a: 64.51±6.37% vs. 82.12±4.25%, P=0.003; OXA
+ NC vs. OXA + siDNMT3a: 46.35±6.91% vs. 75.26±4.00%, P<0.001;
Fig. 4C). Furthermore, p-CHK1
expression induced by GEM and OXA at 6 h apparently decreased after
DNMT3a downregulation in Panc-1 cells (Fig. 4D). These results indicated that
DNMT3a downregulation enhanced the sensitivity of GEM and OXA in
Panc-1 cells by disrupting the activation of AKT, CHK1 and S phase
arrest.
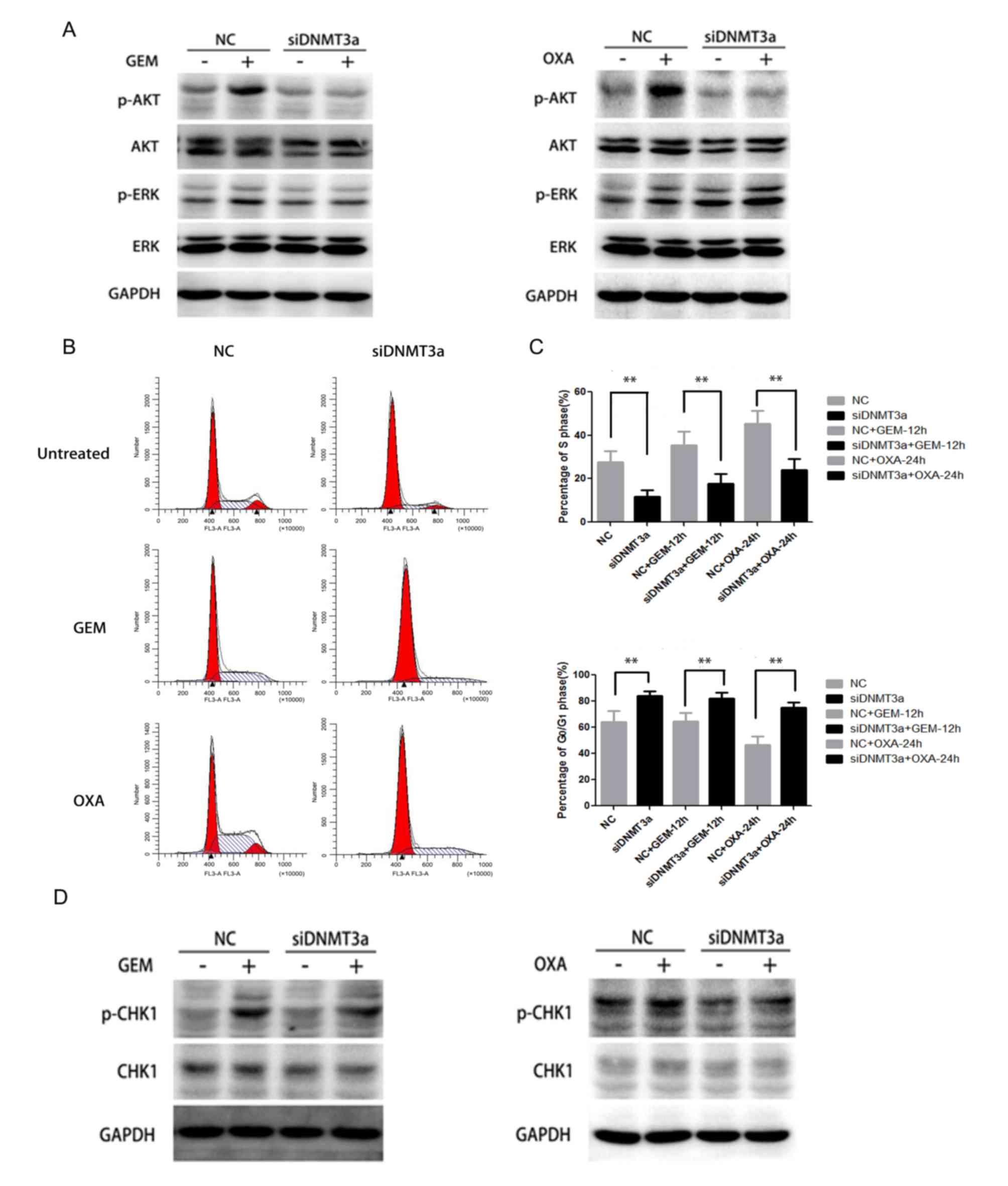 | Figure 4.Effect of DNMT3a downregulation by
siRNA on activation of AKT, CHK1 and cell cycle arrest in Panc-1
cells. (A) Panc-1 cells were transfected with DNMT3a siRNA, then
the cells were treated by GEM (5 µM) and OXA (5 µM) for 6 h. The
expression level of p-AKT, AKT, p-ERK and ERK in negative control
and DNMT3a knockdown cells was assessed by western blot analysis.
(B) Following transfection with DNMT3a siRNA and treatment by GEM
(5 µM) and OXA (5 µM) for 12 and 24 h, flow cytometry was applied
to observe the cell cycle alteration. (C) Percentage of cells in S
and G0/G1 phases. (D) Western blot analysis
of p-CHK1 and CHK1 expression in Panc-1 cells, which were
transfected with DNMT3a siRNA, then treated with GEM (5 µM) and OXA
(5 µM) for 6 h. Data are presented as the mean ± standard
deviation. **P<0.01 vs. corresponding control cells. GEM,
gemcitabine; OXA, oxaliplatin; NC, negative control; si, small
interfering; DNMT3a, DNA methyltransferase 3a; p, phosphorylated;
AKT, protein kinase B; CHK1, cell cycle checkpoint kinase 1; ERK,
extracellular signal-regulated kinase. |
Discussion
Aberrant methylation has been considered to be
involved in pancreatic carcinogenesis and progression. Inhibition
of the function of DNMTs has been proven to be a potential target
for improving survival and reinforcing therapeutic effect (5,9,10).
The present study reported that the cytotoxicity of GEM and OXA was
significantly enhanced in DNMT3a knockdown Panc-1 cells. The
underlying mechanisms suggested DNMT3a downregulation inhibited the
activation of CHK1 and decreased the S phase fraction in Panc-1
cells after drug administration. On the other hand, DNMT3a
downregulation also suppressed AKT activation to inhibit the
responsiveness to DNA damage, and increase cell apoptosis caused by
chemotherapeutic drugs. These data suggested that DNMT3a served an
important role in the chemotherapy sensitivity of p53-deficient
PDAC cells.
CHK1 is the most important serine/threonine kinase
in the cell cycle checkpoint during DNA damage responses. It is
overexpressed in a variety of human tumors, especially in breast,
cervical and gastric carcinomas (21–23).
The activation of the ATR-CHK1 pathway in response to DNA damage
leads to cell cycle arrest for DNA repair during the application of
radiotherapy or anti-cancer therapy agents (24). Accordingly, CHK1 inhibition
potentiates the sensitivity of multiple DNA damage chemotherapy
agents by restraining the DNA damage response, especially
antimetabolites, notably GEM, which is widely used in various of
solid tumors (25,26). In addition, decreased expression of
CHK1 leads to sensitization of mesothelioma cells to platinum, and
hepatocellular carcinoma cells to cisplatin (27,28).
In p53-deficient cells, CHK1 dominates in cell cycle regulation
after DNA damage instead of G1/G0 checkpoint
p53. Therefore, p53-deficient cancer cells are considered to be
more sensitive to therapeutic strategies that combine DNA damaging
agents with CHK1 inhibitors (29).
Encouraging results were obtained with combination of demethylating
agents and classic anticancer chemotherapeutics in colorectal
cancer. DNMTs inhibitors could potentiate the inhibitory effects of
OXA in colorectal cancer cells, while activation of CHK1 respond to
DNA damage response varied in different DNMT inhibitors (30). The role executed by different
members of the DNMT family is still unidentified and needs to be
evaluated (30). In the present
study, it was demonstrated that p-CHK1 and γ-H2AX expression levels
were elevated in p53-deficient Panc-1 cells following DNA damage
caused by GEM and OXA, accompanied by cell accumulation in S phase.
Inhibition of DNMT3a restored the S phase fraction and CHK1
activation, arrested cells in G0/G1 phase and
increased response to chemotherapy treatment. These results
highlighted that DNMT3a downregulation enhanced the sensitivity of
GEM and OXA in p53-deficient Panc-1 cells by disrupting the
activation of CHK1. However, there was no methylation loci in the
promoter of CHK1, and molecules directly regulated by DNMT3a
through epigenetic regulation, which could modify the
phosphorylation of CHK1, were not investigated. Thus, DNMT3a may
regulate CHK1 activation via an indirect effect in Panc-1 cells,
and further study is needed to elucidate the underlying
mechanisms.
AKT serves a critical role in regulating cellular
processes in cancer cells, including cell proliferation,
anti-apoptosis, migration and drug resistance. An excessive
activation of p-AKT may induce multidrug resistance in cancer
(31). In pancreatic cancer,
abnormal AKT activation is an analogous mechanism affecting
chemoresistance to GEM. The combination with an AKT inhibitor and
GEM synergistically inhibited pancreatic cancer cell growth
(32). An excessive activation of
AKT made a great contribution to OXA resistance in hepatocellular
carcinoma (33). In addition,
activation of AKT can also be induced by DNA damage. DNA damage
caused by cisplatin induces activation of AKT in platinum-resistant
ovarian cancer cells, implicating AKT-activation as a resistance
mechanism (34). The present study
observed that AKT/ERK-mediated pro-survival signaling was markedly
activated following treatment with GEM and OXA in Panc-1 cells.
However, downregulation of DNMT3a merely decreased the activation
of AKT in response to cytotoxic agents, and improved the
sensitivity to GEM and OXA. These results revealed that there was a
suppression of the proliferation signal as a synergistic
therapeutic effect of GEM/OXA and DNMT3a inhibition.
According to previous studies, chemotherapeutic
drugs cause anti-tumor effects partly by promoting cell apoptosis.
In the present study, the increased cell apoptosis was detected in
cells combined with DNMT3a downregulation and GEM/OXA, which was
considered to be induced by the effect of irreversible DNA damage
and depressing AKT activation. However, further research is needed
to discover whether there are other factors leading to
apoptosis.
In conclusion, the present study demonstrated that
DNMT3a downregulation enhanced the chemotherapeutic toxicity of GEM
and OXA by suppressing the phosphorylation of CHK1 and AKT,
inhibiting S phase arrest and promoting apoptosis in Panc-1 cells,
suggesting the suppression of DNMT3a sensitized p53-deficient
pancreatic cancer to DNA damage chemotherapeutic agents. Therefore,
the present study implicated DNMT3a as a promising crucial
therapeutic target for p53-deficient PDAC therapy.
Acknowledgements
The present study was supported by Outstanding
Scientific Foundation of Shengjing Hospital (grant no. 201210) and
the National Natural Science Foundation of China (grant no.
81401938).
Glossary
Abbreviations
Abbreviations:
|
PDAC
|
pancreatic ductal adenocarcinoma
|
|
GEM
|
gemcitabine
|
|
OXA
|
oxaliplatin
|
|
DNMTs
|
DNA methyltransferases
|
|
CHK1
|
cell cycle checkpoint kinase 1
|
References
|
1
|
Chen W, Zheng R, Baade PD, Zhang S, Zeng
H, Bray F, Jemal A, Yu XQ and He J: Cancer statistics in China,
2015. CA Cancer J Clin. 66:115–132. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Sinn M, Bahra M, Denecke T, Travis S,
Pelzer U and Riess H: Perioperative treatment options in resectable
pancreatic cancer-how to improve long-term survival. World J
Gastrointest Oncol. 8:248–257. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Delpu Y, Hanoun N, Lulka H, Sicard F,
Selves J, Buscail L, Torrisani J and Cordelier P: Genetic and
epigenetic alterations in pancreatic carcinogenesis. Current
Genomics. 12:15–24. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Biankin AV, Waddell N, Kassahn KS, Gingras
MC, Muthuswamy LB, Johns AL, Miller DK, Wilson PJ, Patch AM, Wu J,
et al: Pancreatic cancer genomes reveal aberrations in axon
guidance pathway genes. Nature. 491:399–405. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Peng DF, Kanai Y, Sawada M, Ushijima S,
Hiraoka N, Kosuge T and Hirohashi S: Increased DNA
methyltransferase 1 (DNMT1) protein expression in precancerous
conditions and ductal carcinomas of the pancreas. Cancer Sci.
96:403–408. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Yang J, Wei X, Wu Q, Xu Z, Gu D, Jin Y,
Shen Y, Huang H, Fan H and Chen J: Clinical significance of the
expression of DNA methyltransferase proteins in gastric cancer. Mol
Med Rep. 4:1139–1143. 2011.PubMed/NCBI
|
|
7
|
He S, Wang F, Yang L, Guo C, Wan R, Ke A,
Xu L, Hu G, Xu X, Shen J and Wang X: Expression of DNMT1 and DNMT3a
are regulated by GLI1 in human pancreatic cancer. PLoS One.
6:e276842011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhang JJ, Zhu Y, Zhu Y, Wu JL, Liang WB,
Zhu R, Xu ZK, Du Q and Miao Y: Association of increased DNA
methyltransferase expression with carcinogenesis and poor prognosis
in pancreatic ductal adenocarcinoma. Clin Transl Oncol. 14:116–124.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Gao J, Wang L, Xu J, Zheng J, Man X, Wu H,
Jin J, Wang K, Xiao H, Li S and Li Z: Aberrant DNA
methyltransferase expression in pancreatic ductal adenocarcinoma
development and progression. J Exp Clin Cancer Res. 32:862013.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ghoshal K and Bai S: DNA
methyltransferases as targets for cancer therapy. Drugs Today
(Barc). 43:395–422. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mutze K, Langer R, Schumacher F, Becker K,
Ott K, Novotny A, Hapfelmeier A, Höfler H and Keller G: DNA
methyltransferase 1 as a predictive biomarker and potential
therapeutic target for chemotherapy in gastric cancer. Eur J
Cancer. 47:1817–1825. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Nagaraju GP, Zhu S, Wen J, Farris AB,
Adsay VN, Diaz R, Snyder JP, Mamoru S and El-Rayes BF: Novel
synthetic curcumin analogues EF31 and UBS109 are potent DNA
hypomethylating agents in pancreatic cancer. Cancer Lett.
341:195–203. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Von Hoff DD, Ervin T, Arena FP, Chiorean
EG, Infante J, Moore M, Seay T, Tjulandin SA, Ma WW, Saleh MN, et
al: Increased survival in pancreatic cancer with nab-paclitaxel
plus gemcitabine. N Engl J Med. 369:1691–1703. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Moore MJ, Goldstein D, Hamm J, Figer A,
Hecht JR, Gallinger S, Au HJ, Murawa P, Walde D, Wolff RA, et al:
Erlotinib plus gemcitabine compared with gemcitabine alone in
patients with advanced pancreatic cancer: A phase III trial of the
National Cancer Institute of Canada Clinical Trials Group. J Clin
Oncol. 25:1960–1966. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Vaccaro V, Sperduti I and Milella M:
FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N
Engl J Med. 365:768–769. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Poplin E, Feng Y, Berlin J, Rothenberg ML,
Hochster H, Mitchell E, Alberts S, O'Dwyer P, Haller D, Catalano P,
et al: Phase III, randomized study of gemcitabine and oxaliplatin
versus gemcitabine (fixed-dose rate infusion) compared with
gemcitabine (30-min infusion) in patients with pancreatic carcinoma
E6201: A trial of the eastern cooperative oncology group. J Clin
Oncol. 27:3778–3785. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen Z, Xiao Z, Gu WZ, Xue J, Bui MH,
Kovar P, Li G, Wang G, Tao ZF, Tong Y, et al: Selective Chk1
inhibitors differentially sensitize p53-deficient cancer cells to
cancer therapeutics. Int J Cancer. 119:2784–2794. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Morgan MA, Parsels LA, Parsels JD,
Mesiwala AK, Maybaum J and Lawrence TS: Role of checkpoint kinase 1
in preventing premature mitosis in response to gemcitabine. Cancer
Res. 65:6835–6842. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Ma CX, Cai S, Li S, Ryan CE, Guo Z,
Schaiff WT, Lin L, Hoog J, Goiffon RJ, Prat A, et al: Targeting
Chk1 in p53-deficient triple-negative breast cancer is
therapeutically beneficial in human-in-mouse tumor models. J Clin
Invest. 122:1541–1552. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
20
|
Gadhikar MA, Sciuto MR, Alves MV,
Pickering CR, Osman AA, Neskey DM, Zhao M, Fitzgerald AL, Myers JN
and Frederick MJ: Chk1/2 inhibition overcomes the cisplatin
resistance of head and neck cancer cells secondary to the loss of
functional p53. Mol Cancer Ther. 12:1860–1873. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Albiges L, Goubar A, Scott V, Vicier C,
Lefèbvre C, Alsafadi S, Commo F, Saghatchian M, Lazar V, Dessen P,
et al: Chk1 as a new therapeutic target in triple-negative breast
cancer. Breast. 23:250–258. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bargiela-Iparraguirre J, Prado-Marchal L,
Fernandez-Fuente M, Gutierrez-González A, Moreno-Rubio J,
Muñoz-Fernandez M, Sereno M, Sanchez-Prieto R, Perona R and
Sanchez-Perez I: CHK1 expression in gastric cancer is modulated by
p53 and RB1/E2F1: Implications in chemo/radiotherapy response. Sci
Rep. 6:215192016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Xu J, Li Y, Wang F, Wang X, Cheng B, Ye F,
Xie X, Zhou C and Lu W: Suppressed miR-424 expression via
upregulation of target gene Chk1 contributes to the progression of
cervical cancer. Oncogene. 32:976–987. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Ben-Yehoyada M, Wang LC, Kozekov ID, Rizzo
CJ, Gottesman ME and Gautier J: Checkpoint signaling from a single
DNA interstrand crosslink. Mol Cell. 35:704–715. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Xiao Y, Ramiscal J, Kowanetz K, Del Nagro
C, Malek S, Evangelista M, Blackwood E, Jackson PK and O'Brien T:
Identification of preferred chemotherapeutics for combining with a
CHK1 inhibitor. Mol Cancer Ther. 12:2285–2295. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Barnard D, Diaz HB, Burke T, Donoho G,
Beckmann R, Jones B, Barda D, King C and Marshall M: LY2603618, a
selective CHK1 inhibitor, enhances the anti-tumor effect of
gemcitabine in xenograft tumor models. Invest New Drugs. 34:49–60.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Røe OD, Szulkin A, Anderssen E, Flatberg
A, Sandeck H, Amundsen T, Erlandsen SE, Dobra K and Sundstrøm SH:
Molecular resistance fingerprint of pemetrexed and platinum in a
long-term survivor of mesothelioma. PLoS One. 7:e405212012.
View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hong J, Hu K, Yuan Y, Sang Y, Bu Q, Chen
G, Yang L, Li B, Huang P, Chen D, et al: CHK1 targets spleen
tyrosine kinase (L) for proteolysis in hepatocellular carcinoma. J
Clin Invest. 122:2165–2175. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Koniaras K, Cuddihy AR, Christopoulos H,
Hogg A and O'Connell MJ: Inhibition of Chk1-dependent G2 DNA damage
checkpoint radiosensitizes p53 mutant human cells. Oncogene.
20:7453–7463. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Flis S, Gnyszka A and Flis K: DNA
methyltransferase inhibitors improve the effect of chemotherapeutic
agents in SW48 and HT-29 colorectal cancer cells. PLoS One.
9:e923052014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Radisavljevic Z: AKT as locus of cancer
multidrug resistance and fragility. J Cell Physiol. 228:671–674.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kim R, Yamauchi T, Husain K, Sebti S and
Malafa M: Triciribine phosphate monohydrate, an AKT inhibitor,
enhances gemcitabine activity in pancreatic cancer cells.
Anticancer Res. 35:4599–4604. 2015.PubMed/NCBI
|
|
33
|
Xiu P, Dong X, Dong X, Xu Z, Zhu H, Liu F,
Wei Z, Zhai B, Kanwar JR, Jiang H, et al: Secretory clusterin
contributes to oxaliplatin resistance by activating Akt pathway in
hepatocellular carcinoma. Cancer Sci. 104:375–382. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Stronach EA, Chen M, Maginn EN, Agarwal R,
Mills GB, Wasan H and Gabra H: DNA-PK mediates AKT activation and
apoptosis inhibition in clinically acquired platinum resistance.
Neoplasia. 13:1069–1080. 2011. View Article : Google Scholar : PubMed/NCBI
|