Introduction
Congenital heart disease (CHD) is a common birth
defect in humans and is a leading cause of non-infectious
mortalities in infancy (1). Heart
development at the embryonic stage is regulated by a developmental
network, including signaling pathways, transcription factors and
epigenetics (2–4). Gene mutations in any of these factors
may cause the development of CHD (5). Ventricular septal defect (VSD) is the
most common type of CHD and accounts for ~30% of diagnoses
(6). However, there have been few
studies that focused on the underlying molecular pathogenetic
mechanism of VSD. Currently, there are several known pathogenetic
genes of VSD, including GATA-binding protein 4 (GATA4), GATA6, NK2
homeobox 5, T-box 1 (TBX1) and TBX5 (7). Mutations in the Ellis-van Creveld
(EVC) gene may lead to EVC syndrome, and ~60% of patients exhibit
the cardiac septal defect phenotype which is comprised of VSD,
atrial septal defect and atrioventricular septal defects (8,9).
Previous studies on EVC gene mutations have focused on the EVC
syndrome lineage and its influence on cartilage development
(10,11). There appear to be no studies on EVC
gene variations in patients with VSD and the potential underlying
pathogenetic mechanism.
The hedgehog (Hh) signaling pathway is a major
pathway that is involved in heart development and members of this
pathway include the 12-transmembrane receptor patched (Ptc), the
7-transmembrane receptor Smoothened (Smo), and the main effector
factors, including members of the cubitus
interruptus/glioma-associated oncogene homolog (Gli) family
(12). The effector molecules in
vertebrates are members of the Gli family. In the absence of Hh
ligands, ligand-free Ptc represses the activity of Smo. In the
presence of Hh, binding of Hh to Ptc relieves Smo repression, and
then Smo translocates to the membrane and is activated by
phosphorylation on its carboxy-terminal tail (13). EVC is a cilia protein expressed in
the second heart field (SHF) of embryos, including the outflow
tract, dorsal mesenchymal protrusion and atrial septum, as well as
in the atrioventricular endocardial cushions (8). In addition, EVC is an activator
molecule of the Hh signaling pathway, acts downstream of Smo after
it is phosphorylated and mediates Hh signal transduction from Smo
to the Gli transcription factors (14,15).
Previous studies have confirmed that regulation of Hh pathway
activity at the embryonic stage primarily participates in cell
differentiation, proliferation and apoptosis in the SHF (13,16,17).
In addition, abnormal SHF development is associated with
atrioventricular septal defect and various types of CHD, including
VSD (16). Mutations in key Hh
signaling genes, such as Ptc or Smo may cause embryos to present
abnormal SHF development and CHD phenotypes (16,18).
In the present study, it was demonstrated that the
c.1727G>A single-nucleotide polymorphism (SNP) of EVC increased
VSD susceptibility in a Chinese Han population. In addition, the
EVC c.343C>G mutation substitution increased apoptosis and
reduced proliferation through downregulation of Hh pathway activity
in NIH3T3 mouse embryonic fibroblast cells, which may be one of the
mechanisms underlying the development of VSD caused by EVC
mutation. The results of the present study may provide novel
targets for the diagnosis and treatment of patients with VSD.
Materials and methods
Study population
A total of 65 patients with VSD (40 males and 25
females) aged from 2 to 71 years and 210 healthy controls (121
males and 89 females) aged from 3 to 68 years were recruited from
January 2008 to December 2010 in The Second Affiliated Hospital of
Nanchang University (Nanchang, China). The patients with VSD were
diagnosed based on physical examination, chest radiography,
electrocardiograms and echocardiography, and confirmed by cardiac
catheterization or cardiac surgery. The healthy controls were
excluded from having CHD by the cardiac echocardiography. All
participants were from the same ethnicity of Chinese Han
population. The present study was approved by The Human Ethics
Committee of the Second Affiliated Hospital of Nanchang University,
and written informed consent was signed by the patients or their
guardians prior to the present study.
Cell culture
NIH3T3 cells were purchased from the Type Culture
Collection of Chinese Academy of Sciences (Shanghai, China) and
cultured in high glucose Dulbecco's modified Eagle's medium (DMEM;
Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA)
supplemented with 10% fetal bovine serum (Gibco; Thermo Fisher
Scientific, Inc.), 100 U/ml penicillin and 100 µg/ml streptomycin
at 37°C in a 5% CO2 incubator.
Reagents and materials
The TIANamp Blood DNA kit (DP332) and 2X PCR Taq
MasterMix (KT201) was obtained from Tiangen Biotech Co., Ltd.
(Beijing, China). Recombinant adenoviruses (CMV-MCS-SV40-GFP)
EVC-wild-type (WT)-green fluorescent protein (GFP) and
EVC-343C>G-GFP (mutation, Mut), and the Gli2-Flag plasmid were
purchased from Shanghai GeneChem Co., Ltd. (Shanghai, China). An
empty vector was used for control (data not shown). The sequence of
the Gli2 was derived from the Nucleotide database at the National
Center for Biotechnology Information (NM_005270.4). The EVC-WT
group was used as the control and treated with the adenoviruses
EVC-wild-type (WT)-green fluorescent protein. The Smo agonist (SAG;
cat. no. 566661) was purchased from Sigma-Aldrich; Merck KGaA
(Darmstadt, Germany). Rabbit anti-B cell lymphoma 2 (Bcl2; cat. no.
12789-1-AP), rabbit anti-B-cell lymphoma 2-associated X protein
(Bax; cat. no. 50599-2-Ig), mouse anti-Flag (cat. no. 66008-2-Ig)
and rabbit anti-β-tubulin antibodies (cat. no. 10068-1-AP) were all
purchased from ProteinTech Group, Inc. (Chicago, IL, USA);
anti-Gli1 (cat. no. 2643) antibody was obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA); rabbit anti-cyclin D1 (cat.
no. ab134175), rabbit anti-Smo (cat. no. ab72130) and rabbit
anti-Ki67 (cat. no. ab15580) antibodies were all purchased from
Abcam (Cambridge, UK); rabbit anti-EVC (cat. no. SAB1405772) was
obtained from Sigma-Aldrich; Merck KGaA. The
5-ethynyl-20-deoxyuridine (EdU) assay was obtained from Guangzhou
RiboBio Co., Ltd. (Guangzhou, China). Terminal deoxynucleotidyl
transferase dUTP nick-end labeling (TUNEL) assay was purchased from
Beyotime Institute of Biotechnology (Haimen, China). Protein A/G
plus-agarose was obtained from Santa Cruz Biotechnology, Inc.
(Dallas, TX, USA).
Genetic screening of EVC
Genomic DNA was extracted from 2 ml blood samples
obtained from all participants with a TIANamp Blood DNA kit. All
exons and exon-intron boundaries of the EVC gene were generated by
polymerase chain reaction (PCR). The specific PCR primers were
designed using Primer Premier 5.0 software (Premier Biosoft
International, Palo Alto, CA, USA). Primers, region, fragment sizes
were given in the Table I. The PCR
was carried out in a total volume of 16 µl solution containing 50
ng template DNA, 200 nM of each primer, 2X Taq PCR MasterMix 8 µl.
The thermocycling conditions were as follows: One cycle of 5 min at
94°C; 20 cycles of 15 sec at 94°C, 30 sec at 65°C (−0.5°C/cycle);
10 cycles of 15 sec at 94°C and 30 sec at 55°C. The amplicons were
bi-directionally sequenced with a 3130 XL DNA analyzer (Applied
Biosystems; Thermo Fisher Scientific, Inc.). Sequences were aligned
and compared with the reference EVC gene (GenBank ID: NG_008843.1).
For the identified gene variations, the dbSNP (https://www.ncbi.nlm.nih.gov/projects/SNP), the 1000
Genomes Project (http://www.1000genomes.org), the Exome Variant Server
(EVS; http://evs.gs.washington.edu/EVS) and the Exome
Aggregation Consortium (ExAC; http://exac.broadinstitute.org) databases were
investigated in the present study. The Polymorphism Phenotyping
version 2 (PolyPhen-2; http://genetics.bwh.harvard.edu/pph2), Sorting
Intolerant from Tolerant (SIFT; http://sift.bii.astar.edu.sg/) and MutationTaster
(http://www.mutationtaster.org) programs
were used to evaluate the disease-causing potential of detected EVC
variants.
 | Table I.Primers used to amplify the exons and
exon-intron boundaries of the EVC gene. |
Table I.
Primers used to amplify the exons and
exon-intron boundaries of the EVC gene.
| Coding exon | Forward primer
(5′-3′) | Reverse primer
(5′-3′) | Size (bp) |
|---|
| 1 |
TCTGTCTCTGGGCATGCTCAG |
AAGTTCCCAACCAGGCTCAAG | 550 |
| 2 |
AGCAGAAGTGGCTGCTGGACTG |
CTATATCTGTCAGAGGCTTGTC | 345 |
| 3 |
TCTGAGGCTGCTATTACAAATG |
CATGTCCTGTTCCTGAGCCTC | 353 |
| 4 |
CTAGCGTGAATCACTGGTAG |
CAGAGTCACTGAACCTCTCTG | 433 |
| 5 |
ATTACCTCAGCATGCTGCAG |
CACTGATGTCTGCCTCTCAAG | 329 |
| 6 |
ATCGGAGTTCCTTTCCTTCAG |
CTCTTTGAGTAGTAGATATGAC | 508 |
| 7 |
GTCAAACCTCATGTACAAC |
CTTCTAACTATCATCTCTC | 380 |
| 8 |
GAGCACGCACCGTTTGGCTTTC |
GATGTCAAGCCTCAGGATTC | 303 |
| 9 |
CTGGTGGCTTCTTCTCAGCTG |
CAGAAGCAGAAAGGCAGCCTG | 435 |
| 10 |
CTGGCTCACAGAGTCACCTC |
CACCAGCTTACAGTCCACTAG | 389 |
| 11 |
CAGCTGTGAAAGCCATGTGAC |
GACACAGCCTGCGTGGATCAC | 413 |
| 12 |
CCTGTCTGTGGATCTCCTTG |
CAGTCATCTCCGTAGAGCACC | 456 |
| 13 |
CCACATGCCTGCTCTGTCC |
CTATGACAGACAACTCTATC | 347 |
| 14 |
CACCCATGCCTAAGAGGATG |
GCACCAAGGGTGATAGGATTC | 472 |
| 15 |
GAGCTTCTCTGTGAGAGGAG |
CAAGTCATGGAGAAGTCAG | 399 |
| 16 |
GATAGGATTCCCATGACTAG |
GCACACATTGCCAGTCTCTTC | 377 |
| 17 |
CACTCTTGGAGAGCTGGTG |
CACAGAGAGATATGCCATGTG | 294 |
| 18 |
CACTTTGATGGAGAGGCAGCCTG |
CTGACGTGTGGTCACAGGCTG | 403 |
| 19 |
GCAGCAGGAGCTGGTAGATG |
GATGCCTAAGGTCACACAGCTAG | 393 |
| 20 |
CCTCATTAGTTGAGTGGCTG |
CAAACCCAGCCACAGGCAACAG | 312 |
| 21 |
GTCATGGCACTTGGATGACCTC |
CTGCCTCTCAGCACTGCAGGAG | 363 |
Cell transfection and treatments
Cells were grown to 80–90% confluence in 35-mm
dishes and transfected with Gli2-Flag plasmids using Lipofectamine™
LTX (A12621; Invitrogen; Thermo Fisher Scientific, Inc.) and
adenovirus harboring EVC-WT-GFP or EVC-343C>G-GFP (MOI=50) were
added directly to cells for 48 h at 37°C. The efficacy of
transfection was determined using immunofluorescence microscopy.
Cells were treated with SAG (200 nM) 24 h post-transfection when
required at 37°C.
Western blotting and
immunoprecipitation
Western blotting was performed as previously
described (19). In brief, when
grown to 80–90% confluence, 200 µl radioimmunoprecipitation assay
buffer (RIPA; P0013B) and 1 mM PMSF (ST506)(both from Beyotime
Institute of Biotechnology, Beijing, China) were used to lyse the
cells. Total protein was extracted and determined by bicinchoninic
acid (BCA) assay before loading the samples into wells. The
proteins (40 µg) were separated via 8% SDS-PAGE and then were
transferred to a polyvinylidene fluoride (PVDF) membrane
(ISEQ00010; EMD Millipore, Billerica, MA, USA) via electroblotting.
Following blocking with the 5% defatted milk, the membrane was
incubated with primary antibody at 4°C over night and then
incubated with the goat anti-mouse immunoglobulin G (IgG)
horseradish peroxidase (HRP)-conjugated antibody and goat
anti-rabbit IgG HRP conjugate (1:6,000, cat. nos. HS201-01,
HS101-01, respectively; Beijing TransGen Biotech Co., Ltd. Beijing,
China). The protein bands were visualized by chemiluminescence
(PA112-01; Tiangen Biotech Co., Ltd.). Primary antibodies used
were: Mouse anti-Gli1 (1:1,000), rabbit anti-Bcl2 (1:500), rabbit
anti-Bax (1:500), mouse anti-Flag (1:1,000), rabbit anti-cyclin D1
(1:1,000), rabbit anti-EVC (1:1,000), rabbit anti-Smo (1:1,000) and
rabbit anti-β-tubulin (1:1,000). Band intensities were analyzed by
Image Lab software version 2.2 (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA).
Immunoprecipitation experiments were as performed as
previously described (19). Cells
were lysed with addition of 0.5 ml RIPA buffer. The supernatant was
obtained following centrifugation at 14,000 × g for 15 min. The
primary antibody Smo (1:200) was added and then, protein A/G
plus-Agarose was added and incubated over night. The
immunoprecipitate was collected by centrifugation at 3,500 × g for
10 min. The pellet was then washed four times and resuspended in 40
µl, 2X loading buffer. All the above steps were performed at 4°C.
The samples were boiled and analyzed by SDS-PAGE. Experiments were
performed in triplicate.
EdU and TUNEL assays
EdU experiments were performed as previously
described (20). NIH3T3 cells were
seeded in 96-well plates at 1×104 cells/well, The cells
were grown to 80–90% and then incubated with EdU for 2 h, and
processed according to the manufacturer's instructions. Following
three washes with PBS, the cells were treated with 300 µl 1X
Apollo® reaction cocktail for 30 min at room
temperature. Then, the DNA contents of the cells in each well were
stained with 100 µl Hoechst 33342 (R11053.4; Guangzhou RiboBio Co.,
Ltd., Guangzhou, China) for 30 min at room temperature and
visualized under a fluorescence microscope (Nikon Eclipse 90i;
Nikon Corporation, Toyko, Japan). A total of five fields were
examined/group.
The TUNEL assay was performed as previously
described (21). NIH3T3 cells were
grown to 70–80% and pretreated with 200 µM
H2O2 for 24 h at 37°C. TUNEL assays were
performed in following the manufacturer's instructions. Positive
apoptotic cells were stained brown for incubation for 30 min at
room temperature using TUNEL kit and nuclei were counterstained
with hematoxylin at room temperature for 2 min. The cells were
visualized with an optical microscope, five fields were
examined/group.
Dual-luciferase reporter assay
A dual luciferase reporter assay was performed as
previously described (22). In
brief, the NIH3T3 cells were seeded at a density of
1×105 cells/well in 6-well plates. Following incubation
for 24 h, the cells were transfected with adenoviruses EVC-WT-GFP
and EVC-Mut-GFP. The cells were incubated for 48 h prior to
transfection with the Gli1-luciferase reporter (5 µg) using
Lipofectamine™ LTX (A12621; Invitrogen; Thermo Fisher Scientific,
Inc.) according to the manufacturer's instructions. SAG was added
and incubated for 24 h before harvesting. Gli1 transcriptional
activity was calculated as the ratio between firefly and
Renilla luciferase units. The assay was performed using the
Dual-Luciferase® Reporter Assay System (E1910; Promega
Corporation, Madison, WI, USA).
Immunofluorescence staining
Immunofluorescence was performed as previously
described (23). In brief, the
NIH3T3 cells were seeded in 6-well plates at a density of
1×105 cells/well. The cells were grown to 70–80% and
fixed with 4% paraformaldehyde at room temperature for 20 min
before blocking with 5% bull serum albumin (BSA; 10099158;
Invitrogen; Thermo Fisher Scientific, Inc.) at room temperature for
20 min. Following incubation with the primary antibody anti-Ki67
(1:50) at 4°C overnight, the cells were then washed and incubated
again with fluorescein isothiocyanate-conjugated donkey anti-rabbit
immunoglobulin G (1:200) and DAPI (0.1 mg/ml) at 37°C for 1 h. The
image was visualized using a fluorescence microscope (Nikon Eclipse
90i; Nikon Corporation) following DAPI staining. A total of five
fields were examined/group. The percentage of Ki67-positive cells
was calculated.
Statistical analysis
Statistical analysis was performed with SPSS version
20.0 statistical software (IBM Corp., Armonk, NY, USA). The genetic
balance of SNP genotypes was tested according to the Hardy-Weinberg
equilibrium (24). Allele and
genotype frequencies in the cases and controls were compared using
χ2 tests. Genetic data were corrected for multiple
comparisons using the Bonferroni correction test. Risk prediction
for VSD was expressed as odds ratio and 95% confidence interval
(CI). Continuous data were presented as the mean ± standard error
of the mean. The differences between groups were analyzed using
Student's t-test or one-way analysis of variance followed by
Tukey's post hoc test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Analysis of EVC SNPs in patients with
VSD
Through sequencing and screening of the EVC gene, 11
EVC SNP sites were detected in patients with VSD, including
c.221A>C (p.Q74P), c.772T>C (p.Y258H), c.969T>C (p.N323N),
c.1026G>C (p.L342L), c.1068A>G (p.L356L), c.1320T>A
(p.F440L), c.1333A>C p.K445Q), c.1346C>A (p.T449K),
c.1727G>A (p.R576Q), c.1854C>T (p.G618G) and c.2279G>A
(p.R760Q). The detection results of the genotype distribution,
allele frequency and Hardy-Weinberg equilibrium of all the sites
are provided in Table II. Four of
the SNPs (c.1026G>C, c.1320T>A, c.1333A>C and
c.1346C>A) were excluded for the association analysis owing to
deviation from the Hardy-Weinberg equilibrium. The remaining seven
SNPs were analyzed under five genetic models, including homozygous,
heterozygous, dominant, recessive and allele (Table III). The results revealed that
the C allele of SNP c.221A>C was negatively associated with the
risk of VSD development (OR=0.543; CI=0.300–0.981; P=0.041). The
risk of VSD development in patients carrying the CC genotype of the
c.1854C>T site was 2.246 times higher compared with the risk in
patients carrying the TT genotype (CI=1.009–4.998; P=0.045). The C
allele was the risk allele of VSD (OR=1.523; CI=1.019–2.278;
P=0.040). Only the c.1727G>A site was significantly positively
associated with the development of VSD following Bonferroni
correction (P<0.007), indicating that the risk of VSD in people
carrying the GA genotype was 4.411 times higher compared with the
risk in people carrying the GG genotype (CI=2.047–9.506). In
addition, in the dominant model, the risk of VSD in people carrying
the GA or AA genotype was 4.2 times higher compared with people
carrying the GG genotype (CI=1.982–8.901). The A allele in
c.1727G>A was the risk allele of VSD (OR=2.039; CI=1.301–3.195;
P=0.002). Differences in the distribution of the four other SNP
sites in the five models did not have statistical significance
(P>0.05; data not shown).
| Table II.Hardy-Weinberg equilibrium test of
Ellis-van Creveld variations in controls and patients with VSD. |
Table II.
Hardy-Weinberg equilibrium test of
Ellis-van Creveld variations in controls and patients with VSD.
| Variation | Genotype | n | Genotype frequency
(%) | Allele frequency
(%) | HWEa (P-value) |
|---|
| c.221A>C | Genotype |
| AA | AC | CC | C | A |
|
| p.Q74P | VSD | 64 | 50 (78.1) | 13 (20.3) | 1 (1.6) | 15 (11.7) | 113 (88.3) | 0.884 |
|
| Control | 201 | 131 (65.2) | 61 (30.3) | 9 (4.5) | 79 (19.7) | 323 (80.3) | 0.580 |
| c.343C>G | Genotype |
| CC | CG | GG | C | G |
|
| p.L115V | VSD | 61 | 60 (98.4) | 1 (1.6) | 0 (0.0) | 121 (99.2) | 1 (0.8) |
|
|
| Control | 210 | 210 (100.0) | 0 (0.0) | 0 (0.0) | 420 (100) | 0 (0) |
|
| c.772T>C | Genotype |
| CC | TC | TT | T | C |
|
| p.Y258H | VSD | 60 | 51 (85.0) | 9 (15.0) | 0 (0.0) | 9 (7.5) | 111 (92.5) | 0.530 |
|
| Control | 200 | 172 (86.0) | 28 (14.0) | 0 (0.0) | 28 (7.0) | 372 (93.0) | 0.287 |
| c.969T>C | Genotype |
| CC | TC | TT | T | C |
|
| p.N323N | VSD | 63 | 26 (41.3) | 31 (49.2) | 6 (95.2) | 43 (34.1) | 83 (65.9) | 0.454 |
|
| Control | 210 | 75 (35.7) | 94 (44.8) | 41 (19.5) | 176 (41.9) | 244 (58.1) | 0.242 |
| c.1026G>C | Genotype |
| CC | GC | GG | G | C |
|
| p.L342L | VSD | 63 | 25 (39.7) | 30 (47.6) | 8 (12.7) | 46 (36.5) | 80 (63.5) | 0.829 |
|
| Control | 210 | 85 (40.5) | 85 (40.5) | 40 (19.0) | 165 (39.3) | 255 (60.7) | 0.028b |
| c.1068A>G | Genotype |
| AA | AG | GG | G | A |
|
| p.L356L | VSD | 63 | 42 (66.7) | 19 (30.2) | 2 (3.2) | 23 (18.3) | 103 (81.7) | 0.933 |
|
| Control | 210 | 128 (61.0) | 66 (31.4) | 16 (7.6) | 98 (23.3) | 322 (76.7) | 0.078 |
| c.1346C>A | Genotype |
| AA | CA | CC | C | A |
|
| p.T449K | VSD | 64 | 45 (70.3) | 19 (29.7) | 0 (0.0) | 19 (14.8) | 109 (85.2) | 0.163 |
|
| Control | 207 | 142 (68.6) | 49 (23.7) | 16 (7.7) | 81 (19.6) | 333 (80.4) |
<0.001b |
| c.1320T>A | Genotype |
| TT | TA | AA | A | T |
|
| p.F440L | VSD | 62 | 56 (90.3) | 6 (9.7) | 0 (0.0) | 6 (4.8) | 118 (95.2) | 0.689 |
|
| Control | 207 | 179 (86.5) | 22 (10.6) | 6 (2.9) | 34 (8.2) | 380 (91.8) |
<0.001b |
| c.1333A>C | Genotype |
| AA | AC | CC | C | A |
|
| p.K445Q | VSD | 63 | 60 (95.2) | 3 (4.8) | 0 (0.0) | 3 (2.4) | 123 (97.6) | 0.847 |
|
| Control | 207 | 196 (94.7) | 8 (3.9) | 3 (1.4) | 14 (3.4) | 400 (96.6) |
<0.001b |
| c.1727G>A | Genotype |
| GG | GA | AA | A | G |
|
| p.R576Q | VSD | 52 | 10 (19.2) | 36 (69.2) | 6 (11.5) | 48 (46.2) | 56 (53.8) | 0.005 |
|
| Control | 174 | 87 (50.0) | 71 (40.8) | 16 (9.2) | 103 (29.6) | 245 (70.4) | 0.783 |
| c.1854C>T | Genotype |
| TT | CT | CC | C | T |
|
| p.G618G | VSD | 65 | 13 (0.2) | 31 (47.7) | 21 (32.3) | 73 (56.2) | 57 (43.8) | 0.780 |
|
| Control | 185 | 57 (30.8) | 87 (47.0) | 41 (22.2) | 169 (45.7) | 201 (54.3) | 0.476 |
| c.2279G>A | Genotype |
| GG | GA | AA | A | G |
|
| p.R760Q | VSD | 62 | 55 (88.7) | 6 (9.7) | 1 (1.6) | 8 (6.5) | 116 (93.5) | 0.119 |
|
| Control | 200 | 167 (83.5) | 33 (16.5) | 0 (0.0) | 33 (8.3) | 367 (92.7) | 0.204 |
| Table III.Associations between EVC SNPs and VSD
risk. |
Table III.
Associations between EVC SNPs and VSD
risk.
| SNP | Model | Comparison |
P-valuea | OR (95% CI) |
|---|
| c.221A>C | Heterozygous | AC vs. AA |
0.091 | 0.559
(0.282–1.104) |
|
| Homozygous | CC vs. AA |
0.390 | 0.291
(0.036–2.358) |
|
| Dominant | AC+CC vs. AA |
0.053 | 0.524
(0.271–1.014) |
|
| Recessive | CC vs. AC+AA |
0.491 | 0.339
(0.042–2.727) |
|
| Allele | C vs. A |
0.041 | 0.543
(0.300–0.981) |
| c.1727G>A | Heterozygous | GA vs. GG | <0.001 | 4.411
(2.047–9.506) |
|
| Homozygous | AA vs. GG |
0.035 | 3.263
(1.039–10.24) |
|
| Dominant | GA+AA vs. GG | <0.001 | 4.200
(1.982–8.901) |
|
| Recessive | AA vs. GA+GG |
0.617 | 1.288
(0.477–3.481) |
|
| Allele | A vs. G |
0.002 | 2.039
(1.301–3.195) |
| c.1854C>T | Heterozygous | CT vs. TT |
0.228 | 1.562
(0.754–3.238) |
|
| Homozygous | CC vs. TT |
0.045 | 2.246
(1.009–4.998) |
|
| Dominant | CC+CT vs. TT |
0.095 | 1.781
(0.899–3.528) |
|
| Recessive | CC vs. CT+TT |
0.103 | 1.676
(0.897–3.132) |
|
| Allele | C vs. T |
0.040 | 1.523
(1.019–2.278) |
The results of gene variants function predictions
using PolyPhen-2, SIFT and MutationTaster software demonstrated
that the SNP c.1727G>A may cause an alteration in the splicing
site, thus resulting in the development of the disease (data not
shown).
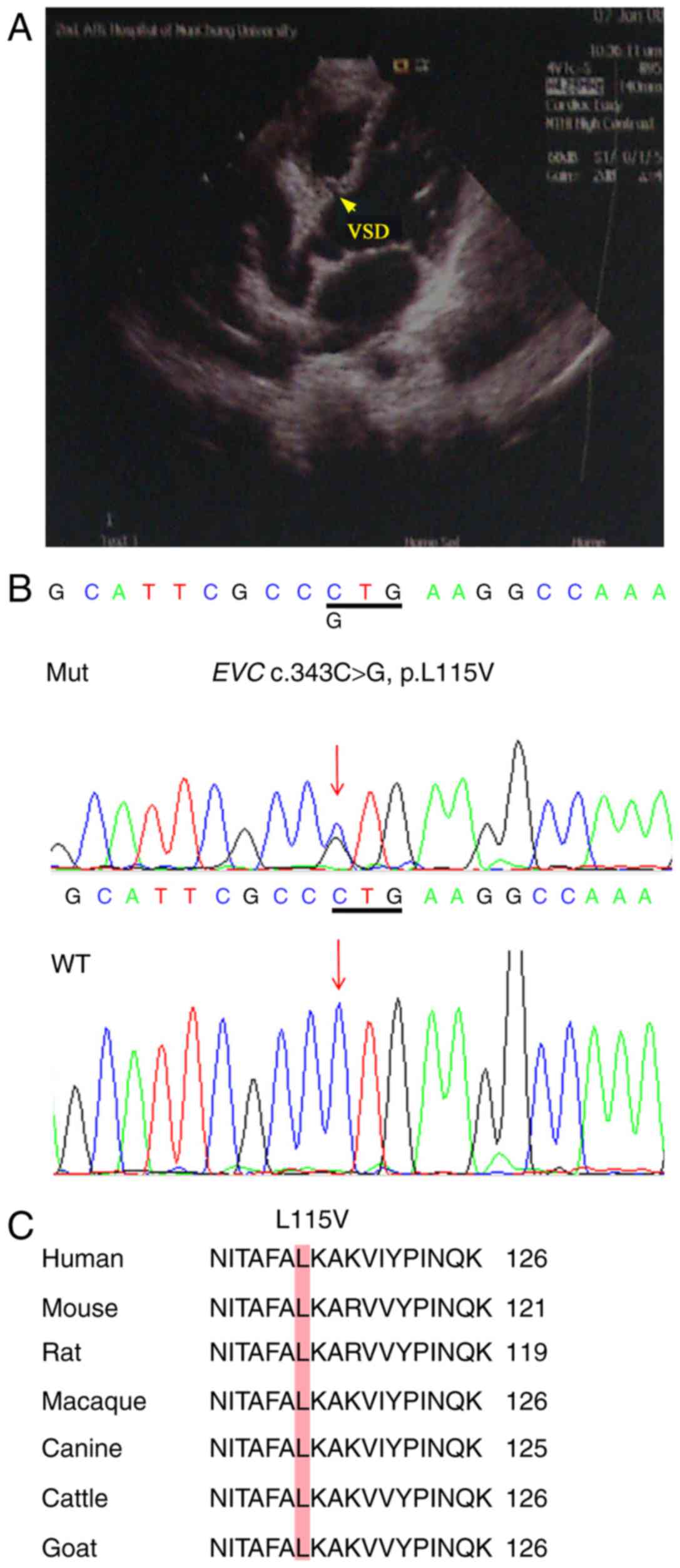
Importantly, a heterozygous nonsynonymous mutation
was identified using the direct DNA sequencing method in a patient
with VSD (Fig. 1); however, DNA
from family members was not available. Sequencing confirmed that it
was c.343C>G, which caused a change from leucine to valine
(p.L115V; Fig. 1B), which was not
found in 420 normal chromosomes. This mutation was not reported in
the 1000 Genomes database and EVS database, whereas in the ExAC
database, its minor allele frequency (MAF) was 0.0002. The
prediction results of the c.343C>G site revealed the possibility
of a pathogenic mutation (data not shown). Comparison of the EVC
coding region sequences among different species demonstrated that
the EVC-L115V substitution site was highly conserved among
different species (Fig. 1C).
Therefore, the EVC c.343C>G SNP was selected for subsequent
functional analysis.
Reduction of NIH3T3 cell proliferation
by the EVC c.343C>G mutation
It has been previously reported that blocking Hh
pathway activity may affect cell proliferation levels in the SHF
(25); following EVC knockout,
proliferation of cartilage cells in mice was also inhibited
(11). To clarify whether the EVC
c.343C>G mutation affected proliferation, the cell proliferation
marker Ki67 was used to observe the proliferation levels of NIH3T3
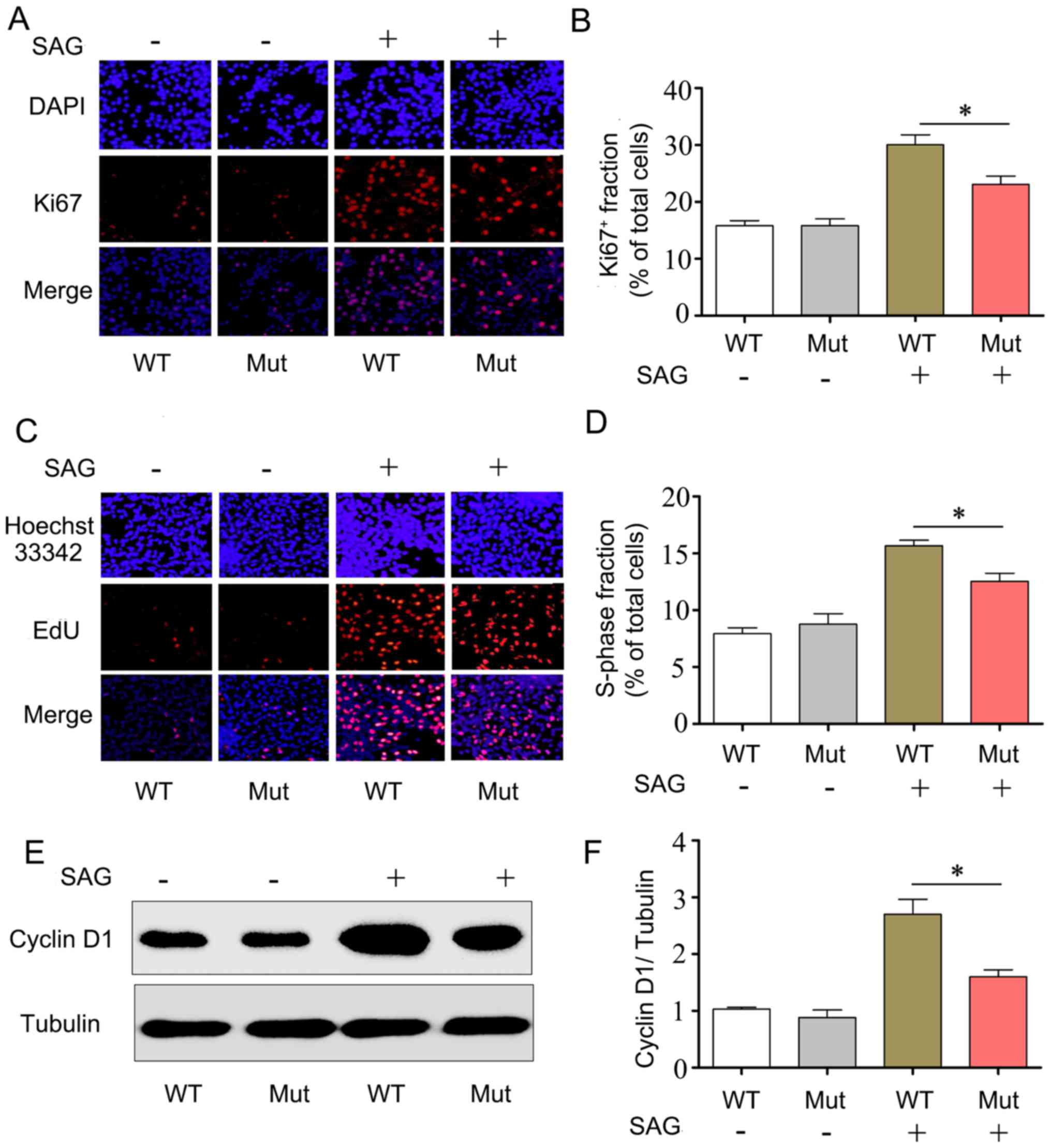
cells. The results indicated that the percentage of Ki67-stained
cells among the total cells in the Mut group was significantly
decreased compared with the WT group in the presence of the Smo
agonist SAG (23±1.3 vs. 30.4±1.8%, respectively; P<0.05;
Fig. 2A and B).
In addition, the effects of the EVC mutation on cell
proliferation were further validated using an EdU assay. The
results revealed that the percentage of EdU-labeled S-phase cells
in the Mut group was significantly decreased compared with that in
the WT group in the presence of SAG (12.5±0.8 vs. 15.6±0.4%,
respectively; P<0.05; Fig. 2C and
D), suggesting that the EVC c.343C>G mutation may cause an
abnormal cell cycle transition from the G1 to the S phase in NIH3T3
cells, therefore affecting their proliferation.
In the Hh signaling pathway, EVC may influence cell
proliferation by regulating the expression of cyclin D1 (26). In the present study, western
blotting results demonstrated that during the activation of the Hh
pathway following SAG treatment, the expression of cyclin D1 in the
Mut group was significantly decreased compared with expression in
the WT group (Fig. 2E and F),
which suggested that the cells were arrested during their
progression from G1 phase to S phase of the cell cycle.
Increase in apoptosis in NIH3T3 cells
by the EVC c.343C>G mutation treated with SAG
A previous study demonstrated that Hh signaling
pathway may protect the astrocytes from oxidative stress by
activating PI3-K/AKT/Bcl-2 pathway (27). NIH3T3 cell were treated with
H2O2 and the cell apoptosis was detected by
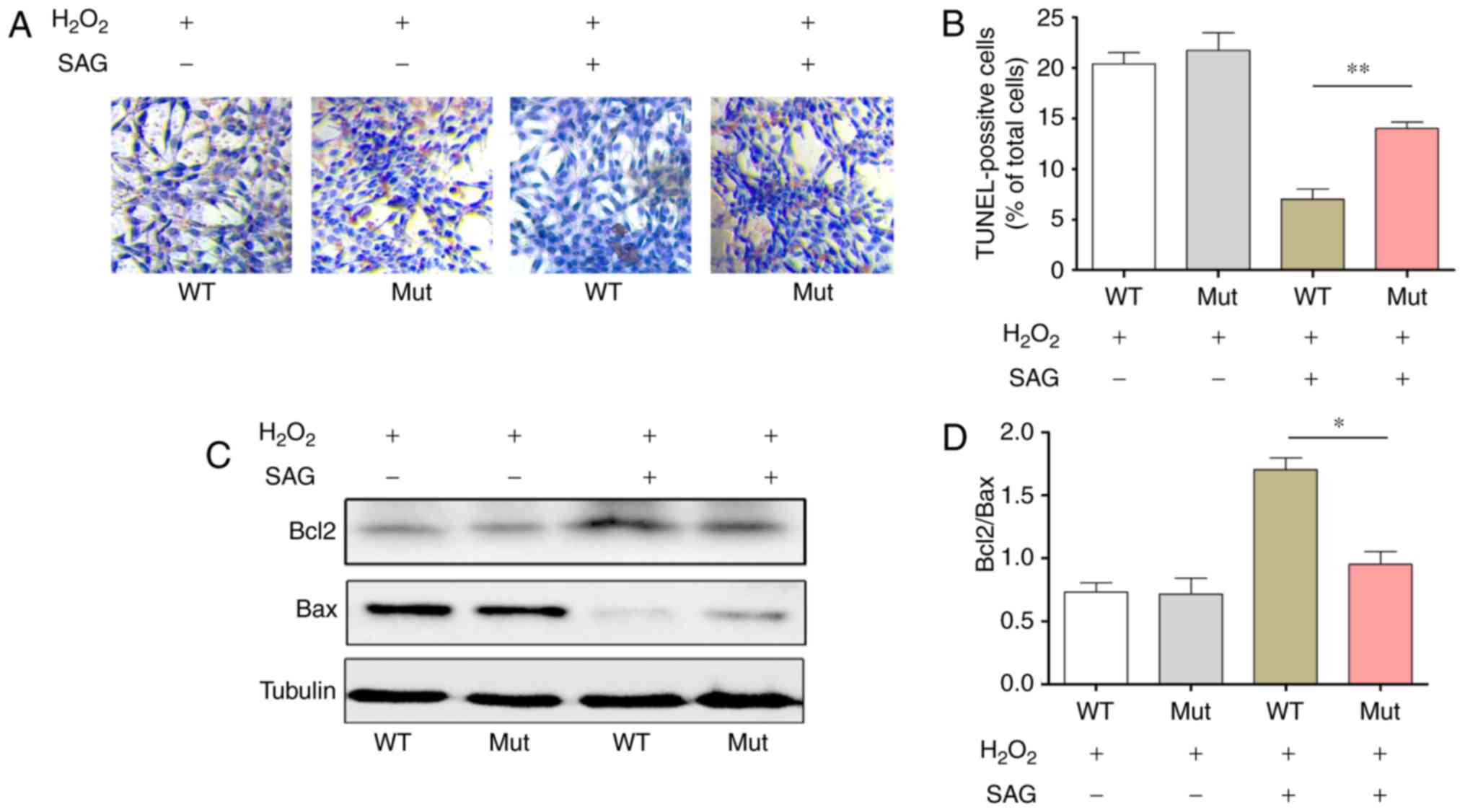
TUNEL to observe whether the EVC mutation affected cell apoptosis.
The results of the present study indicated that NIH3T3 cells
apoptosis in the Mut group was significantly higher compared with
that in the WT group in the presence of SAG (15.0±0.7 vs. 7.5±0.6%;
Fig. 3A and B).
In addition, the protein expression levels of Bcl2
and Bax in cells transfected with either EVC-WT or EVC-Mut was
measured by western blotting following H2O2
induction. The results demonstrated that when the Hh pathway was
activated by SAG treatment, Bcl2 protein expression decreased and
Bax expression increased in the EVC-Mut group compared to the
EVC-WT group (Fig. 3C), and the
protein expression ratio of Bcl2/Bax was significantly lower in the
Mut group compared to the WT group (P<0.05; Fig. 3D), which suggested that EVC
c.343C>G mutation may reduce the anti-apoptotic activity of
NIH3T3 cells.
Downregulation of Hh pathway activity
by the EVC c.343C>G mutation
EVC is an important activator of the Hh pathway
(14–15). To confirm whether the EVC
c.343C>G mutation affected the activity of the Hh pathway and
investigate the association of the decrease in the expression level
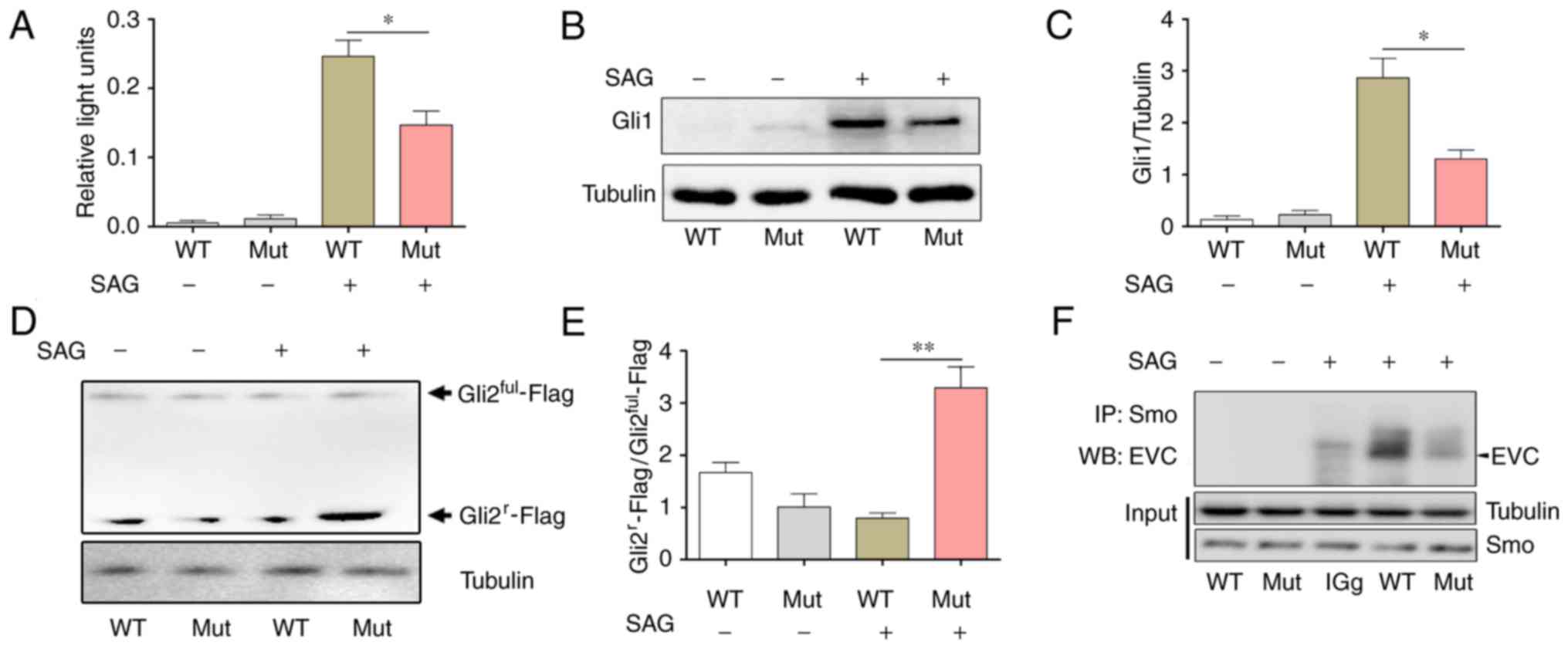
of cyclin D1 and Bcl2 proteins, a Gli1 luciferase reporter gene
plasmid was used to determine the transcription activity of Gli1
which acts as an upstream effector of the Hh pathway. The results
revealed that there was significantly reduced Gli1 luciferase
reporter gene activity in the Mut group following Hh pathway
activation by SAG treatment, compared with activity in the WT
transfection group (Fig. 4A). This
result suggested that EVC c.343C>G mutation decreased the
transcriptional activity of Gli1 following Hh activation. In
addition, western blotting results demonstrated that, compared with
the WT group, the expression levels of the Gli1 protein were
significantly decreased in the Mut group when the Hh pathway was
activated by SAG (Fig. 4B and
C).
Gli2 is the primary effector molecule of the Hh
pathway. When the Hh pathway is in the resting state, full-length
Gli2 (Gli2ful) is cut by the proteasome to generate the
repressor form of Gli2, Gli2r, which inhibits the
transcription of downstream target genes. When the Hh pathway is
activated, Gliful is converted to its activated for
GliA. A previous study demonstrated that EVC was
required for Hh pathway to inhibit Gli2r formation in
cultured cells (15). To determine
whether these processes were affected by EVC c.343C>G mutation,
a Gli2-Flag plasmid was constructed and transfected it into NIH3T3
cells to determine the levels of the two isoforms of Gli2 using
western blotting. The results indicated that in the presence of
SAG, the Gli2r−Flag/Gli2ful−Flag ratio was
significantly increased in the Mut group compared with WT (Fig. 4D and E), which suggested that the
suppressor form Gli2r increased following the
full-length Gli2 was cut and that EVC c.343C>G mutation affected
the Hh pathway.
Previous study has been demonstrated that Smo is
upstream of EVC in the Hh pathway (15). When Hh is activated, EVC may form a
protein complex with Smo to transduce the signal intracellularly,
leading to Gli activation (15).
To investigate whether EVC c.343C>G mutation affected the
interaction between Smo and EVC, co-immunoprecipitation was used
for analysis and the results revealed that the binding between EVC
and Smo in the Mut group was notably decreased compared with that
in the WT group in the presence of SAG (Fig. 4F).
Discussion
Genetic variations serve important roles in the
development of VSD. In the present study, 11 SNPs and one EVC
genetic mutation were discovered in 65 cases of VSD. Following the
Hardy-Weinberg equilibrium examination and Bonferroni correction,
only the c.1727G>A SNP was identified as significantly
positively associated with the development of VSD in a Chinese Han
population. These results suggested that c.1727G>A may be an
important disease-susceptible polymorphism site for the development
of VSD. It has been reported that the c.1727G>A site may be
associated with smoking-associated pancreatic cancer and male
suicidal behavior (28,29). In addition, PolyPhen-2 and
MutationTaster software both predicted that this site was a
pathogenic site and may alter the splicing site. Therefore, it was
hypothesized that c.1727G>A may be a potential susceptible gene
maker for genetic diagnostics and targeted treatment of VSD.
A rare EVC c.343C>G (p.L115V) mutation in a VSD
subpopulation was detected. This mutation had not been reported in
the 1000 Genomes database, the EVS database or the control group in
the present study. Only the ExAC database reported two carriers
among 4,327 East Asians. The frequency was very low, and the MAF
was 0.0002, which supported the judgment of the mutations (30). This mutation was located in the
conserved EVC coding sequences. PolyPhen-2 and MutationTaster
software both predicted this to be a pathogenic site. The present
study revealed that the EVC c.343C>G mutation may affect Hh
pathway activity by downregulating the binding between EVC and Smo,
reducing the activity of the downstream Gli1 transcription factor,
increasing the production of the Gli2 repressor form
Gli2r and downregulating the expression levels of the
downstream cyclin D1 and Bcl2 proteins. As a result, this mutation
also reduced the anti-apoptotic ability of cells, caused cell cycle
disorder in the transition from G1 to S phase in cells and reduced
the proliferation level. Results from the present study
demonstrated the association between Chinese Han patients with VSD
and the EVC gene mutation and may be the first to systematically
describe the molecular mechanism underlying the development of VSD
caused by EVC mutations.
The development of VSD at the embryonic stage is
associated with various mechanisms, of which abnormal SHF
development is one of the primary mechanisms (31). Previous studies have revealed that
inhibition of the Hh pathway may cause reduction of the Hh ligand
Ptc in the SHF to inhibit cardiomyocyte proliferation and cause CHD
phenotypes such as pulmonary vein atresia (16). In a cilia protein Meckel syndrome
type 1 gene homozygous mutation mouse model, Hh pathway activity in
the SHF of an embryonic mouse heart significantly decreased and
reduced Gli1 expression led to abnormal development of the SHF
region and caused atrioventricular septal defect (32). Results from these studies suggested
that the Hh signaling pathway and development of the SHF in an
embryonic stage heart were closely associated. A number of other
studies also demonstrated that when the Hh pathway was activated,
interference with or inhibition of EVC expression significantly
decreased Gli1 expression and blocked Hh pathway activity to reduce
cell proliferation in in vivo and in vitro
experiments (11,33). These results suggested that EVC was
an activator molecule of the Hh pathway. Therefore, combined with
the in vitro experiment results, the present study
hypothesized that EVC mutations may cause a reduction in Hh pathway
activity to inhibit the proliferation of SHF cells at the embryonic
stage, thus causing abnormal heart development. These alterations
may be one of the mechanisms underlying the development of VSD
caused by EVC mutations.
As early as 2003, it was reported that the Hh
pathway had anti-apoptotic functions and promoted cell survival
during vertebrate development (34). Subsequent study revealed that the
anti-apoptotic functions of Hh were through the inhibition of the
mitochondrial pathway, which caused neuronal apoptosis resistance
by regulating the expression ratio between the anti-apoptotic
protein Bcl2 and the pro-apoptotic protein Bax (27). The present study demonstrated that
the EVC c.343C>G mutation may block the activity of Hh
signaling, thus reducing the Bcl2/Bax ratio, activating the
mitochondrial apoptosis pathway and reducing the anti-apoptotic
functions of cells.
Smo is an indispensable key member in the signal
transduction of the Hh pathway. Previous studies indicated that the
Hh pathway promoted the formation of a protein complex between
phosphorylated Smo and EVC to activate downstream Gli family
transcription factors and promote the transcription of downstream
target genes (13,15). Binding between EVC and Smo depends
on the result of Hh pathway activation, which is consistent with
the results of the present study in which there was a reduction in
the binding between EVC-L115V and Smo that led to an increase in
generation of the Hh pathway downstream effector factor
Gli2r and reduced the expression of genes associated
with proliferation and anti-apoptosis.
In conclusion, results from the present study
suggested that the EVC gene was the pathogenic gene in patients
with VSD. The c.1727G>A polymorphism was a susceptible gene
locus for the development of VSD. By affecting the activities of
the Hh pathway, the EVC c.343C>G mutation may decrease Bcl2
expression, reduce the anti-apoptosis activity of cells, induce
G1-to-S phase transition disorders in cells and reduce cell
proliferation levels. The results may partially reveal the
pathogenesis of cardiac defects in patients with EVC syndrome and
provide new targets for the diagnosis and treatment of patients
with VSD.
Acknowledgements
The authors would like to thank Xin Liu and Menglu
Liu (Nanchang University, China) for assisting in laboratory
work.
References
|
1
|
Pierpont ME, Basson CT, Benson DW Jr, Gelb
BD, Giglia TM, Goldmuntz E, McGee G, Sable CA, Srivastava D and
Webb CL: American Heart Association Congenital Cardiac Defects
Committee, Council on Cardiovascular Disease in the Young: Genetic
basis for congenital heart defects: Current knowledge: A scientific
statement from the American Heart Association Congenital Cardiac
Defects Committee, Council on Cardiovascular Disease in the Young:
Endorsed by the American Academy of Pediatrics. Circulation.
115:3015–3038. 2007. View Article : Google Scholar
|
|
2
|
Serra-Juhé C, Cuscó I, Homs A, Flores R,
Torán N and Pérez-Jurado LA: DNA methylation abnormalities in
congenital heart disease. Epigenetics. 10:167–177. 2015. View Article : Google Scholar :
|
|
3
|
Koefoed K, Veland IR, Pedersen LB, Larsen
LA and Christensen ST: Cilia and coordination of signaling networks
during heart development. Organogenesis. 10:108–125. 2014.
View Article : Google Scholar
|
|
4
|
Muntean I, Toganel R and Benedek T:
Genetics of congenital heart disease: Past and present. Biochem
Genet. 55:105–123. 2017. View Article : Google Scholar
|
|
5
|
Chen D, Qiao Y, Meng H, Pang S, Huang W,
Zhang H and Yan B: Genetic analysis of the TBX3 gene promoter in
ventricular septal defects. Gene. 512:185–188. 2013. View Article : Google Scholar
|
|
6
|
Leirgul E, Fomina T, Brodwall K, Greve G,
Holmstrøm H, Vollset SE, Tell GS and Øyen N: Birth prevalence of
congenital heart defects in Norway 1994-2009-a nationwide study. Am
Heart J. 168:956–964. 2014. View Article : Google Scholar
|
|
7
|
Gittenberger-de Groot AC, Calkoen EE,
Poelmann RE, Bartelings MM and Jongbloed MR: Morphogenesis and
molecular considerations on congenital cardiac septal defects. Ann
Med. 46:640–652. 2014. View Article : Google Scholar
|
|
8
|
Sund KL, Roelker S, Ramachandran V, Durbin
L and Benson DW: Analysis of Ellis van Creveld syndrome gene
products: Implications for cardiovascular development and disease.
Hum Mol Genet. 18:1813–1824. 2009. View Article : Google Scholar :
|
|
9
|
Hills CB, Kochilas L, Schimmenti LA and
Moller JH: Ellis-van Creveld syndrome and congenital heart defects:
Presentation of an additional 32 cases. Pediatr Cardiol.
32:977–982. 2011. View Article : Google Scholar
|
|
10
|
Caparrós-Martín JA, De Luca A, Cartault F,
Aglan M, Temtamy S, Otaify GA, Mehrez M, Valencia M, Vázquez L,
Alessandri JL, et al: Specific variants in WDR35 cause a
distinctive form of Ellis-van Creveld syndrome by disrupting the
recruitment of the EvC complex and SMO into the cilium. Hum Mol
Genet. 24:4126–4137. 2015. View Article : Google Scholar :
|
|
11
|
Pacheco M, Valencia M, Caparrós-Martín JA,
Mulero F, Goodship JA and Ruiz-Perez VL: Evc works in chondrocytes
and osteoblasts to regulate multiple aspects of growth plate
development in the appendicular skeleton and cranial base. Bone.
50:28–41. 2012. View Article : Google Scholar
|
|
12
|
Hui C and Angers S: Gli proteins in
development and disease. Annu Rev Cell Dev Bi. 27:513–537. 2011.
View Article : Google Scholar
|
|
13
|
Briscoe J and Thérond PP: The mechanisms
of Hedgehog signalling and its roles in development and disease.
Nat Rev Mol Cell Bio. 14:416–429. 2013. View Article : Google Scholar
|
|
14
|
Ruiz-Perez VL, Blair HJ, Rodriguez-Andres
ME, Blanco MJ, Wilson A, Liu YN, Miles C, Peters H and Goodship JA:
Evc is a positive mediator of Ihh-regulated bone growth that
localises at the base of chondrocyte cilia. Development.
134:2903–2912. 2007. View Article : Google Scholar
|
|
15
|
Yang C, Chen W, Chen Y and Jiang J:
Smoothened transduces Hedgehog signal by forming a complex with
Evc/Evc2. Cell Res. 22:1593–1604. 2012. View Article : Google Scholar :
|
|
16
|
Dyer LA and Kirby ML: Sonic hedgehog
maintains proliferation in secondary heart field progenitors and is
required for normal arterial pole formation. Dev Biol. 330:305–317.
2009. View Article : Google Scholar :
|
|
17
|
Liu X, Li M, Peng Y, Hu X, Xu J, Zhu S, Yu
Z and Han S: miR-30c regulates proliferation, apoptosis and
differentiation via the Shh signaling pathway in P19 cells. Exp Mol
Med. 48:e2482016. View Article : Google Scholar :
|
|
18
|
Smoak Washington I, Byrd NA, Abu-Issa R,
Goddeeris MM, Anderson R, Morris J, Yamamura K, Klingensmith J and
Meyers EN: Sonic hedgehog is required for cardiac outflow tract and
neural crest cell development. Dev Biol. 283:357–372. 2005.
View Article : Google Scholar
|
|
19
|
Wan R, Guo R, Chen C, Jin L, Zhu C, Zhang
Q, Xu Y and Li S: Urocortin increased LPS-induced endothelial
permeability by regulating the cadherin-catenin complex via
corticotrophin-releasing hormone receptor 2. J Cell Physiol.
228:1295–1303. 2013. View Article : Google Scholar
|
|
20
|
Liu X, Chen L, Ge J, Yan C, Huang Z, Hu J,
Wen C, Li M, Huang D, Qiu Y, et al: The Ubiquitin-like protein
FAT10 stabilizes eEF1A1 expression to promote tumor proliferation
in a complex manner. Cancer Res. 76:4897–4907. 2016. View Article : Google Scholar
|
|
21
|
Peng X, Shao J, Shen Y, Zhou Y, Cao Q, Hu
J, He W, Yu X, Liu X, Marian AJ and Hong K: FAT10 protects cardiac
myocytes against apoptosis. J Mol Cell Cardiol. 59:1–10. 2013.
View Article : Google Scholar :
|
|
22
|
Yuan R, Wang K, Hu J, Yan C, Li M, Yu X,
Liu X, Lei J, Guo W, Wu L, et al: Ubiquitin-like protein FAT10
promotes the invasion and metastasis of hepatocellular carcinoma by
modifying β-catenin degradation. Cancer Res. 74:5287–5300. 2014.
View Article : Google Scholar
|
|
23
|
Liu T, Yu X, Li G, Yuan R, Wang Q, Tang P,
Wu L, Liu X, Peng X and Shao J: Rock2 regulates Cdc25A through
ubiquitin proteasome system in hepatocellular carcinoma cells. Exp
Cell Res. 318:1994–2003. 2012. View Article : Google Scholar
|
|
24
|
Nuño-Arana I, Sahagún-Núñez Vdel R,
Muñoz-Valle JF, Sandoval L, Pinto-Escalante D, Páez-Riberos LA,
Lazalde B, Maldonado-González M and Rangel-Villalobos H:
Distribution of three SNPs related to low bone mineral density in
Amerindian groups and Mestizos from Mexico. Am J Hum Biol.
24:569–572. 2012. View Article : Google Scholar
|
|
25
|
Goddeeris MM, Schwartz R, Klingensmith J
and Meyers EN: Independent requirements for Hedgehog signaling by
both the anterior heart field and neural crest cells for outflow
tract development. Development. 134:1593–1604. 2007. View Article : Google Scholar
|
|
26
|
Shahi MH, Afzal M, Sinha S, Eberhart CG,
Rey JA, Fan X and Castresana JS: Regulation of sonic hedgehog-GLI1
downstream target genes PTCH1, Cyclin D2, Plakoglobin, PAX6 and
NKX2.2 and their epigenetic status in medulloblastoma and
astrocytoma. Bmc Cancer. 10:6142010. View Article : Google Scholar :
|
|
27
|
Xia YP, Dai RL, Li YN, Mao L, Xue YM, He
QW, Huang M, Huang Y, Mei YW and Hu B: The protective effect of
sonic hedgehog is mediated by the phosphoinositide [corrected]
3-kinase/AKT/Bcl-2 pathway in cultured rat astrocytes under
oxidative stress. Neuroscience. 209:1–11. 2012. View Article : Google Scholar
|
|
28
|
Tang H, Wei P, Duell EJ, Risch HA, Olson
SH, Bueno-de-Mesquita HB, Gallinger S, Holly EA, Petersen G, Bracci
PM, et al: Axonal guidance signaling pathway interacting with
smoking in modifying the risk of pancreatic cancer: A gene- and
pathway-based interaction analysis of GWAS data. Carcinogenesis.
35:1039–1045. 2014. View Article : Google Scholar :
|
|
29
|
Must A, Kõks S, Vasar E, Tasa G, Lang A,
Maron E and Väli M: Common variations in 4p locus are related to
male completed suicide. Neuromol Med. 11:13–19. 2009. View Article : Google Scholar
|
|
30
|
Karki R, Pandya D, Elston RC and Ferlini
C: Defining ‘mutation’ and ‘polymorphism’ in the era of personal
genomics. BMC Med Genomics. 8:372015. View Article : Google Scholar :
|
|
31
|
Meilhac SM, Esner M, Kelly RG, Nicolas JF
and Buckingham ME: The clonal origin of myocardial cells in
different regions of the embryonic mouse heart. Dev Cell.
6:685–698. 2004. View Article : Google Scholar
|
|
32
|
Burnicka-Turek O, Steimle JD, Huang W,
Felker L, Kamp A, Kweon J, Peterson M, Reeves RH, Maslen CL, Gruber
PJ, et al: Cilia gene mutations cause atrioventricular septal
defects by multiple mechanisms. Hum Mol Genet. 25:3011–3028.
2016.
|
|
33
|
Nakatomi M, Hovorakova M, Gritli-Linde A,
Blair HJ, MacArthur K, Peterka M, Lesot H, Peterkova R, Ruiz-Perez
VL, Goodship JA and Peters H: Evc regulates a symmetrical response
to Shh signaling in molar development. J Dent Res. 92:222–228.
2013. View Article : Google Scholar
|
|
34
|
Thibert C, Teillet MA, Lapointe F, Mazelin
L, Le Douarin NM and Mehlen P: Inhibition of neuroepithelial
patched-induced apoptosis by sonic hedgehog. Science. 301:843–846.
2003. View Article : Google Scholar
|