Introduction
Hearing loss can be present in >400 genetic
syndromes. Waardenburg syndrome (WS) is the most common type of
autosomal dominant syndromic hearing loss (1). It accounts for 2% of congenital
deafness, is most commonly described in western populations and
affects ~1 in 42,000 people. WS is characterized by congenital
sensorineural hearing loss, and pigmentation defects of the eyes,
hair and skin (for example, heterochromia iridum, white forelock
and patchy hypopigmented skin), and dystopia canthorum (2). WS is commonly hypothesized to be
caused by the absence of neuralcrest-derived melanocytes (3–5).
WS has been classified into four subtypes (WS1-4)
based on the presence or absence of additional symptoms. WS1 and
WS2 are more common than WS3 and WS4. Type 1 [Mendelian inheritance
in man (MIM) no. 193500] is characterized by congenital
sensorineural hearing loss, heterochromia iridis or vivid blue
eyes, and depigmented patches of the skin and hair. Type 2 (MIM no.
193510) is distinguished from WS1 by the absence of dystopia
canthorum. The presence of limb abnormalities separates type III WS
(Klein-Waardenburg syndrome; MIM no. 148820) from type I. Type IV
WS (Shah-Waardenburg syndrome disease; MIM no. 277580) exhibits
features of Hirschsprung disease (an aganglionic megacolon) in
addition to WS2 (5).
Currently, there are six genes, paired box 3
(PAX3), SRY-box 10 (SOX10), melanogenesis associated
transcription factor (MITF), endothelin-2, endothelin
receptor type B (EDNRB) and snail family transcriptional
repressor 2 (SNAI2), reported to be associated with WS
(6). PAX3 on chromosome
2q35 is a member of the mammalian PAX gene family and
encodes a DNA-binding transcription factor expressed in neural
crest cells. Human PAX3, which is 98% homologous to the murine
PAX3, has four structural motifs, including a paired domain, an
octapeptide sequence, a homeodomain and a Pro-Ser-Thr-rich-COOH
terminus (7). PAX3 serves an
important role in the migration and differentiation of melanocytes,
which originate from the embryonic neural crest. The dysfunction of
melanocyte migration and differentiation affects pigmentation of
skin, hair and eyes, and hearing function in the cochlea. Almost
all patients with WS1 and WS3 present with heterozygous mutations
of PAX3 (8,9). Researchers have proposed that 15% of
WS2 patients possess heterozygous mutations of MITF, which
is also a transcription factor with a basic helix-loop-helix
leucine zipper (b-HLH-Zip) motif. MITF has differentially
expressed isoforms. The M-isoform, which is composed of nine exons
(2–9) and is routinely studied in the WS
field, encodes a protein of 419 amino acids (10). MITF is vital to the
development and survival of melanocytes, osteoclasts and mast cells
(11). It is estimated that the
SOX10 gene accounts for ~15% of WS2 cases and 50% of WS4
cases (12). A homozygous deletion
in the SNAI2 gene was described in two unrelated patients
with WS2 (13) and a heterozygous
EDNRB mutation has been identified in patients with WS2
(14).
Little is known about the genetic background of WS
in China. In the present study, detailed analyses of the clinical
manifestations and molecular mechanisms of patients with WS1/2 from
five Chinese families were conducted. Prenatal diagnosis and
counseling for WS was performed, as there is no specific treatment
for the disease.
Materials and methods
Patients and samples
All the patients with WS were identified at Henan
Provincial People's Hospital (Zhengzhou, China; March 2014 to
December 2015). There were 11 patients with WS (from five
families), aged 33 months-44 years old, and unaffected 10 family
members who agreed to take part in the study following audiological
and general physical examinations. Among the five families, family
05 was a sporadic case and the remaining families had multiple
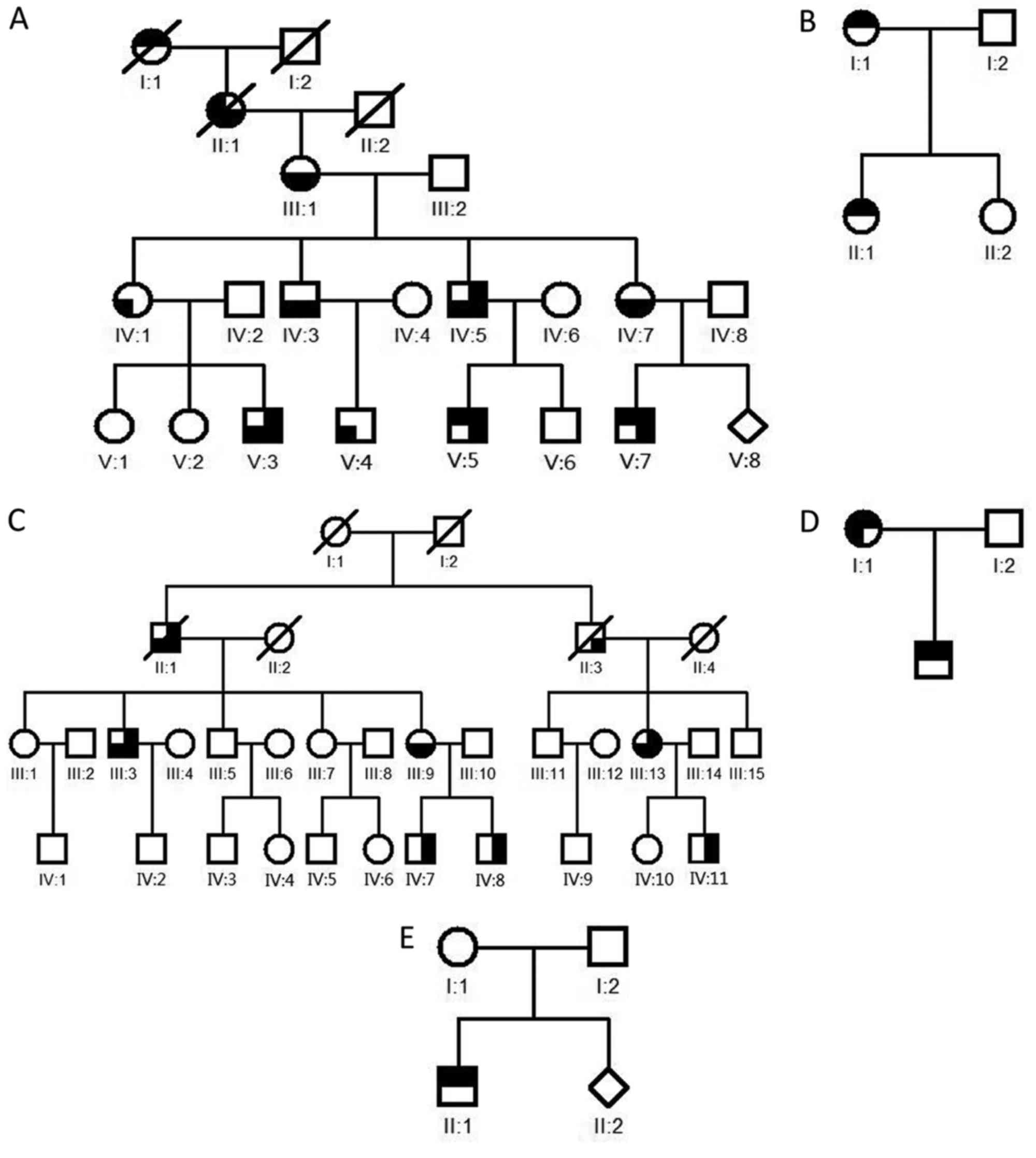
affected patients (Fig. 1).
Patient characteristics are presented in Table I. The present study was approved by
the ethics committee of Henan Provincial People's Hospital. Written
informed consent was obtained from all adult subjects and guardians
on behalf of the children, prior to clinical evaluation and blood
sample collection.
 | Table I.Clinical characteristics and gene
variants of Waardenburg syndrome families. |
Table I.
Clinical characteristics and gene
variants of Waardenburg syndrome families.
|
| Severity of HL |
|
|
|---|
|
|
|
|
|
|---|
| Pedigree | Gender | Age | Iris | Skin | W | Left ear | Right ear | Gene | Variant |
|---|
| 01-IV:5 | M | 44 | − | − | 1.85 | Moderate | Severe | PAX3 | c.583C>T |
| 01-IV:7 | F | 39 | − | − | 1.87 | Normal | Normal | PAX3 | c.583C>T |
| 01-V:7 | M | 10 | + | − | 1.85 | Profound | Moderate | PAX3 | c.583C>T |
| 02-I:1 | F | 32 | + | − | 1.83 | Profound | Moderate | PAX3 | − |
| 02-II:1 | F | 5 | + | − | 1.86 | Profound | Profound | PAX3 | − |
| 03-III:3 | M | 48 | − | + | 1.75 | Severe | Severe | MITF | c.909G>A |
| 03-III:9 | F | 41 | − | + | 1.73 | Normal | Normal | MITF | c.909G>A |
| 03- IV:8 | M | 18 | − | + | 1.71 | Profound | Profound | MITF | c.909G>A |
| 04-I:1 | F | 28 | + | − | 1.75 | Profound | Profound | MITF | c.1060C>A |
| 04-II:1 | M | 3 | + | − | 1.42 | Severe | Profound | MITF | c.1060C>A |
| 05-II:1 | M | 2 | + | − | 1.41 | Profound | Profound | MITF | c.649_651del |
EDTA-K2 peripheral venous blood was taken
from the 11 proposituses and the 10 unaffected family members
(Table II). The chorionic villi
sampling with ultrasonic guidance was performed for the high-risk
women at 11 weeks of pregnancy. Genomic DNA was extracted using the
DNA extraction kit (Tiangen Biotech Co., Ltd., Beijing, China),
according to the manufacturer's protocols. The
PowerPlex® 16 HS System kit (Promega Corporation,
Madison, WI, USA) was used to exclude genetic contamination from
the mother. The results were analyzed using ABI3130xl genetic
analyzer and GeneMapper version 3.2 (both from Thermo Fisher
Scientific, Inc., Waltham, MA, USA) software.
| Table II.Characteristics of the normal
controls. |
Table II.
Characteristics of the normal
controls.
| Pedigree | Gender | Age |
|---|
| 01-IV8 | M | 40 |
| 01-V6 | M | 13 |
| 02-I2 | M | 31 |
| 02-II2 | F | 3 |
| 03-III5 | M | 46 |
| 03-III7 | F | 43 |
| 03-III10 | M | 43 |
| 04-I2 | M | 30 |
| 05-I1 | F | 27 |
| 05-I2 | M | 29 |
Clinical evaluation
Patients with WS were diagnosed according to the
criteria proposed by the WS consortium (15). The diagnosis of WS can be made when
≥two major, or one major and two minor, phenotypic criteria are
met. A comprehensive clinical history was taken, and audiological,
neurological, ophthalmological and dermatological examinations were
performed on all the subjects. The audiological and neurological
examination included otoscopy, pure-tone audiometry and auditory
steady-state response, immittance testing, and auditory brainstem
response. Attention was paid to the color of skin, hair and iris,
as well as developmental defects including dystopia canthorum and
limb abnormalities. The degree of hearing loss (HL) was defined
according to the pure-tone average, which was based on three
frequencies (500, 1,000 and 2,000 Hz) as follows: Normal, <26 dB
HL; mild, 26–40 dB HL; moderate, 41–70 dB HL; severe, 71–90 dB HL;
and profound, >90 dB HL. The audiological follow-ups in infants
were performed using Transient Evoked Otoacoustic Emissions and
Automated Auditory Brainstem Response at 0 and 3 months.
Mutational analysis
Polymerase chain reaction (PCR) was carried out
using the DNA from the patients with specific primers to amplify
all exons and intron/exon boundaries of the PAX3 [National
Center for Biotechnology Information reference number
(NM)_181458.2], MITF-M isoform (NM_000248.2), SOX10
(NM_006941.3), and SNAI2 (NM_003068.3) and EDNRB
(NM_001201397.1) genes. The primers (Table III) were designed using the
online program PRIMER3 (biotools.umassmed.edu/bioapps/primer3_www.cgi).
The PCR reactions were carried out in a total volume of 25 µl
reaction mixture containing 40 ng genomic DNA, 2 pmol primer, 150
µmol dNTP, 0.125 units HotstarTaqDNA polymerase and 2.5 mmol
MgCl2. PCR conditions consisted of an initial
denaturation at 94°C for 4 min; 30 cycles of denaturation at 94°C
for 30 sec, annealing at various temperatures (Table III) for 45 sec for the different
primers and extension at 72°C for 45 sec; followed by a 7 min final
extension at 72°C.
| Table III.Amplification and sequencing
primers. |
Table III.
Amplification and sequencing
primers.
| Gene | Exon | Forward primer
sequence (5′-3′) | Reverse primer
sequence (5′-3′) | Product size
(bp) | Annealing
temperature (°C) |
|---|
| PAX3 | 1 |
TCACCACAGGAGGAGACTCA |
GAGGCCCTCCCTTACCTTC | 472 | 57 |
|
| 2 |
TACGTGCTGCTGTTCTTTGC |
TTACGCACCTTCACAAACCTC | 442 | 58 |
|
| 3 |
TCTGGTCTGCCCCTTTCTAA |
ATTGGGGTGATTACGTCTGG | 388 | 58 |
|
| 4 |
GCTGGAGAAGGATGAGGATG |
CTCCAAGTGACCCAGCAAGT | 351 | 57 |
|
| 5 |
TGTCTTGCAGTCGGAGAGAG |
GGTGGACTTCTGTGTGTCGT | 492 | 58 |
|
| 6 |
AATTCGCCCAAACAACACA |
CAGAGAAATCGCCTGGAAGT | 368 | 58 |
|
| 7 |
TGGCGATGAACTTTTGCAC |
GGGTGGAGAGAAAGGAAACC | 451 | 58 |
|
| 8 |
TCGTCGGGCATGATGTAATA |
AGGAGAAATTGCCCCCTAAA | 359 | 58 |
|
| 9 |
GAATTGTCCCAGCATGACCT |
TGCTCCAGGTCTTCCTCTTC | 311 | 60 |
|
| 10a |
ACTGGCCCTGTTTCTGGTCT |
TGGCAAACATCACTGCACTC | 943 | 58 |
|
| 10b |
CCAGTTCACATTTATTTGG |
CTCATAGAAAGGGTCCAC | 887 | 58 |
| MITF | 1 |
TGGTGTCTCGGGATACCTTG |
TGGCATCAAATAATAAACAGCA | 304 | 57 |
|
| 2 |
CGTTAGCACAGTGCCTGGTA |
GTGGCCACAAGGACAAACTA | 482 | 57 |
|
| 3 |
CATCTTGTTGCTCTGTGCCATC |
AAGGTGATCCACCACAAA | 253 | 56 |
|
| 4 |
GACCATTATTGCTTTGGGTAAAA |
TGTGATCCTGAGATAATTCTCCATT | 343 | 57 |
|
| 5 |
TGAGGAGATCCTGTACCTCTCTT |
AAAAGTTACGTCCATGAGTTGGA | 425 | 57 |
|
| 6 |
GCTTTTGAAAACATGCAAGC |
GCTGTAGGAATCAACTCTCCTCT | 350 | 58 |
|
| 7 |
CATGACCTGGAGAAGTTAATATGC |
AGTGTCCAACAATCCTTTTGC | 398 | 56 |
|
| 8 |
CACCTGTTCCCCAAAACTA |
GTCAACTCCCCTATGGCTCA | 372 | 58 |
|
| 9 |
CTAATGACGCGCATCTACCA |
TCAAGAAAACCCCTTAGGT | 594 | 57 |
| SOX10 | 1 |
GAGTGTTGGGGATGAAGGAA |
CCTGGAATTTCCCACCTTTT | 500 | 58 |
|
| 2 |
AGATGGGTTTAGCTGGAGCA |
ACCTGGTCTTCCAGCCCTAT | 765 | 58 |
|
| 3 |
GTTATTCCTTGGGCCTCACA |
CTTTGCCCAGTAGGATCAGC | 686 | 57 |
|
| 4-a |
CATGCTGCCAAAATGTGAAA |
ATAGGGTCCTGAGGGCTGAT | 678 | 56 |
|
| 4-b |
AGCCCAGGTGAAGACAGAGA |
TCTGTCCAGCCTGTTCTCCT | 561 | 57 |
| EDNRB | 1 |
CATTTCCTGGTTCCCTGACT |
ACCAAAACCAAAGTGCCAAT | 361 | 58 |
|
| 2 |
CTTTTGAGCGTGGATACTGG |
AGGGAGCTAAAGGGAAGCTC | 748 | 58 |
|
| 3/4 |
CCCAACACACTTTCCTGTCC |
TTCTTGCAGCTTGAGTCATTG | 816 | 57 |
|
| 5 |
TGTTCAGTAAGTGTGGCCTGA |
CAAGAAAAAGGAAATATGCTCTGG | 432 | 56 |
|
| 6 |
CACTTCGGTTCCACTTCACA |
CTTCCCTGTCCCTCTCAACA | 466 | 58 |
|
| 7 |
GAGGGGGACACAGACAGAGA |
GCAGTAGGGAGTGGCTGACT | 493 | 57 |
|
| 8 |
AAGAGGGAAAATAAAAGAGCACTG |
TTCTTTCCATGCCGTAAACA | 466 | 57 |
| SNAI2 | 1 |
GCTGTGATTGGATCTTTCTTGC |
TGTAAGCTCCCTTTCAGGACAC | 449 | 56 |
|
| 2 |
TGTGTGTATACTTGCGTGTGG |
CTTCATGCAAATCCAACAGC | 700 | 58 |
|
| 3 |
ATTTCTGTATGATTGGCAGCAG |
GCTTCGGAGTGAAGAAATGC | 471 | 56 |
PCR fragments were purified and sequenced in each
direction on an ABI3130xl DNA sequencer, with a BigDye™
Terminator Cycle Sequencing kit (Thermo Fisher Scientific, Inc.),
with the same primers used for PCR. The raw sequence data were
aligned with the wild-type sequence using the GeneTool program
version 2.0 (Syngene Europe, Cambridge, UK). Following the
identification of mutations of the WS genes in the propositus,
samples from the related family members and normal controls were
further screened for the mutations.
Array-based comparative genomic
hybridization (aCGH)
a-CGH was performed on patients whose sequence
results were normal, a-CGH was carried out by using the SurePrint
G3 Human CGH Microarray 8×60K kit (Agilent Technologies, Inc.,
Santa Clara, CA, USA). For each sample, 500 ng genomic DNA was used
for each a-CGH experiment. DNA was labeled by direct incorporation
of Cy5-deoxyuridine triphosphate (dUTP) and Cy3-dUTP, according to
the Agilent protocol. Labeled DNA was purified and then hybridized
to the arrays. Following performance of the hybridization step and
the recommended washes, the arrays were scanned with a SureScan Dx
Microarray scanner and analyzed using CytoGenomics 2.9 (both from
Agilent Technologies, Inc., Santa Clara, CA, USA).
Results
Clinical findings
According to the diagnostic criteria for WS, five
cases (IV:5, IV:7 and V:7 in family 01, I:1 and II:1 in family 02)
were diagnosed as WS1, while the other six cases (III:3, III:9 and
IV:8 in family 03, I:1 and II:1 in family 04, II:1 in family 05)
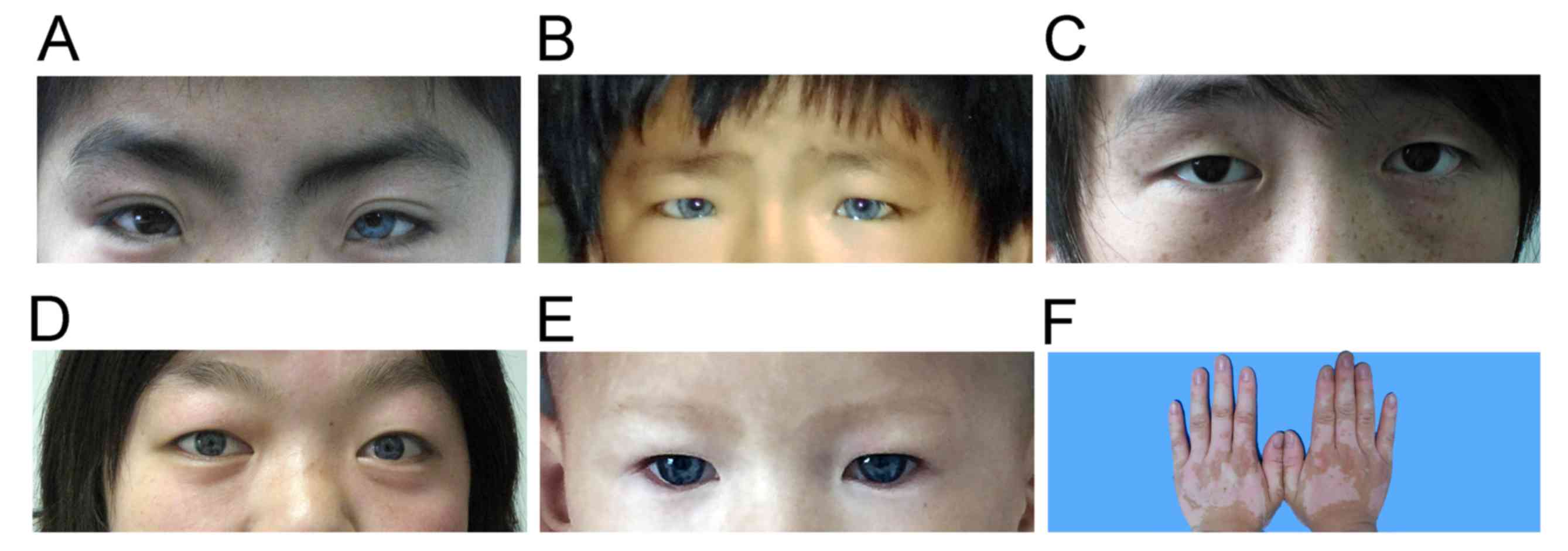
were WS2. Among these patients, deafness and heterochromia iridum
were the most frequent features, and nine patients exhibited
sensorineural hearing impairment varying from moderate to profound.
Additionally, the characteristic brilliant blue irides (unilateral
or bilateral heterochromia irides), were observed in 6 patients.
Each of the WS1 cases exhibited dystopia canthorum, and three of
them (01-V7, IV7 and 02-II1) also presented with broad nasal roots.
A total of 3 patients in family 03 had numerous brown freckles on
the faces. Patchy skin depigmentation was only observed in family
05. Other clinical features, a white forelock and hypopsia for
example, were not observed in the present study (Table I). Representative clinical findings
and the typical characteristics of these WS cases were illustrated
in Fig. 2.
Identification of mutations
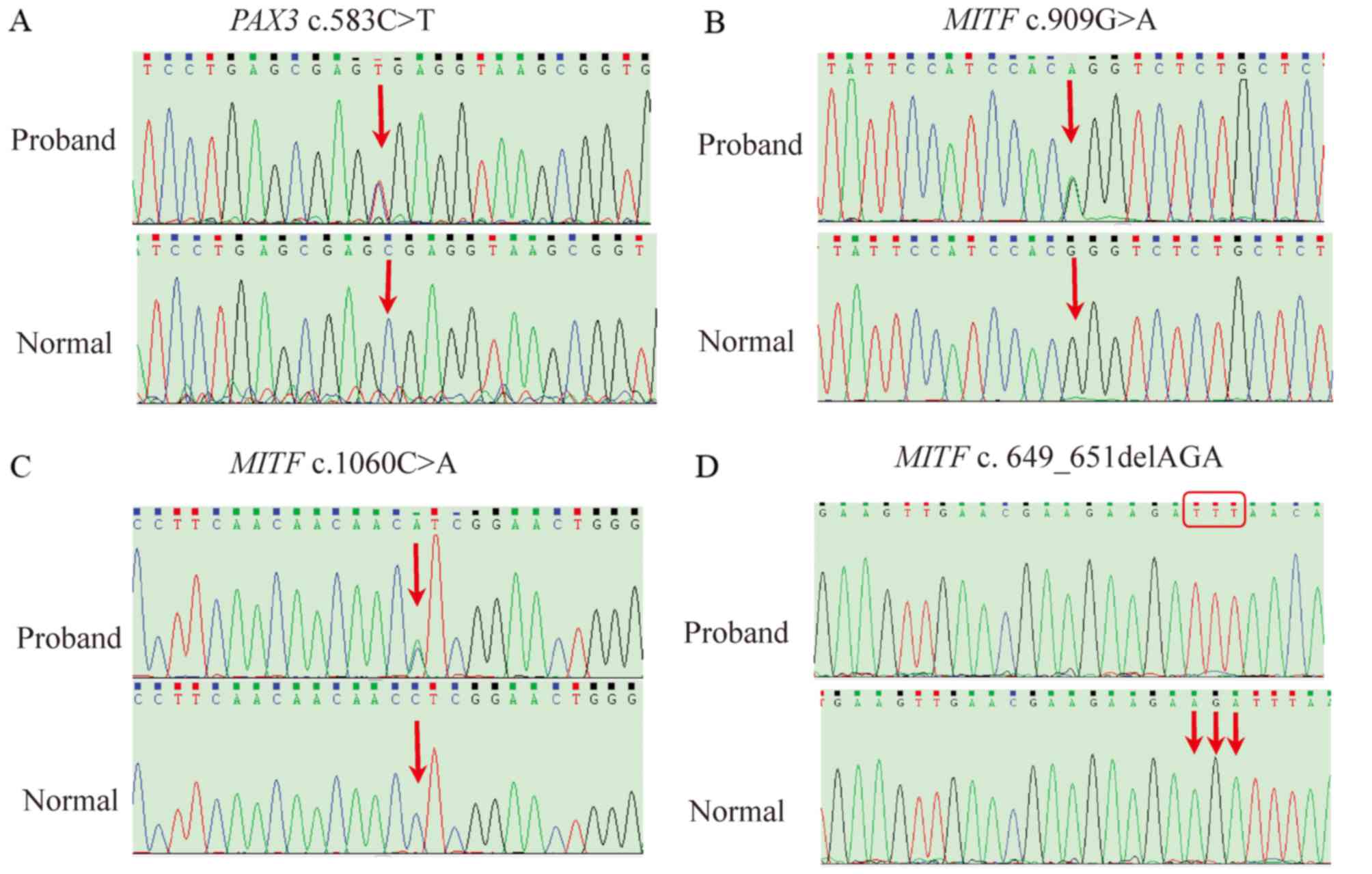
Six variants were identified in these proposituses.
A heterozygous nonsense mutation c.583C>T in PAX3 exon 4
was identified in 3 affected members of family 01, resulting in a
premature termination codon at the 195th nucleotide within the
octapeptide domain of the PAX3 protein (Fig. 3A). The sequencing and a-CGH assays
failed to detect the mutation in family 02.
Three heterozygous mutations in MITF were
identified in these WS2 patients, including splice-site, missense
and deletion mutations. The splice-site mutation c.909G>A was
identified in family 03 (Fig. 3B),
which has been reported by Brenner et al (16). Unlike the previous splicing
variants reported, the mutation did not destroy the acceptor or
donor splicing sites, but created a new splice acceptor site. The
variant c.1060C>A (p.L354I) in exon 9 was found in family 04,
which led to a substitution from Leu to Ile at the 354th amino acid
(Fig. 3C). An in-frame deletion
c.649_651delGAA, del (R217) mutation was identified in family WS05,
which removed one of a run of four arginines in the basic domain
(Fig. 3D). The mutations
(c.583C>T in PAX3, c.909G>A and c.649_651delGAA in
MITF) identified in these patients were not observed in any
other unaffected family members. Other synonymous substitutions
were, c.927T>C in SOX10 (family 03) and c.831A>G in
EDNRB (family 04).
Prenatal diagnosis and follow-up
Following identification of the genotype of the
probands and their parents, prenatal genetic diagnosis was
performed for the two fetuses of family 01 and family 05, as a
carrier, the couples have a 50% chance of affected offspring. DNA
was extracted from chorionic villi samples and mutations of the
PAX3/MITF gene were detected. The prenatal genetic diagnosis
revealed the two babies were normal for the genes examined (data
not shown). Furthermore, the audiological follow-ups between 0 and
6 months revealed hearing was within normal thresholds.
Discussion
WS may not be a rare disease in China as reported
previously, up to now, >50 cases had been reported (17–19).
More patients may be identified in special schools for children
with hearing loss, although a large-scale, population-based study
is required to confirm this. The syndrome involves a number of
highly variable clinical manifestations. A previous study suggested
that sensorineural deafness is the most frequent feature of WS (60%
in WS1 and 90% in WS2) (20). In
the present study, the majority of the patients (81.81%; 9/11)
exhibited bilateral sensorineural hearing loss, ranging from
moderate deafness to a progressive profound hearing loss. The
syndrome exhibits a highly variable phenotype within families and
incomplete penetrance. The affected individuals may exhibit
multiple symptoms; however, certain patients exhibit few symptoms
of WS, including IV:7 and V:7 in family 01. In addition, the
propositus's father in family 05 was a healthy subject, despite the
fact that he carried the same mutation as propositus. The father
exhibited only a skin pigmentary disorder on both hands and is not
a patient with WS according to the diagnostic criteria. It was
speculated that the symptom may be associated with the genetic
abnormalities. The skin pigmentation disorder is a rare clinical
feature in WS families in China. This supported the results of the
study by Liu et al (21)
that skin pigmentation disorders are less frequent. Conversely,
features including deafness or facial freckles are relatively
common, which makes the clinical diagnosis difficult, particularly
for WS2 (IV:8 from family 03). Therefore, molecular genetic
analyses of WS genes are important diagnostic steps to explain the
molecular cause of WS, and facilitate genetic counseling of
affected patients and their families.
Currently six genes have been identified to be
associated with WS. To date, >280 mutations in these genes have
been reported in patients with WS (grenada.lumc.nl/LOVD2/WS).
PAX3 mutations account for the majority of WS1 and WS3;
however, few of them are recurrent. Missense and nonsense
mutations, frameshifts, small in-frame insertions or deletions, and
splice alterations of the PAX3 gene have been reported in
patients with WS1. Altogether, ~50% of the mutations are missense
and 50% are truncating variations. The majority of the novel
mutations in PAX3 are localized in exons 2–6 and therefore
influence functionally relevant domains. In the present study, the
PAX3 mutation c.583C>T in exon 4 was detected within
family 01 cases, which led to premature termination at the 195th
amino acid and was void of functional domains. This mutation has
been previously reported by Read et al (2). No mutation was identified in family
02 by PAX3 exon sequencing which cannot reliably detect
whole exon or whole gene copy number alterations. Drozniewska et
al (22) reported a patient
with a de novo 2q36.1 deletion of 862 kb, including the
PAX3 gene, which indicates that deletion or duplication
screening is indispensable for the molecular genetic diagnosis of
WS. Neither deletion nor duplication was observed in family 02 by
a-CGH. It's possible that the mutation remains to be identified as:
i) Mutations deep in introns or promoter regions were not
sequenced; and ii) mutations in other genes may be involved in
pathogenesis (23,24).
Currently, ~30% of WS2 cases can be explained at the
molecular level. It is estimated that MITF mutations occur
in ~15% of WS2. Through binding to DNA sequences, MITF
regulates melanocyte differentiation and the transcription of
several melanocyte-specific genes. Mutant MITF proteins are thought
to possess defects in homo- or heterodimerization, and DNA binding
through their basic regions. Point mutations are not evenly
scattered along the gene, the majority of them are located in exons
7 and 8 that correspond to the b-HLH-Zip motifs. Interruption of
the b-HLH-Zip domain decreases the ability of the mutant MITF
protein to bind to the CATGTG core DNA sequence in the human
tyrosinase promoter (25).
Wildhardt et al (26)
reported a patient with WS2 carrying two MITF mutations (missense
mutation and small deletion) within the same copy of the gene. In
the present study, three MITF mutations, c.909G>A
(p.Thr303Thr), c.649_651delGAA and c.1060C>A (p.L354I) were
identified. The synonymous mutation c.909G>A was demonstrated to
create a novel splice acceptor site that removes the 52 bps from
the mRNA and results in the introduction of seven novel amino acids
with premature termination and loss of the terminal 133 amino acids
(16). The in-frame deletion
c.649_651delGAA, del (R217) mutation in exon 7 was identified in
family WS05, which removes one of four arginines in the basic
domain. Conservation analyses revealed that the Arg residues at 217
in MITF are conserved across humans, mice, chickens, cows, and
dogs. The mutation had been previously reported in a Caucasian
family (8). In the family, the
mother and son exhibited severe congenital sensorineural hearing
loss and pigmentary disorder. The phenotype resembles the
albinism-deafness syndrome of Tietz (MIM no. 103500) to an
increased extent compared with classical Waardenburg syndrome,
which is different from the patients in the present study. It can
be speculated that there is interplay between genetic modifiers,
environmental factors and stochastic events, in addition to the
mutation itself. The mutation c.649_651delGAA in the MITF
gene may be a hot spot for 7 cases reported in the mutation
database.
Variant c.1060C>A (L354I) was identified in WS04.
The mutation p.L354I fell outside of the important MITF domain, and
was predicted to exhibit a ‘benign’ or ‘tolerated’ effect by the
PolyPhen-2 (http://tiddlyspace.com/bags/icgc_public/tiddlers/PolyPhen2),
and SIFT (http://sift.jcvi.org) software,
respectively.
Simultaneously, the child inherited it from his
unaffected father, while his affected mother did not carry the same
mutation. Other variants of MITF have been reported, including:
c.20A>G(p.Tyr7Cys), c.332C>T(p.Ala111Val),
c.483A>T(p.Gln161His), c.608G>A (p.Arg203 Lys) and
c.892T>C (p.Ser298Pro), all of which are thought to be likely
neutral variants or the Single Nucleotide Polymorphism database
(5,8,19).
The above mutations (c.583C>T in PAX3 and
c.909G>A in MITF) were first identified in the Chinese
population. These mutations in the WS genes enable prenatal
diagnosis to be performed for the high-risk fetus. If one of the
parents carries a dominant altered WS gene, there is a 50% chance
that the children will inherit the disease. Preimplantation genetic
diagnosis (PGD) is an alternative to prenatal diagnosis and is an
almost safe, harmless, non-invasive and ethically acceptable
procedure (27). However, PGD
could not be easily obtained by the families for economic reasons.
Invasive prenatal diagnosis may be another effective means of
preventing recurrence of genetic disease. Therefore, prenatal
diagnosis for the two families (01 and 05) was performed. The
results demonstrated that the two fetuses were normal, which was
confirmed by audiological follow-ups. However, a positive test
result may be derived from a patient exhibiting a normal phenotype
despite the presence of mutations as there may be reduced
penetrance. For this reason, it is important to discuss these
issues during genetic counseling. In the future, non-invasive
prenatal diagnosis of WS by examination of cell-free fetal DNA in
maternal blood will be performed.
In conclusion, the results of the present study
expand the database of known mutations in Chinese patients with WS.
Certain cases remain unexplained at the molecular level, which
require further study. The exact description of the mutations
responsible for the WS type is of importance in genetic counseling
of patients with WS and their families.
Acknowledgements
The present study was supported by the National
Natural Science Foundation of China (grant no. 81170581), the
Science and Technology Project of Henan Province (grant no.
2010031114), and the Medical Science and Technology Project of
Henan Province (grant no. 87).
References
|
1
|
Zaman A, Capper R and Baddoo W:
Waardenburg syndrome: More common than you think! Clin Otolaryngol.
40:44–48. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Read AP and Newton VE: Waardenburg
syndrome. J Med Genet. 34:656–665. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Baxter LL, Hou L, Loftus SK and Pavan WJ:
Spotlight on spotted mice: A review of white spotting mouse mutants
and associated human pigmentation disorders. Pigment Cell Res.
17:215–224. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Mollaaghababa R and Pavan WJ: The
importance of having your SOX on: Role of SOX10 in the development
of neural crest-derived melanocytes and glia. Oncogene.
22:3024–3034. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Pingault V, Ente D, Dastot-Le Moal F,
Goossens M, Marlin S and Bondurand N: Review and update of
mutations causing Waardenburg syndrome. Hum Mutat. 31:391–406.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Song J, Feng Y, Acke FR, Coucke P,
Vleminckx K and Dhooge IJ: Hearing loss in Waardenburg syndrome: A
systematic review. Clin Genet. Jun 22–2016.(Epub ahead of print).
View Article : Google Scholar
|
|
7
|
DeStefano AL, Cupples LA, Arnos KS, Asher
JH Jr, Baldwin CT, Blanton S, Carey ML, da Silva EO, Friedman TB,
Greenberg J, et al: Correlation on between Waardenburg syndrome
phenotype and genotype in a population of individuals with
identified PAX3 mutations. Hum Genet. 102:499–506. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Tassabehji M, Newton VE, Liu XZ, Brady A,
Donnai DK, ajewska-Walasek M, Murday V, Norman A, Obersztyn E,
Reardon W, et al: The mutational spectrum in Waardenburg syndrome.
Hum Mol Genet. 4:2131–2137. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wollnik B, Tukel T, Uyguner O, Ghanbari A,
Kayserili H, Emiroglu M and Yuksel-Apak M: Homozygous and
heterozygous inheritance of PAX3 mutations causes different types
of Waardenburg syndrome. Am J Med Genet A. 122A:42–45. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Shibahara S, Takeda K, Yasumoto K, Udono
T, Watanabe K, Saito H and Takahashi K: Microphthalmia-associated
transcription factor (MITF): Multiplicity in structure, function,
and regulation. J Invest Derm Symp Proc. 6:99–104. 2001. View Article : Google Scholar
|
|
11
|
Grill C, Bergsteinsdottir K, Ogmundsdóttir
MH, Pogenberg V, Schepsky A, Wilmanns M, Pingault V and
Steingrimsson E: MITF mutations associated with pigment deficiency
syndromes and melanoma have different effects on protein function.
Hum Mol Genet. 22:357–4367. 2013. View Article : Google Scholar
|
|
12
|
Tassabehji M, Newton VE and Read AP:
Waardenburg syndrome type 2 caused by mutations in the human
microphthalmia (MITF) gene. Nat Genet. 8:251–255. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Sánchez-Martín M, Rodríguez-García A,
Pérez-Losada J, Sagrera A, Read AP and Sánchez-García I: SLUG
(SNAI2) deletions in patients with Waardenburg disease. Hum Mol
Genet. 11:3231–3236. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Jabeen R, Babar ME, Ahmad J and Awan AR:
Novel mutations of endothelin-B receptor gene in Pakistani patients
with Waardenburg syndrome. Mol Biol Rep. 39:785–788. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Farrer LA, Grundfast KM, Amos J, Arnos KS,
Asher JH Jr, Beighton P, Diehl SR, Fex J, Foy C, Friedman TB, et
al: Waardenburg syndrome (Ws) type-I is caused by defects at
multiple Loci, one of which is near alpp on chromosome-2-1st report
of the Ws consortium. Am J Hum Genet. 50:902–913. 1992.PubMed/NCBI
|
|
16
|
Brenner L, Burke K, Leduc CA, Guha S, Guo
J and Chung WK: Novel splice mutation in microthalmia-associated
transcription factor in Waardenburg Syndrome. Genet Test Mol
Biomarkers. 15:525–529. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Chen H, Jiang L, Xie Z, Mei L, He C, Hu Z,
Xia K and Feng Y: Novel mutations of PAX3, MITF, and SOX10 genes in
Chinese patients with type I or type II Waardenburg syndrome.
Biochem Biophys Res Commun. 397:70–74. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Jiang L, Chen H, Jiang W, Hu Z, Mei L, Xue
J, He C, Liu Y, Xia K and Feng Y: Novel mutations in the SOX10 gene
in the first two Chinese cases of type IV Waardenburg syndrome.
Biochem Biophys Res Commun. 408:620–624. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Yang S, Dai P, Liu X, Kang D, Zhang X,
Yang W, Zhou C, Yang S and Yuan H: Genetic and phenotypic
heterogeneity in Chinese patients with Waardenburg syndrome type
II. PLoS One. 8:e771492013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Nayak CS and Isaacson G: Worldwide
distribution of Waardenburg syndrome. Ann Oto Rhinol Laryn.
112:817–820. 2003. View Article : Google Scholar
|
|
21
|
Liu X, Newton V and Read A: Hearing loss
and pigmentary disturbances in Waardenburg syndrome with reference
to WS type II. J Laryngol Otol. 109:96–100. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Drozniewska M and Haus O: PAX3 gene
deletion detected by microarray analysis in a girl with hearing
loss. Mol Cytogenet. 7:302014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Berlin I, Denat L, Steunou AL, Puig I,
Champeval D, Colombo S, Roberts K, Bonvin E, Bourgeois Y, Davidson
I, et al: Phosphorylation of BRN2 modulates its interaction with
the Pax3 promoter to control melanocyte migration and
proliferation. Mol Cell Biol. 32:1237–1247. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Degenhardt KR, Milewski RC, Padmanabhan A,
Miller M, Singh MK, Lang D, Engleka KA, Wu ML, Li J, Zhou D, et al:
Distinct enhancers at the Pax3 locus can function redundantly to
regulate neural tube and neural crest expressions. Dev Biol.
339:519–527. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Takeda K, Takemoto C, Kobayashi I,
Watanabe A, Nobukuni Y, Fisher DE and Tachibana M: Ser298 of MITF,
a mutation site in Waardenburg syndrome type 2, is a
phosphorylation site with functional significance. Hum Mol Genet.
9:125–132. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Wildhardt G, Zirn B, Graul-Neumann LM,
Wechtenbruch J, Suckfüll M, Buske A, Bohring A, Kubisch C, Vogt S,
Strobl-Wildemann G, et al: Spectrum of novel mutations found in
Waardenburg syndrome types 1 and 2: Implications for molecular
genetic diagnostics. Bmj Open. 3:pii: e001917. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Millan JM, Aller E, Jaijo T, Grau E,
Beneyto M and Najera C: Genetic counselling in visual and auditory
disorders. Arch Soc Esp Oftalmol. 83:689–702. 2008.(In Spanish).
PubMed/NCBI
|