Introduction
Osteoarthritis (OA) is one of the most common types
of arthritis and is a primary cause of disability (1). The prevalence of knee and hip OA
ranges from 20–30% in the general population worldwide (1). OA is primarily characterized by focal
cartilage degradation, which may be caused by the imbalance between
anabolic and catabolic factors in the joints. Furthermore, the
affected joints exhibit osteophyte formation and sub-chondral bone
sclerosis (2,3). OA is also a multifactorial disease
with strong heritability ranging from 40 to 65%, which depends on
the joint sites (4–7). The identification of genes associated
with OA would aid with revealing the underlying molecular
pathogenic mechanisms and pathways, and lead to development of the
gene diagnosis and targeted therapy of OA.
Currently, the association between cartilage biology
and OA in numerous studies has been concentrated on microRNAs
(miRNAs). miRNA, a class of non-coding small RNAs, is widely
accepted as one of the post-transcriptional modifications that
influences entire intracellular molecular cascades, such as
intracellular signaling (8,9).
Therefore, miRNAs perform critical and diverse activities in
biological functions, such as apoptosis, differentiation, stem cell
maintenance and metabolism. Recently, miRNA was suggested as an
integral aspect of the regulatory network of chondrocyte
differentiation and cartilage function, which indicates miRNA may
contribute to the development of OA (10–12).
The expression levels of one or a few miRNAs between OA cartilage
and healthy control samples have been compared by various studies
(10–12). These expression-profiling studies,
which often reported conflicting results, are dispersed and have
never been systematically assessed. Consequently, the potential of
miRNAs serving as diagnostic markers or even therapeutic targets of
OA remains unclear and requires analysis.
In the present study, a comprehensive meta-analysis
was performed based on published studies, and compared miRNA
expression profiles between OA cartilage and normal cartilage to
overcome the limitations of these miRNA expression-profiling
studies. The robust rank aggregation (RRA) method was applied in
order to reduce the influences of heterogeneity (13–15)
and establish the key miRNAs in OA. Various crucial miRNA target
genes were predicted using bioinformatics tools. Based on this,
consensus targets were combined for further analysis in
corresponding databases, such as the Kyoto Encyclopedia of Genes
and Genomes (KEGG) and Gene Ontology (GO) databases. The present
study focused upon identifying the consistence of differently
expressed miRNAs, which may provide a novel insight into the
differentially expressed miRNA profiling studies of OA. This is
considered to be of great value in improving the diagnostics,
therapeutics and prognosis of OA.
Materials and methods
Literature review
A two-step literature searching strategy was used to
identify the OA miRNA expression profiling studies. First,
literature reviews were performed using the PubMed database
(www.ncbi.nlm.nih.gov/pubmed/), Gene
Expression Omnibus (www.ncbi.nlm.nih.gov/geo/) and ArrayExpress
(www.ebi.ac.uk/arrayexpress) according
to the following search terms: ‘microRNA’ OR ‘miRNA’ AND
‘osteoarthritis’ OR ‘arthritis’ AND ‘expression’ OR ‘profile’ OR
‘profiling’. In the second step, relevant references that meet the
above-mentioned criteria were carefully screened manually for
further studies. The latest year of publication was 2016.
Inclusion and exclusion criteria
The following studies that: i) Profiled miRNA
expression levels of patients with OA; ii) compared the expression
profile of cartilage collected by surgery from OA patients with
that of normal cartilage from healthy control subjects; iii)
reported cut-off criteria for determining whether miRNAs were
differentially expressed between the OA and normal control; and iv)
reported their method for validating the profiling results were
included in the meta-analysis. Therefore, miRNA profiling studies
we excluded by analyzing plasma or serum sample collected from OA
patients or cell lines. Review articles were also excluded.
Data extraction
Two reviewers independently extracted the following
information from the included studies: Author, publication year,
country of subjects, sample size and characteristics of patients,
author-defined cut-off criteria of differential miRNA expression
profiling, and the direction (up- or down regulated) and
fold-change of the differential expression of each miRNA (if
available). Subsequently, the authors aimed to reach a consensus on
these items through discussion. All of the miRNA names were
standardized depending on the miRBase version 21 (www.mirbase.org). miRNAs that were considered as a
‘dead entry’ due to re-annotation in miRBase (version 21) were
omitted in subsequent meta-analysis.
RRA method for meta-analysis
The RRA method, which is a free R software package
(https://cran.r-project.org/mirrors.html) was used for
this meta-analysis. The gene lists of miRNAs from selected articles
were ranked according to P-values (P<0.05) according to the RRA
method. The leave-one-out cross-validation algorithm was applied in
this method. A ten thousand times repeating analysis permutation
procedure was performed to calculate an average P-value from the
random gene lists, which represents the best P-value of each of the
miRNAs. To avoid false-positive results, the Bonferroni correction
of the P-values was calculated.
Prediction and filtering of
dysregulated miRNAs targets
The targets of miRNA were predicted using TargetScan
version 6.2 (www.targetscan.org) (16), PicTar (http://pictar.mdc-berlin.de/cgi-bin/PicTar_vertebrate.cgi)
(17) and miRDB (http://www.mirdb.org/mining.html) (18). The TarBasev7.0 database (http://diana.imis.athena-innovation.gr/DianaTools/index.php?r=tarbase/index/)
(19) and starBase (http://starbase.sysu.edu.cn/targetSite.php) (20) were also used to facilitate with the
screening and provide experimental proof for predicted targets.
Considering the reliability, only overlapping targets predicted by
more than one algorithm were selected for further
investigation.
Enrichment analysis
The Database for Annotation, Visualization and
Integrated Discovery (DAVID) web tool (21,22)
(http://david.abcc.ncifcrf.gov/) was used
for pathway identification and enrichment analysis. The consensus
targets of each miRNA were entered into the DAVID web tool to
screen the following database Gene Ontology terms, such as KEGG,
Panther and REACTOME signaling pathways.
Results
Study selection and data
extraction
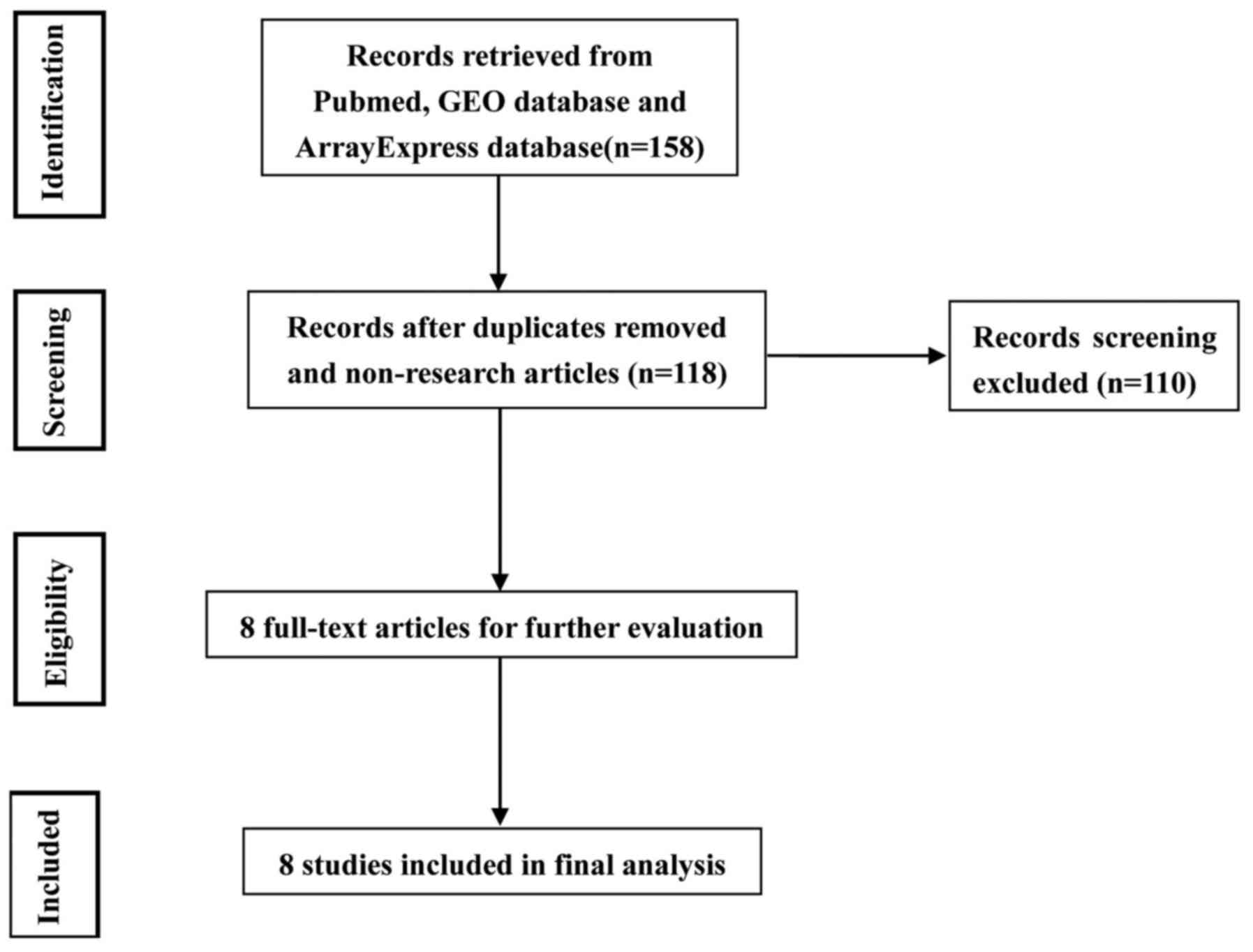
A total of 139 studies were identified in PubMed,
and only eight of these met the inclusion criteria (Fig. 1; Table
I) (23–30). The included studies were published
between 2009 and 2016. Three of these were conducted with samples
from Europe (United Kingdom, Greece, Spain and Germany), another
two studies with samples from Asian countries (Japan and Korea) and
the other two studies with samples from North America (Canada). The
average number of miRNA probes was 799 (range, 157–2,100). A total
of 121 human cartilage samples were used for this meta-analysis.
The significant data from these studies are presented in Table I. There were 87 differentially
expressed miRNAs reported in eight studies in total, out of which
46 were reported to demonstrate increased expression and 41
demonstrated decreased expression (Table I).
 | Table I.Characteristics of studies included in
the meta-analysis on microRNA expression levels in cartilage tissue
samples of OA. |
Table I.
Characteristics of studies included in
the meta-analysis on microRNA expression levels in cartilage tissue
samples of OA.
|
|
|
| Differential miRNA
expression |
|
|
|---|
|
|
|
|
|
|
|
|---|
| Author | Year | Samples, n
(OA/control) | Upregulated, n | Downregulated, n | Cut-off criteria | Platform | (Refs.) |
|---|
| Jones et
al | 2009 | 6 (3/3) | 11 | 5 | FC>2.0 | TaqmanqPCR
assays | (24) |
| Iliopoulos et
al | 2008 | 43 (33/10) | 9 | 7 | FC>1.5 | Taqman miRNA
assays | (23) |
| Miyaki et
al | 2009 | 19 (11/8) | 10 | 0 | FC>1.5 | mirVana miRNA
Labeling Kit | (25) |
| Diaz-Prado et
al | 2012 | 10 (6/4) | 1 | 6 | FC>1.5 | miRNA PCR system | (26) |
| Song et
al | 2013 | 15 (10/5) | 7 | 11 | P<0.05 | TaqmanqPCR
assays | (27) |
| Li et
al | 2016 | 8 (4/4) | 6 | 5 | FC>1.5 | TaqmanqPCR
assays | (28) |
| Beyer et
al | 2015 | 26 (13/13) | 0 | 2 | FC>1.5 | TaqmanqPCR
assays | (29) |
| Nakamura et
al | 2016 | 4 (2/2) | 2 | 5 | FC>2.0 | miRCURY LNA miRNA
Array | (30) |
| Total |
| 121 (82/39) | 46 | 41 |
|
|
|
miRNA meta-signature
A meta-signature consisted of six dysregulated
miRNAs that were significantly identified in OA samples, which
included four increased and two decreased expression levels when
compared with controls according to the permutation P-value. The
most significantly dysregulated miRNA was miR-23b-3p, which was
reported by three datasets. Two miRNAs, miR-23b-3p and miR-27b-3p
were observed none cluster located at9q22.3 (Table II), and the cluster was identified
in MirBase within a distance <10 kb. Other miRNAs were scattered
on different chromosomal locations.
| Table II.Summary of six miRNAs that were
significantly up- and downregulated in osteoarthritis cartilage
tissue samples compared with normal cartilage tissue samples. |
Table II.
Summary of six miRNAs that were
significantly up- and downregulated in osteoarthritis cartilage
tissue samples compared with normal cartilage tissue samples.
|
|
| P-value |
|
|---|
|
|
|
|
|
|---|
| miRNA | Studies, n | Robust rank
aggregation | Corrected | Permutation | Chromosome |
|---|
| Upregulated |
|
|
|
|
|
|
miR-23b-3p | 3 | 8.53E-11 | 0.00000955 | 0.024 | 9q22.3 |
|
miR-27b-3p | 2 | 0.000000179 | 0.020079 | 0.031 | 9q22.3 |
|
miR-211-5p | 1 | 0.000000258 | 0.028913 | 0.01 | 15q13.3 |
|
miR-16-5p | 2 | 0.000000257 | 0.028924 | P<0.05 | 13q14.2 |
| Downregulated |
|
|
|
|
|
|
miR-25-3p | 2 | 0.000000105 | 0.011807 | 0.03 | 7q22.1 |
|
miR-149-5p | 3 | 0.000000421 | 0.047223 | 0.000228 | 2q37.3 |
Target prediction of meta-signature
miRNAs
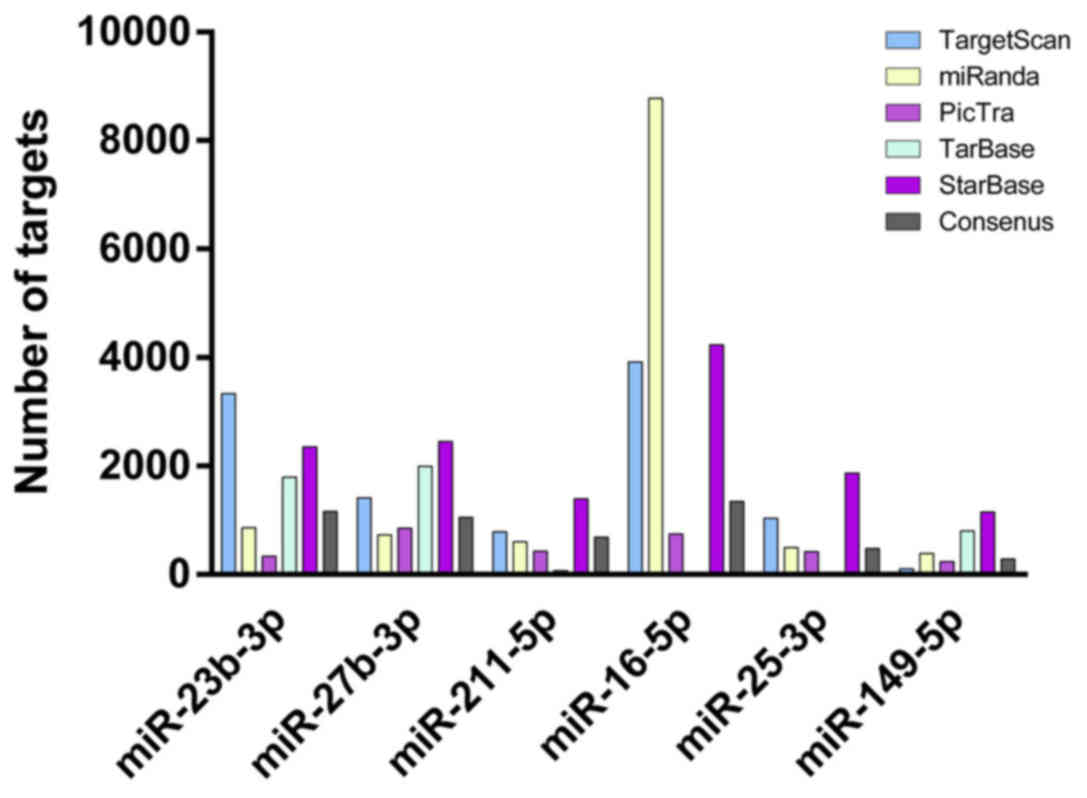
The numbers of targets were presented in Fig. 2. The overlapping consensus targets
of meta-signature miRNAs identified by RRA were extracted,
predicted by at least two different algorithms and validated by two
databases, TarBase and StarBase. miR-23b-3p and miR-16-5p were
observed to have more targets than the other miRs.
Enrichment analysis for predicted
target of meta-signature miRNAs
The enrichment analysis for predicted targets of
meta-miRNAs was completed using the DAVID web tool. Numerous
significant results were screened through the enrichment of KEGG
and Panther signaling pathways, most of which were associated with
cell signaling and cell regulation, as presented in Table III.
| Table III.GO process, KEGG pathway, Panther
pathway and REACTOME pathway enriched by meta-signature miRNA
targets. |
Table III.
GO process, KEGG pathway, Panther
pathway and REACTOME pathway enriched by meta-signature miRNA
targets.
| Pathway enrichment
analysis | Targets | P-value |
|---|
| GO process |
|
|
|
GO:0044260: Cellular
macromolecule metabolic process | 286 | 8.64962E-2 |
|
GO:0043170: Macromolecule
metabolic process | 290 | 1.05878E-2 |
|
GO:0006464: Protein
modification process | 110 | 2.70971E-2 |
|
GO:0043687: Post-translational
protein modification | 94 | 2.16068E-2 |
|
GO:0043412: Biopolymer
modification | 111 | 2.96494E-2 |
|
GO:0044267: Cellular protein
metabolic process | 149 | 5.41348E-2 |
|
GO:0044237: Cellular metabolic
process | 315 | 3.20433E-2 |
|
GO:0009987: Cellular
process | 440 | 8.45760E-2 |
|
GO:0060255: Regulation of
macromolecule metabolic process | 184 | 9.34049E-2 |
|
GO:0080090: Regulation of
primary metabolic process | 183 | 4.36152E-2 |
|
GO:0019222: Regulation of
metabolic process | 196 | 6.01876E-2 |
| KEGG pathway |
|
|
| 04115:
p53 signaling pathway | 10 | 4.92024E-4 |
| 05215:
Prostate cancer | 11 | 9.08174E-4 |
| 05200:
Pathways in cancer | 23 | 1.91181E-3 |
| 05210:
Colorectal cancer | 10 | 2.28645E-3 |
| 05222:
Small cell lung cancer | 9 | 8.12637E-3 |
| 00562:
Inositol phosphate metabolism | 7 | 1.02755E-2 |
| 04110:
Cell cycle | 11 | 1.08581E-2 |
| 04070:
Phosphatidylinositol signaling system | 8 | 1.34821E-2 |
| 04310:
Wnt signaling pathway | 12 | 1.49651E-2 |
| 04120:
Ubiquitin mediated proteolysis | 11 | 1.96911E-2 |
| Panther
pathway |
|
|
| P00030:
Hypoxia response via hypoxia-inducible factor activation | 6 | 1.28214E-2 |
| P00034:
Integrin signalling pathway | 17 | 1.50999E-2 |
| P00033:
Insulin/insulin-like growth factor pathway-protein kinase B
signaling cascade | 9 | 2.58629E-2 |
| P04398:
p53 pathway feedback loops 2 | 7 | 3.93284E-2 |
| P00048:
Phosphoinositide 3 kinase pathway | 10 | 4.35708E-2 |
| P00046:
Oxidative stress response | 7 | 4.49204E-2 |
| P00052:
TGF-βsignaling pathway | 12 | 4.59938E-2 |
| REACTOME
pathway |
|
|
|
REACT_12034: Signaling by bone
morphogenetic proteins | 4 | 3.08411E-2 |
|
REACT_6844: Signaling by
TGF-β | 3 | 4.39359E-2 |
Discussion
In recent years, with the realization of the
importance of miRNAs in different biological processes, numerous
studies have focused on the association between miRNA expression
and the development of OA. However, certain studies have shown
inconsistent miRNA expression in OA cartilage samples when compared
with those of healthy controls, which may be due to the following
factors: i) Different platforms of profiling; ii) small sample
size; and iii) inconsistent methods for data analysis. To overcome
these defects, a meta-analysis using the RRA method was performed
for analyzing particular miRNAs of OA from eight independent
profiling experiments. When compared to the classical vote-counting
method, the RRA algorithm has certain advantages, including being
robust to noise, incomplete rankings, assigns scores to each
element for ranking, and is efficient to compute (31). In addition, two different
meta-analysis methods for differential expression of miRNAs in
pancreatic ductal adenocarcinoma have been performed. In the
present study, the results of the different methods included the
potential prognostic biomarkers, which were detected by
experimental validation. Therefore, the RRA method was more
accurate than the vote-counting method (32).
In the current study, six overlapping significantly
dysregulated miRNAs were demonstrated with four that were
upregulated and two that were downregulated. Among the identified
miRNAs, miR-23b-3p demonstrated consistent up regulation in three
studies. Ham et al (33)
demonstrated that miR-23b-3p over expression induced chondrogenic
differentiation by downregulating protein kinase A (PKA) signaling
based on targeting protein kinase cAMP-activated catalytic subunit
β, which encodes a catalytic subunit of PKA in synovial
fluid-derived mesenchymal stem cells (33,34).
In 2015, Gabler et al identified that miR-23b-3p was
expressed at significantly higher levels in hypertrophic
chondrocytes (35). As to
miR-16-5p, although the function of miR-16-5p in chondrocyte has
yet to be determined, it is present at high levels in the majority
of cell types and proposed to be potentially a master miRNA
involved in determining mRNA stability via adenylate-uridylate-rich
element sites (36). Murata et
al identified that synovial fluid miR-16-5p levels of
rheumatoid arthritis (RA) patients were higher than those of OA
patients, and plasma miR-16-5p levels of OA patients were lower
than that of healthy control subjects (37). Certain studies identified that the
expression levels of miR-16-5p were decreased in the sera of early
RA patients in comparison with advanced RA, which indicate
miR-16-5p as a possible predictor for RA (38). miR-16-5p was reported to be over
expressed in peripheral blood mononuclear cells of RA patients who
were active when compared with RA patients who were relatively
static, or with healthy control subjects (39). In the current meta-analysis,
miR-23b-3p was significantly increased in OA cartilage tissue.
These results indicate that miR-23b-3p and miR-16-5p are potential
molecular markers for diagnostics, therapeutics and prognosis of
OA. However, further investigation of these miRNAs in OA is
considered to be necessary.
miR-149-5p was identified to be downregulated in OA
chondrocytes in the study by Santini et al (40), and this decrease was correlated
with increased expression levels of pro-inflammatory cytokines,
such as tumor necrosis factor-α, interleukin (IL)-β, and IL-6.
Furthermore, miR-149-5p was significantly decreased in the current
meta-analysis, and these results supported the hypothesis that
miR-149-5p is an important regulatory molecule in the progression
of OA.
The pathway enrichment analysis identified that many
signaling pathways are involved in the regulation of miRNAs.
Through the KEGG pathway analysis (Table III), the Wnt signaling pathway
was identified to be significantly associated with development of
chondrocytes, which are involved in the maintenance of fully
differentiated chondrocyte phenotypes, and may, therefore, be
crucial in cartilage homeostasis throughout life (41).
There are certain limitations associated with the
meta-analysis on miRNA expression profiles. The included studies
depended on a variety of microarray platforms and validation
criteria. Hence, it is important to apply a more objective and
comprehensive methodological standardization for maximum
reliability. Three of the included studies in the current
meta-analysis involved Asian subjects, three involved European
subjects and the other two involved North America subjects, which
could explain the inconsistency of miRNA expression directions.
Therefore, future studies that validate profiling results in the
same ethnicities may provide improved results. In addition, certain
studies did not confirm whether their microarray results were in
vitro or in vivo, nor did they assess the prognostic
association between miRNAs and OA, which is important to validate
OA biomarkers. Furthermore, the inconsistent profiling results of
miRNA expression maybe due to various potential reasons, including
differences in samples, the genetic and environmental background of
tissue donors, the clinical pathological characteristics of tissue
donors, and the expression profiling platforms.
In conclusion, six significant dysregulated miRNAs
were identified across eight independent studies in OA. The
meta-signature miRNAs and associated pathways may present as
promising markers for clinical intervention. Further investigations
should focus upon the molecular mechanisms that miRNAs may exert in
the occurrence and progression of OA.
Acknowledgements
This study was funded by grants from the National
Natural Science Foundation of China (grant no. 81472924).
References
|
1
|
Taylor AM: Metabolic and endocrine
diseases, cartilage calcification and arthritis. Curr Opin
Rheumatol. 25:198–203. 2013. View Article : Google Scholar
|
|
2
|
Bian Q, Wang YJ, Liu SF and Li YP:
Osteoarthritis: Genetic factors, animal models, mechanisms, and
therapies. Front Biosci (Elite Ed). 4:74–100. 2012. View Article : Google Scholar
|
|
3
|
Loeser RF, Goldring SR, Scanzello CR and
Goldring MB: Osteoarthritis: A disease of the joint as an organ.
Arthritis Rheum. 64:1697–1707. 2012. View Article : Google Scholar :
|
|
4
|
Cicuttini FM and Spector TD: Genetics of
osteoarthritis. Ann Rheum Dis. 55:665–667. 1996. View Article : Google Scholar :
|
|
5
|
Spector TD, Cicuttini F, Baker J, Loughlin
J and Hart D: Genetic influences on osteoarthritis in women: A twin
study. BMJ. 312:940–943. 1996. View Article : Google Scholar :
|
|
6
|
Kraus VB, Jordan JM, Doherty M, Wilson AG,
Moskowitz R, Hochberg M, Loeser R, Hooper M, Renner JB, Crane MM,
et al: The genetics of generalized osteoarthritis (GOGO) study:
Study design and evaluation of osteoarthritis phenotypes.
Osteoarthritis Cartilage. 15:120–127. 2007. View Article : Google Scholar
|
|
7
|
Tsezou A: Osteoarthritis year in review
2014: Genetics and genomics. Osteoarthritis Cartilage.
22:2017–2024. 2014. View Article : Google Scholar
|
|
8
|
Berezikov E: Evolution of microRNA
diversity and regulation in animals. Nat Rev Genet. 12:846–860.
2011. View
Article : Google Scholar
|
|
9
|
Jingsheng S, Yibing W, Jun X, Siqun W,
Jianguo W, Feiyan C, Gangyong H and Jie C: MicroRNAs are potential
prognostic and therapeutic targets in diabetic osteoarthritis. J
Bone Miner Metab. 33:1–8. 2015. View Article : Google Scholar
|
|
10
|
Baxter D, McInnes IB and
Kurowska-Stolarska M: Novel regulatory mechanisms in inflammatory
arthritis: A role for microRNA. Immunol Cell Biol. 90:288–292.
2012. View Article : Google Scholar
|
|
11
|
Hong E and Reddi AH: MicroRNAs in
chondrogenesis, articular cartilage, and osteoarthritis:
Implications for tissue engineering. Tissue Eng Part B Rev.
18:445–453. 2012. View Article : Google Scholar
|
|
12
|
Le LT, Swingler TE and Clark IM: Review:
The role of microRNAs in osteoarthritis and chondrogenesis.
Arthritis Rheum. 65:1963–1974. 2013. View Article : Google Scholar
|
|
13
|
Griffith OL, Melck A, Jones SJ and Wiseman
SM: Meta-analysis and meta-review of thyroid cancer gene expression
profiling studies identifies important diagnostic biomarkers. J
Clin Oncol. 24:5043–5051. 2006. View Article : Google Scholar
|
|
14
|
Guan P, Yin Z, Li X, Wu W and Zhou B:
Meta-analysis of human lung cancer microRNA expression profiling
studies comparing cancer tissues with normal tissues. J Exp Clin
Cancer Res. 31:542012. View Article : Google Scholar :
|
|
15
|
Li MY and Hu XX: Meta-analysis of microRNA
expression profiling studies in human cervical cancer. Med Oncol.
32:5102015. View Article : Google Scholar
|
|
16
|
Grimson A, Farh KK, Johnston WK,
Garrett-Engele P, Lim LP and Bartel DP: MicroRNA targeting
specificity in mammals: Determinants beyond seed pairing. Mol Cell.
27:91–105. 2007. View Article : Google Scholar :
|
|
17
|
Blin K, Dieterich C, Wurmus R, Rajewsky N,
Landthaler M and Akalin A: DoRiNA 2.0-upgrading the doRiNA database
of RNA interactions in post-transcriptional regulation. Nucleic
Acids Res. 43:(Database issue). D160–D167. 2015. View Article : Google Scholar
|
|
18
|
Wong N and Wang X: miRDB: An online
resource for microRNA target prediction and functional annotations.
Nucleic Acids Res. 43:(Database issue). D146–D152. 2015. View Article : Google Scholar
|
|
19
|
Vlachos IS, Paraskevopoulou MD, Karagkouni
D, Georgakilas G, Vergoulis T, Kanellos I, Anastasopoulos IL,
Maniou S, Karathanou K, Kalfakakou D, et al: DIANA-TarBase v7.0:
Indexing more than half a million experimentally supported
miRNA:mRNA interactions. Nucleic Acids Res. 43:(Database issue).
D153–D159. 2015. View Article : Google Scholar
|
|
20
|
Yang JH, Li JH, Shao P, Zhou H, Chen YQ
and Qu LH: starBase: A database for exploring microRNA-mRNA
interaction maps from Argonaute CLIP-Seq and Degradome-Seq data.
Nucleic Acids Res. 39:(Database issue). D202–D209. 2011. View Article : Google Scholar
|
|
21
|
da Huang W, Sherman BT and Lempicki RA:
Bioinformatics enrichment tools: Paths toward the comprehensive
functional analysis of large gene lists. Nucleic Acids Res.
37:1D–13D. 2009. View Article : Google Scholar
|
|
22
|
da Huang W, Sherman BT and Lempicki RA:
Systematic and integrative analysis of large gene lists using DAVID
bioinformatics resources. Nat Protoc. 4:44–57. 2009. View Article : Google Scholar
|
|
23
|
Iliopoulos D, Malizos KN, Oikonomou P and
Tsezou A: Integrative microRNA and proteomic approaches identify
novel osteoarthritis genes and their collaborative metabolic and
inflammatory networks. PLoS One. 3:e37402008. View Article : Google Scholar :
|
|
24
|
Jones SW, Watkins G, Le Good N, Roberts S,
Murphy CL, Brockbank SM, Needham MR, Read SJ and Newham P: The
identification of differentially expressed microRNA in
osteoarthritic tissue that modulate the production of TNF-alpha and
MMP13. Osteoarthritis Cartilage. 17:464–472. 2009. View Article : Google Scholar
|
|
25
|
Miyaki S, Nakasa T, Otsuki S, Grogan SP,
Higashiyama R, Inoue A, Kato Y, Sato T, Lotz MK and Asahara H:
MicroRNA-140 is expressed in differentiated human articular
chondrocytes and modulates interleukin-1 responses. Arthritis
Rheum. 60:2723–2730. 2009. View Article : Google Scholar :
|
|
26
|
Diaz-Prado S, Cicione C, Muiños-López E,
Hermida-Gómez T, Oreiro N, Fernández-López C and Blanco FJ:
Characterization of microRNA expression profiles in normal and
osteoarthritic human chondrocytes. BMC Musculoskelet Disord.
13:1442012. View Article : Google Scholar :
|
|
27
|
Song J, Kim D, Lee CH, Lee MS, Chun CH and
Jin EJ: MicroRNA-488 regulates zinc transporter SLC39A8/ZIP8 during
pathogenesis of osteoarthritis. J Biomed Sci. 20:312013. View Article : Google Scholar :
|
|
28
|
Li YH, Tavallaee G, Tokar T, Nakamura A,
Sundararajan K, Weston A, Sharma A, Mahomed NN, Gandhi R, Jurisica
I and Kapoor M: Identification of synovial fluid microRNA signature
in knee osteoarthritis: Differentiating early- and late-stage knee
osteoarthritis. Osteoarthritis Cartilage. 24:1577–1586. 2016.
View Article : Google Scholar
|
|
29
|
Beyer C, Zampetaki A, Lin NY, Kleyer A,
Perricone C, Iagnocco A, Distler A, Langley SR, Gelse K, Sesselmann
S, et al: Signature of circulating microRNAs in osteoarthritis. Ann
Rheum Dis. 74:e182015. View Article : Google Scholar
|
|
30
|
Nakamura A, Rampersaud YR, Sharma A, Lewis
SJ, Wu B, Datta P, Sundararajan K, Endisha H, Rossomacha E, Rockel
JS, et al: Identification of microRNA-181a-5p and microRNA-4454 as
mediators of facet cartilage degeneration. JCI Insight.
1:e868202016. View Article : Google Scholar :
|
|
31
|
Kolde R, Laur S, Adler P and Vilo J:
Robust rank aggregation for gene list integration and
meta-analysis. Bioinformatics. 28:573–580. 2012. View Article : Google Scholar :
|
|
32
|
Ma MZ, Kong X, Weng MZ, Cheng K, Gong W,
Quan ZW and Peng CH: Candidate microRNA biomarkers of pancreatic
ductal adenocarcinoma: Meta-analysis, experimental validation and
clinical significance. J Exp Clin Cancer Res. 32:712013. View Article : Google Scholar :
|
|
33
|
Ham O, Lee CY, Song BW, Lee SY, Kim R,
Park JH, Lee J, Seo HH, Lee CY, Chung YA, et al: Upregulation of
miR-23b enhances the autologous therapeutic potential for
degenerative arthritis by targeting PRKACB in synovial
fluid-derived mesenchymal stem cells from patients. Mol Cells.
37:449–456. 2014. View Article : Google Scholar :
|
|
34
|
Mirzamohammadi F, Papaioannou G and
Kobayashi T: MicroRNAs in cartilage development, homeostasis, and
disease. Curr Osteoporos Rep. 12:410–419. 2014. View Article : Google Scholar :
|
|
35
|
Gabler J, Ruetze M, Kynast KL, Grossner T,
Diederichs S and Richter W: Stage-Specific miRs in chondrocyte
maturation: Differentiation-dependent and hypertrophy-related miR
clusters and the miR-181 family. Tissue Eng Part A. 21:2840–2851.
2015. View Article : Google Scholar
|
|
36
|
Asirvatham AJ, Magner WJ and Tomasi TB:
miRNA regulation of cytokine genes. Cytokine. 45:58–69. 2009.
View Article : Google Scholar :
|
|
37
|
Murata K, Yoshitomi H, Tanida S, Ishikawa
M, Nishitani K, Ito H and Nakamura T: Plasma and synovial fluid
microRNAs as potential biomarkers of rheumatoid arthritis and
osteoarthritis. Arthritis Res Ther. 12:R862010. View Article : Google Scholar :
|
|
38
|
Filková M, Aradi B, Senolt L, Ospelt C,
Vettori S, Mann H, Filer A, Raza K, Buckley CD, Snow M, et al:
Association of circulating miR-223 and miR-16 with disease activity
in patients with early rheumatoid arthritis. Ann Rheum Dis.
73:1898–1904. 2014. View Article : Google Scholar
|
|
39
|
Pauley KM, Satoh M, Chan AL, Bubb MR,
Reeves WH and Chan EK: Upregulated miR-146a expression in
peripheral blood mononuclear cells from rheumatoid arthritis
patients. Arthritis Res Ther. 10:R1012008. View Article : Google Scholar :
|
|
40
|
Santini P, Politi L, Vedova PD, Scandurra
R and d'Abusco Scotto A: The inflammatory circuitry of miR-149 as a
pathological mechanism in osteoarthritis. Rheumatol Int.
34:711–716. 2014. View Article : Google Scholar
|
|
41
|
Ma B, Landman EB, Miclea RL, Wit JM,
Robanus-Maandag EC, Post JN and Karperien M: WNT signaling and
cartilage: Of mice and men. Calcif Tissue Int. 92:399–411. 2013.
View Article : Google Scholar
|