Introduction
The dynamic balance between cell proliferation and
apoptosis is of importance in the maintenance of homeostasis
(1–3). Once the balance is destroyed, cell
apoptosis occurs, resulting in secondary damage. It has been
verified that various genes are associated with cell apoptosis
(4). B cell lymphoma (Bcl)-2
associated X, apoptosis regulator (Bax) (5), p53 (6,7), and
Fas (8,9) promote cell apoptosis, whereas Bcl-2
(5,10) and MYC Proto-Oncogene, BHLH
Transcription Factor (myc) (11–13)
inhibit cell apoptosis.
It has previously been demonstrated that tumor
resistance to chemotherapy is closely associated with inactivation
of apoptotic pathways (14–16).
Therefore, a better understanding of the molecular mechanisms
underlying tumor resistance is helpful to predict responses to
drugs and assist in the design of tailored therapeutic regimens to
overcome drug resistance.
Apoptosis is a well-organized process of programmed
cell death. It may be initiated either by activation of death
receptors on the cell surface membranes (extrinsic pathway)
(17) or through a series of
cellular events primarily processed in the mitochondria (intrinsic
pathway) (18).
Previous studies have demonstrated that cell
apoptosis is important to tumorigenesis (19–21).
Defects in cell apoptosis result in a population expansion of
neoplastic cells. Chemotherapy or radiotherapy-induced tumor cell
death is largely mediated by the activation of apoptosis, and the
inhibition of apoptosis enables the tumor cells to become resistant
to these treatments.
In the present study, the inhibitory effects of
gemcitabine (GEM), a commonly used therapeutic reagent in clinic,
on the proliferation and induction of apoptosis of the human
esophageal cancer cell line Eca-109, were assayed. Furthermore, the
morphological alterations in the treated cancer cells were observed
under a transmission electron microscope (TEM). Two-dimensional gel
electrophoresis (2-DE), combined with matrix-assisted laser
desorption/ionization time of flight mass spectrometry
(MALDI-TOF-MS) were used to validate the differentially expressed
proteins in the treated and non-treated Eca-109 cells. Western
blotting was then used to quantify the differential proteins in the
treated cancer cells. The present study therefore aimed to clarify
the primary targets of GEM in the Eca-109 cells.
Materials and methods
Cell line and culture conditions
Human esophageal cancer cell line Eca-109 was
provided by the Cell Resource Center of Shanghai Life Sciences
Institute, Chinese Academy of Sciences (Shanghai, China). The cells
were cultured in Roswell Park Memorial Institute (RPMI)-1640 medium
(Thermo Fisher Scientific, Inc., Waltham, MA, USA), supplemented
with 10% fetal bovine serum (Zhejiang Kangchen Biotech Co., Ltd.,
Wuhan, China), at 37°C in an atmosphere containing 5%
CO2. When the cells reached a confluence of ~90% (~every
three days), they were digested and passaged. The cells in passages
3–5 were used for experimental analyses.
MTT assay
The Eca-109 cells in logarithmic phase were prepared
to a single cell suspension (2×104/ml) and 100 µl of the
cells were added in 96-well culture plates. Then, 50 µl of GEM
(≥98%, high performance liquid chromatography; Sigma-Aldrich; Merck
KGaA, Darmstadt, Germany) was added in the wells to reach various
final concentrations (1, 2, 4, 8, 16 µg/ml). Next, the treated
cells were cultured in an atmosphere containing 5% CO2
at 37°C for 24 or 48 h. Following this, the cells were collected
and centrifuged at (150 × g) for 10 min. The supernatant was
discarded, and the precipitate was washed with PBS once. A total of
200 µl of serum-free RPMI-1640 medium was added, followed by
addition of 10 µl of MTT (5 mg/ml). Following culturing for a
further 4 h, 100 µl of dimethyl sulfoxide solution was added to
dissolve the formazan. The cell viability was assessed by measuring
the optical density (OD) at a wavelength of 450 nm in a Multiskan
SPECTRUM full spectrum microplate spectrophotometer (Thermo Fisher
Scientific, Inc., Waltham, MA, USA). The untreated wells also
served as a control. Data from triplicate samples were averaged.
The cell inhibition
rate=(ODcontrol-ODtreated)/ODcontrol
× 100%.
Analysis of cell apoptosis
The cells were seeded at a concentration of
2×104 cells/ml, then cells were treated with GEM (4
µg/ml). After co-cultured for 12 and 24 h, the cells were removed
from the plates by using 0.25% trypsin. The cells were centrifuged
at 150 × g for 10 min and then collected. Following washing with
PBS twice, 1×105−5×105 cells were resuspended
in 500 µl binding buffer and sequentially mixed with 5 µl of
Annexin V-FITC (Nanjing KeyGEN Biotech Co., Ltd., Nanjing, China)
in the dark for 15 min at room temperature, followed by 5 µl of
propidium iodide (PI) (Nanjing KeyGen Biotech Co., Ltd., Nanjing,
China) for 5 min. The cells were incubated at room temperature in
the dark for 5–10 min. Apoptotic cells were detected using a flow
cytometer (Beckman Coulter, Inc., Brea, CA, USA) with an excitation
wavelength at 488 nm and an emission wavelength at 530 nm and
observed using a fluorescence microscope (Olympus Corporation,
Tokyo, Japan). The data was analyzed using kaluza version 1.20
software (Beckman Coulter, Inc.).
Terminal
deoxynucleotidyl-transferase-mediated dUTP nick-end labeling
(TUNEL) assay
The treated cells were seeded in 24-well plates
(2×105/ml). Following a culture period of 72 h, the
cells were fixed using 3.7% neutral formalin for 10 min at room
temperature. Then, they were rehydrated with a descending gradient
of ethanol (100, 95, and 70%, 5 min each). Then, they were washed
with PBS for 10 min, and 50 µl of cell suspension was added on a
poly-l-lysine-pretreated slide. A total of 0.01 M PBS was then used
to wash the cells, followed by the TdT labeling at room temperature
for 5 min. The reaction was stopped with a stop buffer at room
temperature for 5 min, followed by an incubation with 50 µl of
FITC-labeled antibody (cat.: 129–10684; R&D Systems Inc.,
Minneapolis, MN, USA) at room temperature for 30 min, and then
washed with PBS twice. The cell nuclei were examined under a laser
scanning confocal microscope (at 490 nm excitation wavelength and
520 nm emission wavelength). A total of 20 random fields of view
were selected for analysis.
Morphology assay of treated Eca-109
cells
The cells were treated with GEM (8 µg/ml) as
previously described. Then, the cells were collected and prepared
into specimens ~3.0×1.0 mm, and then fixed in 3% glutaraldehyde and
osmic acid for 24 h at 4°C. These specimens were dehydrated using
graded ethanol at 37°C for 30 min, followed by embedding in epoxy
resin. Then, the specimens were cut into 50 nm-thick sections
consecutively. They were doubly stained with 2% uranyl acetate and
lead citrate for 12 h at room temperature. The sections were washed
and then the alterations in endocardium, nucleus, cytoplasm, and
matrix components were examined using a HT7700 transmission
electron microscope (Hitachi, Ltd., Tokyo, Japan).
Two-dimensional electrophoresis (2-DE)
assay of differentially expressed proteins in the Eca-109
cells
The cells were treated as previously described.
Following a culture period of 24 h, the cells were harvested and
collected. Then, a cell lysis solution was added containing 8 mol/l
urea, 40 g/l CHAPS, 2 mmol/l TBP, and 2 ml/l Bio-Lyte. The cell
lysates were collected and centrifuged at 13,400 × g at 4°C for 15
min. The supernatant was then harvested and the proteins were
quantified using the Bradford method.
The protein samples (~400 µg) were subjected to
immobilized pH gradients (IPG) isoelectric focusing (IEF) and then
run on sodium dodecyl sulfate polyacrylamide gel electrophoresis
(SDS-PAGE). Next, the proteins were isolated by vertical
electrophoresis. The gel was stained with coomassie brilliant blue
R-250 for 2 h and destained for 2 h until the protein spots were
clear. The stained proteins were scanned using a FXMolecular Imager
(Bio-Rad Laboratories Inc., Hercules, CA, USA).
The differential protein spots (>than 3-fold
alteration in OD) were cut off from the gel and digested with 0.25%
trypsin for 20 h. The digested peptide fragments were isolated and
desalinated with ZipTipTM (EMD Millipore, Billerica, MA, USA).
Amino acid sequence analysis was then performed. According to the
mass of the digested peptide fragment, the Mascot score was
automatically obtained to verify the potential characteristics and
names of the differential proteins by checking the Swiss-Prot
protein database.
Western blotting of the differential
proteins
The cells were treated and lysed using the
aforementioned procedure. The proteins were separated by 12%
SDS-PAGE and then transferred onto a sheet of polyvinylidene
fluoride membrane. Following blocking with 5% skim milk for 4 h and
washing with Tris-buffered saline (TBS ×3, 5 min), the membrane was
respectively incubated overnight with anti-human ASC (cat no.
sc-33958), Mcl-1(cat no. sc-53951) and Bax-a (cat no. sc-70408)
polyclonal antibody (1:200; Santa Cruz Biotechnology, Inc., Dallas,
TX, USA) at 4°C, followed by incubation with horseradish
peroxidase-conjugated rabbit anti-goat IgG (cat no. TA130028;
1:3,000; Origene technologies Inc., Beijing, China) for 2 h at room
temperature. Immunoreactive bands were detected by an enhanced
chemiluminescence reagent (Pierce; Thermo Fisher Scientific, Inc.),
visualized by autoradiography, and quantified by the QuantityOne
analysis system (Bio-Rad Laboratories, Inc.). β-actin (primary
antibody dilution 1:200; cat no. sc-130656; Santa Cruz
Biotechnology, Inc.) served as an internal control.
Statistical analysis
Data are expressed as the mean ± standard error of
the mean, and statistical comparisons were carried out by Student's
t-test or one-way analysis of variance followed by Tukey's post hoc
test, with the SPSS statistical software, version 17.0 (SPSS Inc.,
Chicago, IL, USA). P<0.05 was considered to indicate a
statistically significant difference.
Results
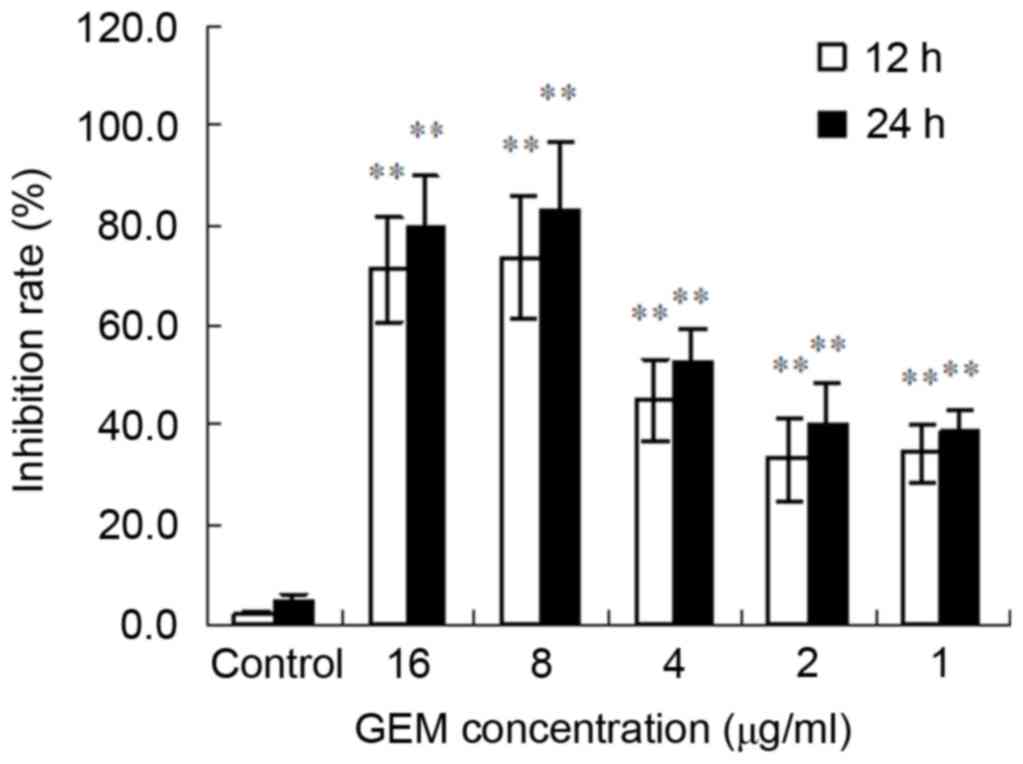
GEM inhibits proliferation of Eca-109
cells
The MTT results demonstrated that GEM significantly
inhibited the proliferation of Eca-109 cells. Furthermore, the
inhibitory effect was observed to have been exhibited in a time-
and concentration-dependent manner. The greatest inhibitory effect
of GEM was at 8 µg/ml for 24 h (Fig.
1).
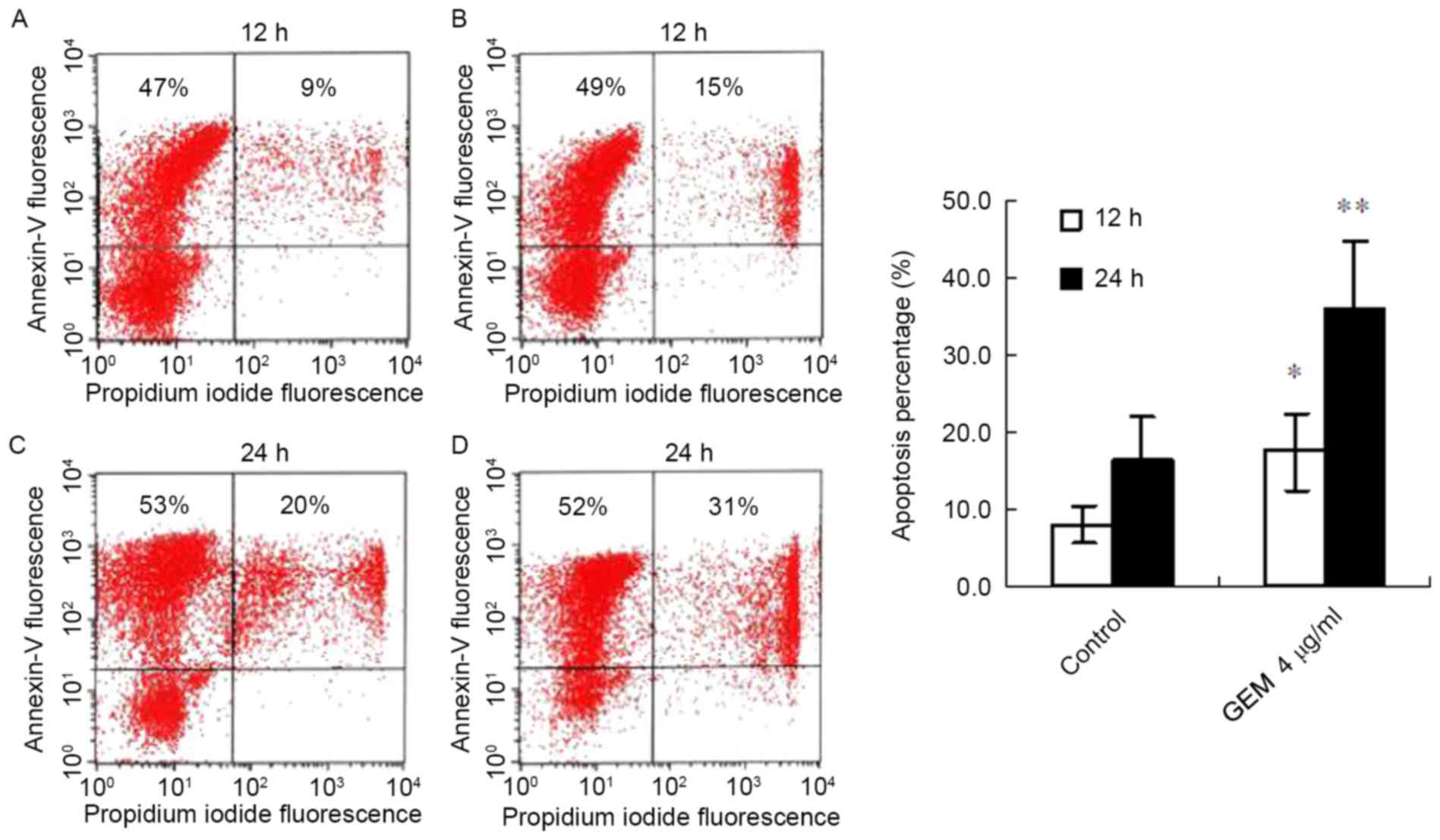
GEM induces apoptosis of Eca-109
cells
Following treatment with GEM (4 µg/ml) for 12 h, the
apoptosis proportion (the cells doubly stained with AnnexinV-FITC
and PI represent apoptotic cells) of the cancer cells significantly
increased compared with the control (17.5±5.1% vs. 8.1±2.4%,
P<0.05; Fig. 2A and B). With
the prolongation of the treatment time, the apoptosis proportion
was significantly elevated in the treated cancer cells. The FACS
result demonstrated that the apoptosis proportion in the
GEM-treated cells was increased compared with controls (36.1±8.8%
vs. 16.4±5.8%, P<0.01; Fig. 2C and
D).
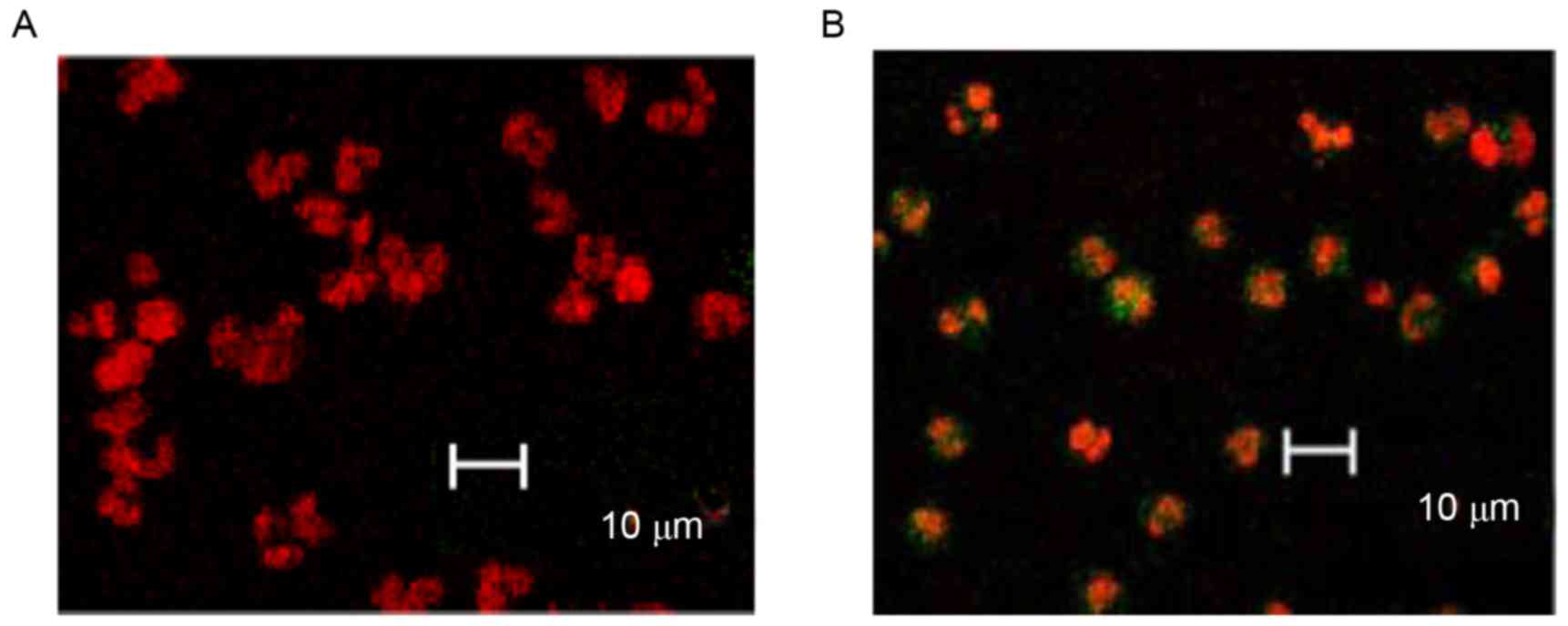
Furthermore, TUNEL combined with laser scanning
confocal microscope observations, demonstrated that GEM (4 µg/ml)
significantly induced the apoptosis of the cancer cells. The
majority of control cells were only stained with PI and few with
green fluorescence (Fig. 3A),
whereas the apoptotic cells (AnnexinV-FITC and PI double staining)
were stained with green fluorescence (Fig. 3B). In addition, nuclear staining
density of the untreated cells was lower and the nuclear shapes
were larger (Fig. 3A), whereas the
treated cells had dense chromatins and comparatively smaller
nuclear shapes (Fig. 3B).
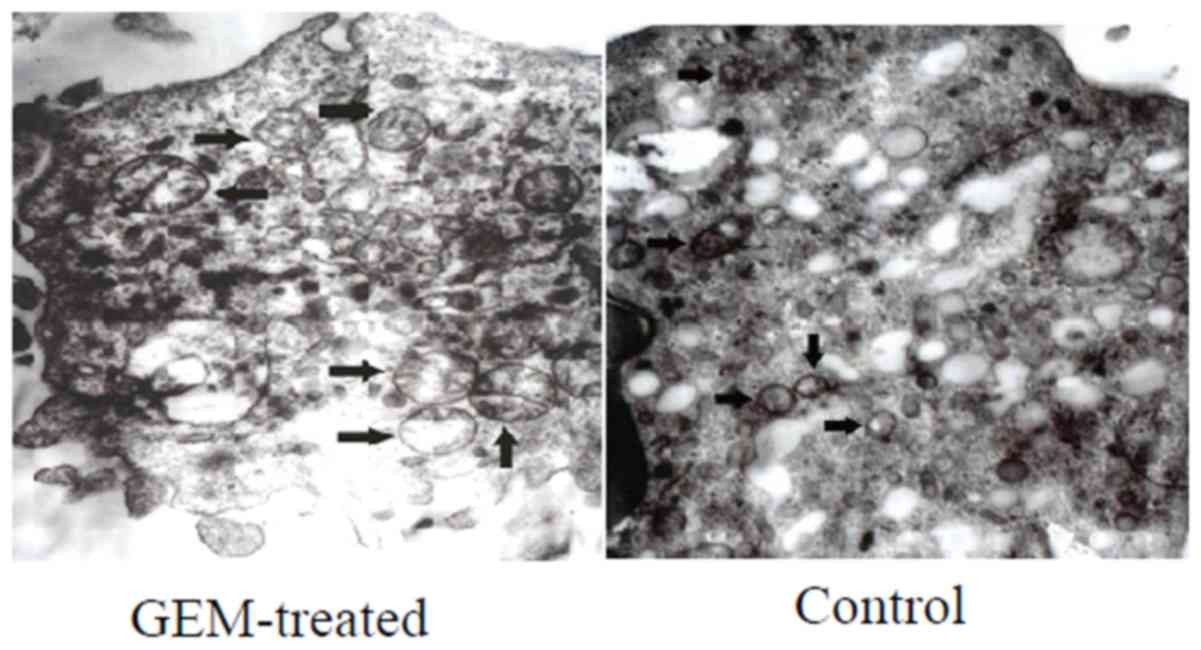
The alterations in the mitochondrial ultrastructure
of the treated Eca-109 cells were notable (Fig. 4). Following treatment with GEM for
24 h, swelling mitochondria, fading matrix and a lack of
mitochondrial crista were observed in the Eca-109 cells (Fig. 4).
Assay of the differential proteins in
Eca-109 cells
The 2-DE assay result demonstrated that there were
12 protein spots between the acidic region (pH 4.7–6.5). These
protein spots were cut off and digested with trypsin (Fig. 5). The MALDI-TOF-MS assay was
performed in the 12 protein spots to gain access to peptide
fingerprints and charge to mass ratio. Then, three differentially
expressed proteins including spots 3, 7 and 12 (Fig. 5) were identified by Mascot and a
ProteinProspector peptide fingerprint matching software. The three
differential proteins were validated to be Bax-α (spot 3), myeloid
cell leukemia sequence (Mcl-1; spot 7), and apoptosis-associated
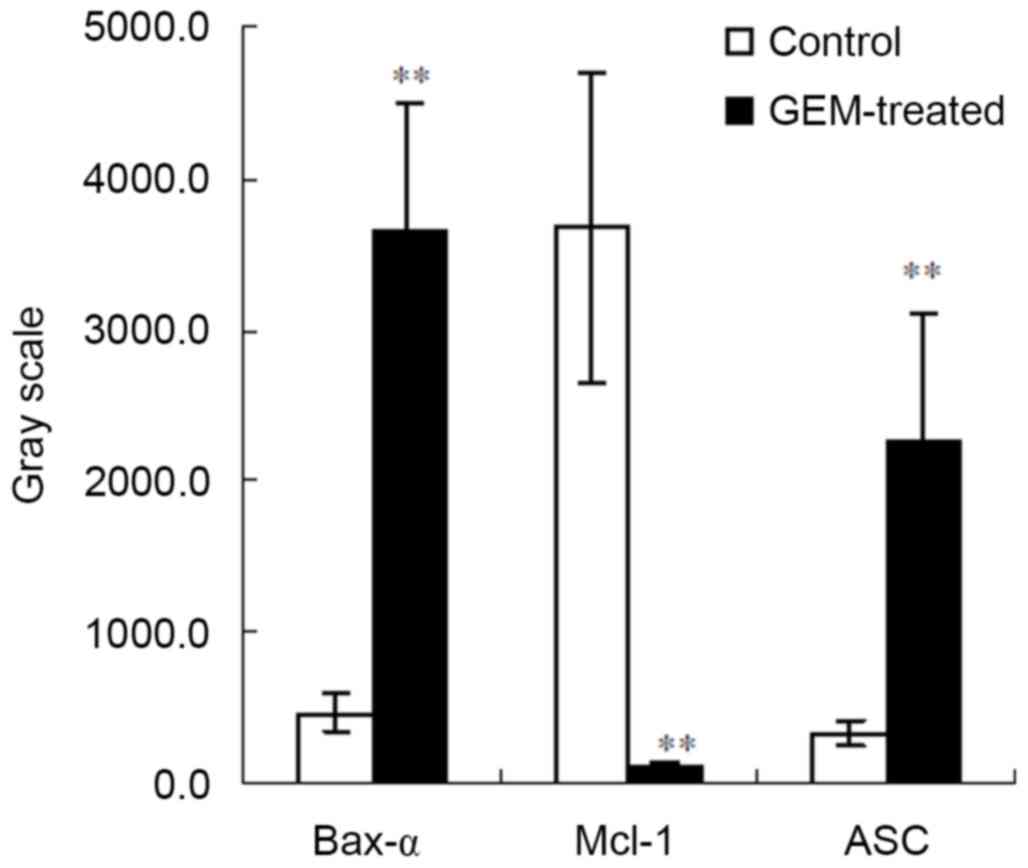
speck-like protein containing a CARD (ASC) (spot 12). The gray
scales were increased in Bax-α and ASC and decreased in Mcl-1 in
the treated cells compared with the controls (P<0.01; Fig. 6).
GEM upregulates Bax-α and ASC and
downregulates Mcl-1 expression levels in Eca-109 cells
The western blotting result demonstrated that Bax-α
and ASC protein levels increased in the treated cells at 12 and 24
h following the GEM treatment (Fig.
7). However, the Mcl-1 protein levels in the treated cells
significantly decreased compared with the control (Fig. 7)
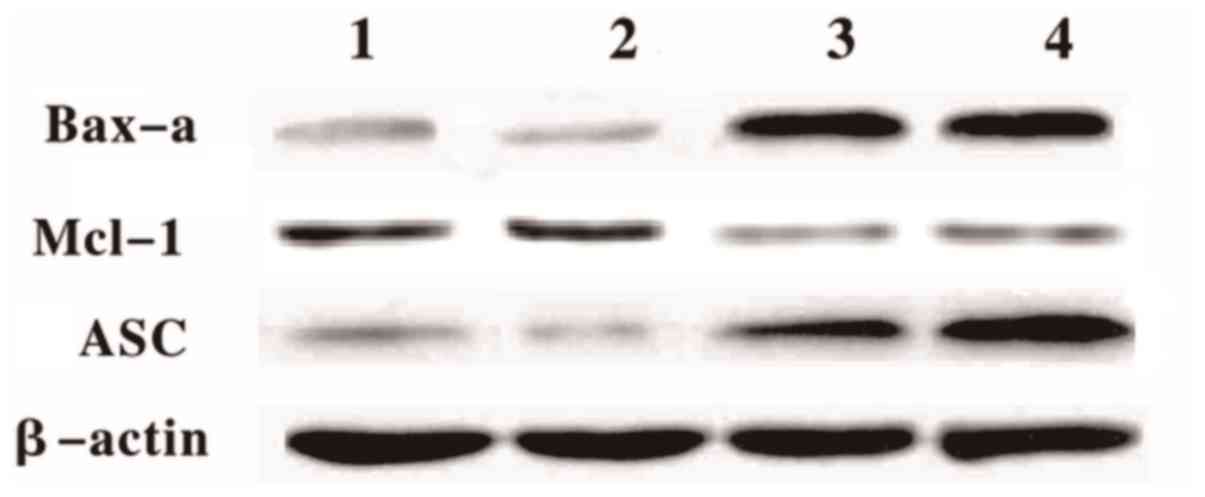 | Figure 7.Differential protein expression levels
in treated and untreated cells. Western blotting analyses of Bax-α,
ASC, and Mcl-1 protein expression levels in the Eca-109 cells. Lane
1, Eca-109 untreated (12 h); Lane 2, Eca-109 untreated (24 h); Lane
3, Eca-109 + GEM (12 h); Lane 4, Eca-109 + GEM (24 h). GEM,
gemcitabine Bax-α, B cell lymphoma-2 associated X, apoptosis
regulator; ASC, apoptosis-associated speck-like protein containing
a CARD; Mcl-1, myeloid cell leukemia sequence. |
Discussion
Esophageal carcinoma is one of the most frequently
occurring cancers worldwide. According to the data from World
Cancer Research Fund, the incidence of esophageal cancer is the
seventh greatest cause of mortality in the world and the survival
rate is low.
Chemotherapy is one of the palliative methods for
the treatment of esophageal cancer. GEM, a deoxycytidine antitumor
drug, has previously been demonstrated to exhibit anti-tumor
properties against solid tumors (22–24).
However, tumor resistance to GEM is becoming a primary issue of
concern affecting chemotherapy. Studies on genesis in the
development and prognosis of esophageal cancer provided certain
evidence for the association between chemotherapeutic sensitivity
and molecular targets (25,26).
The results of the present study demonstrated that
GEM inhibited the proliferation of the esophageal cancer cells in a
time-and concentration-dependent manner. In other tumors, Toyota
et al (27) demonstrated
that GEM inhibited cell cycle progression in HuCCT-1 cells from
G0/G1 to S phase, which resulted in
G1 cell cycle arrest for decreased expression of cyclin
D1.
In addition, a study revealed that the level of
ribonucleotide reductase subunit M1 (RRM1) is correlated with the
therapeutic sensitivity to GEM. Cancer cells expressing a lower
RRM1 level are more sensitive to GEM (28). The present study indicated that GEM
significantly induced cell apoptosis, and this was supported by the
TEM result. Following this, 2-DE combined with MALDI-TOF-MS was
used to assay the differentially expressed proteins in the treated
and untreated Eca-109 cells. Results demonstrated that there were
three differentially expressed proteins including Bax-α, Mcl-1 and
ASC in the treated Eca-109 cells compared with controls.
The Bcl-2 family is a group of apoptosis-associated
genes, which have an important role in inhibiting or promoting cell
apoptosis. Bax-α and Mcl-1 are important members of the Bcl-2
family. It has previously been demonstrated that Mcl-1 exhibits an
inhibitory role, and Bax-α exhibits the opposite effect in
apoptosis. In the present study, the western blotting results
revealed that GEM significantly upregulated ASC and Bax-α protein
expression levels. As a receptor of Bax, ASC exhibits its role via
the p53-Bax mitochondrial apoptotic pathway (29). ASC is a connexin containing caspase
recruitment domain, termed CARD, and contains a pyrin domain,
termed PYD. It is located on human chromosome 16pl1.2–12. The CARD
and PYD belong to the death domain family and exhibit key roles in
cell apoptosis, inflammation, mediated immunity and tumor formation
(30,31).
In conclusion, the results of the present study
demonstrated that GEM inhibited the growth and promoted the
apoptosis of the Eca-109 cells. The alterations in the levels of
various differentially expressed proteins, including ASC, Mcl-1 and
Bax-α, were responsible for this effect.
References
|
1
|
Schutte B and Ramaekers FC: Molecular
switches that govern the balance between proliferation and
apoptosis. Prog Cell Cycle Res. 4:207–217. 2000. View Article : Google Scholar
|
|
2
|
Jaleco S, Swainson L, Dardalhon V,
Burjanadze M, Kinet S and Taylor N: Homeostasis of naive and memory
CD4+ T cells: IL-2 and IL-7 differentially regulate the balance
between proliferation and Fas-mediated apoptosis. J Immunol.
171:61–68. 2003. View Article : Google Scholar
|
|
3
|
Mommers EC, van Diest PJ, Leonhart AM,
Meijer CJ and Baak JP: Balance of cell proliferation and apoptosis
in breast carcinogenesis. Breast Cancer Res Treat. 58:163–169.
1999. View Article : Google Scholar
|
|
4
|
Colitti M, Stefanon B and Wilde CJ:
Apoptotic cell death, bax and bcl-2 expression during sheep mammary
gland involution. Anat Histol Embryol. 28:257–264. 1999. View Article : Google Scholar
|
|
5
|
Heermeier K, Benedict M, Li M, Furth P,
Nuñez G and Hennighausen L: Bax and Bcl-xs are induced at the onset
of apoptosis in involuting mammary epithelial cells. Mech Dev.
56:197–207. 1996. View Article : Google Scholar
|
|
6
|
Kastan MB, Canman CE and Leonard CJ: P53,
cell cycle control and apoptosis: Implications for cancer. Cancer
Metastasis Rev. 14:3–15. 1995. View Article : Google Scholar
|
|
7
|
Leonard CJ, Canman CE and Kastan MB: The
role of p53 in cell-cycle control and apoptosis: Implications for
cancer. Important Adv Oncol. 33–42. 1995.
|
|
8
|
Van Parijs L, Ibraghimov A and Abbas AK:
The roles of costimulation and Fas in T cell apoptosis and
peripheral tolerance. Immunity. 4:321–328. 1996. View Article : Google Scholar
|
|
9
|
Akiyama K, Chen C, Wang D, Xu X, Qu C,
Yamaza T, Cai T, Chen W, Sun L and Shi S:
Mesenchymal-stem-cell-induced immunoregulation involves
FAS-ligand-/FAS-mediated T cell apoptosis. Cell Stem Cell.
10:544–555. 2012. View Article : Google Scholar :
|
|
10
|
Schorr K, Li M, Krajewski S, Reed JC and
Furth PA: Bcl-2 gene family and related proteins in mammary gland
involution and breast cancer. J Mammary Gland Biol Neoplasia.
4:153–164. 1999. View Article : Google Scholar
|
|
11
|
Evan G, Harrington E, Fanidi A, Land H,
Amati B and Bennett M: Integrated control of cell proliferation and
cell death by the c-myc oncogene. Philos Trans R Soc Lond B Biol
Sci. 345:269–275. 1994. View Article : Google Scholar
|
|
12
|
Hoffman B and Liebermann DA: The
proto-oncogene c-myc and apoptosis. Oncogene. 17:3351–3357. 1998.
View Article : Google Scholar
|
|
13
|
Evan GI, Wyllie AH, Gilbert CS, Littlewood
TD, Land H, Brooks M, Waters CM, Penn LZ and Hancock DC: Induction
of apoptosis in fibroblasts by c-myc protein. Cell. 69:119–128.
1992. View Article : Google Scholar
|
|
14
|
Igney FH and Krammer PH: Immune escape of
tumors: Apoptosis resistance and tumor counterattack. J Leukoc
Biol. 71:907–920. 2002.
|
|
15
|
El Hage F, Abouzahr-Rifai S, Meslin F,
Mami-Chouaib F and Chouaib S: Immune response and cancer. Bull
Cancer. 95:57–67. 2008.(In French).
|
|
16
|
Zhao Y and Rangnekar VM: Apoptosis and
tumor resistance conferred by Par-4. Cancer Biol Ther. 7:1867–1874.
2008. View Article : Google Scholar :
|
|
17
|
Li J, Yu W, Tiwary R, Park SK, Xiong A,
Sanders BG and Kline K: α-TEA-induced death receptor dependent
apoptosis involves activation of acid sphingomyelinase and elevated
ceramide-enriched cell surface membranes. Cancer Cell Int.
10:402010. View Article : Google Scholar :
|
|
18
|
Olson M and Kornbluth S: Mitochondria in
apoptosis and human disease. Curr Mol Med. 1:91–122. 2001.
View Article : Google Scholar
|
|
19
|
Cappello F, Bellafiore M, Palma A and
Bucchieri F: Defective apoptosis and tumorigenesis: Role of p53
mutation and Fas/FasL system dysregulation. Eur J Histochem.
46:199–208. 2002. View
Article : Google Scholar
|
|
20
|
Ding HF and Fisher DE: Induction of
apoptosis in cancer: New therapeutic opportunities. Ann Med.
34:451–469. 2002. View Article : Google Scholar
|
|
21
|
Pan G, Vickers SM, Pickens A, Phillips JO,
Ying W, Thompson JA, Siegal GP and McDonald JM: Apoptosis and
tumorigenesis in human cholangiocarcinoma cells. Involvement of
Fas/APO-1 (CD95) and calmodulin. Am J Pathol. 155:193–203. 1999.
View Article : Google Scholar :
|
|
22
|
Fuxius S, Mross K, Mansouri K and Unger C:
Gemcitabine and interferon-alpha 2b in solid tumors: A phase I
study in patients with advanced or metastatic non-small cell lung,
ovarian, pancreatic or renal cancer. Anticancer Drugs. 13:899–905.
2002. View Article : Google Scholar
|
|
23
|
Morisaki T, Hirano T, Koya N, Kiyota A,
Tanaka H, Umebayashi M, Onishi H and Katano M: NKG2D-directed
cytokine-activated killer lymphocyte therapy combined with
gemcitabine for patients with chemoresistant metastatic solid
tumors. Anticancer Res. 34:4529–4538. 2014.
|
|
24
|
Li Q, Yuan Z, Yan H, Wen Z, Zhang R and
Cao B: Comparison of gemcitabine combined with targeted agent
therapy versus gemcitabine monotherapy in the management of
advanced pancreatic cancer. Clin Ther. 36:1054–1063. 2014.
View Article : Google Scholar
|
|
25
|
Sanna V, Pala N and Sechi M: Targeted
therapy using nanotechnology: Focus on cancer. Int J Nanomedicine.
9:467–483. 2014.
|
|
26
|
Xiao JY, Lee JY, Tokuhiro S, Nagataki M,
Jarilla BR, Nomura H, Kim TI, Hong SJ and Agatsuma T: Molecular
cloning and characterization of taurocyamine kinase from Clonorchis
sinensis: A candidate chemotherapeutic target. PLoS Negl Trop Dis.
7:e25482013. View Article : Google Scholar :
|
|
27
|
Toyota Y, Iwama H, Kato K, Tani J, Katsura
A, Miyata M, Fujiwara S, Fujita K, Sakamoto T, Fujimori T, et al:
Mechanism of gemcitabine-induced suppression of human
cholangiocellular carcinoma cell growth. Int J Oncol. 47:1293–1302.
2015.
|
|
28
|
Luo Y, Lin C, Zhang XY, Liang X, Fu M and
Feng FY: Relationship between the level of RRM1 expression and the
sensitivity to gemcitabine in the esophageal squamous cell
carcinoma cell lines. Zhonghua Zhong Liu Za Zhi. 31:660–663.
2009.(In Chinese).
|
|
29
|
Ohtsuka T, Ryu H, Minamishima YA, Macip S,
Sagara J, Nakayama KI, Aaronson SA and Lee SW: ASC is a Bax adaptor
and regulates the p53-Bax mitochondrial apoptosis pathway. Nat Cell
Biol. 6:121–128. 2004. View
Article : Google Scholar
|
|
30
|
Liu W, Luo Y, Dunn JH, Norris DA,
Dinarello CA and Fujita M: Dual role of apoptosis-associated
speck-like protein containing a CARD (ASC) in tumorigenesis of
human melanoma. J Invest Dermatol. 133:518–527. 2013. View Article : Google Scholar
|
|
31
|
Proell M, Gerlic M, Mace PD, Reed JC and
Riedl SJ: The CARD plays a critical role in ASC foci formation and
inflammasome signalling. Biochem J. 449:613–621. 2013. View Article : Google Scholar :
|