Introduction
Serine-arginine protein kinases (SRPKs) family
represents a class of enzymes that can phosphorylate a defined
region in the RS domain in each SR protein. Phosphorylation and
dephosphorylation cycle of SR proteins is essential for pre-mRNA
splicing in cells (1). As a SR
splicing factor phosphorylation protein, SRPK1 plays an important
role in accumulating function signal and regulating the other
members of SRPKs family. Recent study demonstrated that some SR
proteins reveal altered expression in human cancers,
over-expression of a specific SR protein, SF2/ASF, is sufficient to
attract cellular transformation. Mammalian cells express two SRPKs
and four members of the Clk/Sty family of kinases (2). It revealed that knocking down SRPK1
tumors grew significantly more slowly in comparison with SRPK1
expression counterpart tumors (3).
The knockdown of SRPK1 expression through siRNA upregulates
pro-apoptotic proteins, which subsequently sensitise breast,
colorectal and pancreatic tumor cells to undergo apoptosis
(4,5). Therefore, the expression level of
SRPK1 may cause a molecular shift to influence tumorigenesis.
Small interfering RNA (siRNA)-mediated gene
silencing shows an enhanced specificity and effectiveness and thus
provides a powerful tool for many logical and therapeutic
applications. In humans, several efficient siRNA sequences against
members of apoptotic pathways, such as caspase-1, −2, −3, −8 and
Fas, have been designed to examine the regulation of apoptosis
(6–9).
Cell apoptosis plays a key role in the regulation of
tissue turnover that integrates multiple physiological and
pathological death signals. Many apoptotic signalling pathways,
including Fas/FasL, caspase family, cytochrome c signalling
and mitochondrial pathways, have been recognized (10–13).
p53, a tumor suppressor, is involved in the apoptotic effects of
cancer cells; it induces multiple cell death pathways. One key
endpoint in this cascade is the activation of caspase-3, which
cleaves several substrates, such as DNA repair enzyme poly
(ADP-ribose) polymerase (PARP) or DNA fragmentation factor (DFF).
Consequently, DNA strand breaks during apoptosis (14). B-cell lymphoma (Bcl)-2 family
members are important regulators in the apoptotic pathway
associated with individual components, such as Bcl-2 and Bcl-xL,
which can suppress apoptosis, or other factors, including Bax and
Bad, which can promote apoptosis.
In this study, the roles of caspase-3, PARP, p53 and
Bcl-2/Bax proteins in the apoptosis of human chronic myeloid
leukemia cell lines (K562 cells) were evaluated through
interference by SRPK1-siRNA. First, the effect of downregulating
SRPK1 gene expression in K562 cells by RNAi was analyzed and the
proliferation inhibition and apoptosis induction of SRPK1 in K562
cells was confirmed. Second, the roles of caspase-3, PARP, p53 and
Bcl-2/Bax proteins in the apoptosis of human K562 cells were
investigated through western blot analysis. This study provided
novel insights into the mechanism of apoptosis induced by human
K562 cells via the PARP-caspase3 pathway.
Materials and methods
Subjects
K562 cells and HL-60 cells were purchased from the
Cell Bank of the Chinese Academy of Sciences (Shanghai, China). The
cells were maintained in Roswell Park Memorial Institute
(RPMI)-1640 medium (StemCell Technologies, Vancouver, BC, Canada)
containing 10 ml of 1% penicillin/streptomycin (Invitrogen,
Carlsbad, CA, USA). Cultures were incubated at of 37°C in a fully
humidified atmosphere with 5% CO2.
SRPK1 knockdown in K562 and HL-60
cells
SRPK1-siRNA was synthesised by RiboBio Biological
Technology Co., Ltd. (Guangzhou, China). All of the transfections
were conducted by using a C10511-1 riboFect™ CP transfection kit
(RiboBio, Biological Technology Co., Ltd.) according to the
manufacturer's instructions. The cells without the insert gene were
used as control. The same volume-specific siRNA and control RNA
were dissolved in RPMI-1640 medium at 100 µl and a certain volume
of C10511-1 riboFect™ was dissolved in 100 µl of RPMI-1640 medium
for 5 min at room temperature. After 5 min, RNA and C10511-1
riboFect™ were diluted, blended and incubated for 20 min at room
temperature. The compound (200 µl) was added to the cell culture
plate containing a serum-free medium, gently shaken and thoroughly
incorporated. The cells were harvested after 4–6 h and replaced
with a fresh medium. siRNA transfection efficiency was analysed by
using a flow cytometer (FCM; Guava easyCyte 8HT) and Guava Incyte
version 2.8 (both from EMD Millipore, Billerica, MA, USA).
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA was extracted from the sample by using a
TRIzol reagent (Invitrogen) and reversed-transcribed into cDNA by
utilising an M-MLV reverse transcriptase kit (Promega, Madison, WI,
USA) according to the manufacturer's instructions. The primer
sequences were as follows: Human SRPK1 forward,
5′-CACGGCATGCATGGCCTTTGA-3′ and reverse,
5′-CGGCGGCAGTGGCTCTCTTC-3′. Quantitative PCR was performed with an
iCycleriQ real-time PCR system (Bio-Rad Laboratories, Inc.,
Hercules, CA, USA). Triplicate PCRs (20 µl) were performed using
SYBR-Green Supermix (Toyobo Co., Ltd., Osaka, Japan). The reaction
mixture was initially denatured at 95°C for 10 min and then
subjected to 45 PCR cycles of 95°C for 30 sec, 60°C for 30 sec and
72°C for 30 sec. mRNA levels were normalised to β-actin levels.
MTT test
The K562 and HL-60 cells were seeded in 96-well
plates at a density of 1×105/ml. Each plate was
transfected with 50 and 100 nM siRNA according to the introduction
above. Cultures were incubated at 37°C in a fully humidified
atmosphere with 5% CO2 for 24 h. The plates were added
with 10 µl of MTT (5 mg/ml) after 48 h and this procedure was
continuously performed for another 4 h. After the supernatant was
centrifuged, each plate was added with 100 µl of DMSO. The
proliferation of K562 cells was quantified at an OD of 490 nM. Cell
survival was calculated as the percentage of MTT inhibition, using
the following formula: Inhibition rate (%) = (1 – treatment
group/control group) × 100%.
Apoptosis detection assay
The cell apoptotic rate was evaluated by using a
fluorescein isothiocyanate (FITC) Annexin V apoptosis detection kit
(Becton-Dickinson and Co., Franklin Lakes, NJ, USA). The cells in
the logarithmic phase were collected, centrifuged at 1,000 rpm for
5 min and washed with precooled PBS. The cells were suspended with
200 µl of buffer. Each pipe was added with 4 µl of Annexin V/FITC
and 8 µl of 20 µg/ml PI for 15 min at room temperature. The sample
was analysed with a flow cytometer.
Cell protein extraction
Total proteins were extracted from the cells by
using a lysis buffer (20 mM Tris-HCl, 150 mM NaCl, 1 mM
Na3VO4, 1 mM EDTA, 1 mM PMSF, 50 mM NaF, 1%
NP-40, and 1 mM EGTA). The cells were lysed for 30 min in an ice
bath and centrifuged at 13,000 rpm for 15 min at 4°C. The
supernatant was collected to quantify the proteins.
Western blot analysis
Whole-cell extracts were mixed in Laemmli loading
buffer, boiled for 5 min and subjected to SDS-PAGE. After SDS-PAGE
was performed, proteins were transferred to polyvinylidene fluoride
(PVDF) membranes and then incubated with specific antibodies. The
membranes were washed with Tris-buffered saline and Tween-20 (TBST)
and incubated with HRP-conjugated second antibody for 1 h at room
temperature (25°C). Immune complexes were visualised using an
Anmbilon™ Western chemiluminescent HRP substrate system (EMD
Millipore). The primary antibodies were partly from Cell Signaling
Technology, Inc. (Beverly, MA, USA) including caspase-3 (1:1,000),
cleaved caspase-3 (1:1,000), PARP (1:1,000), cleaved PARP
(1:1,000), p53 (1:1,000), Bax (1:1,000), Bcl-2 (1:1,000) while
β-actin antibodies (1:500) were purchased from Wuhan Boster
Biological Technology, Ltd. (Wuhan, China).
Statistical analysis
Results were expressed as mean ± standard error (SE)
of independent experiments performed in triplicate. SPSS (version
14.0; SPSS Inc., Chicago, IL, USA) was used for statistical
analysis. The results were compared via one-way ANOVA followed by
Tukey's post hoc test. Differences between values were considered
significant at P<0.05.
Results
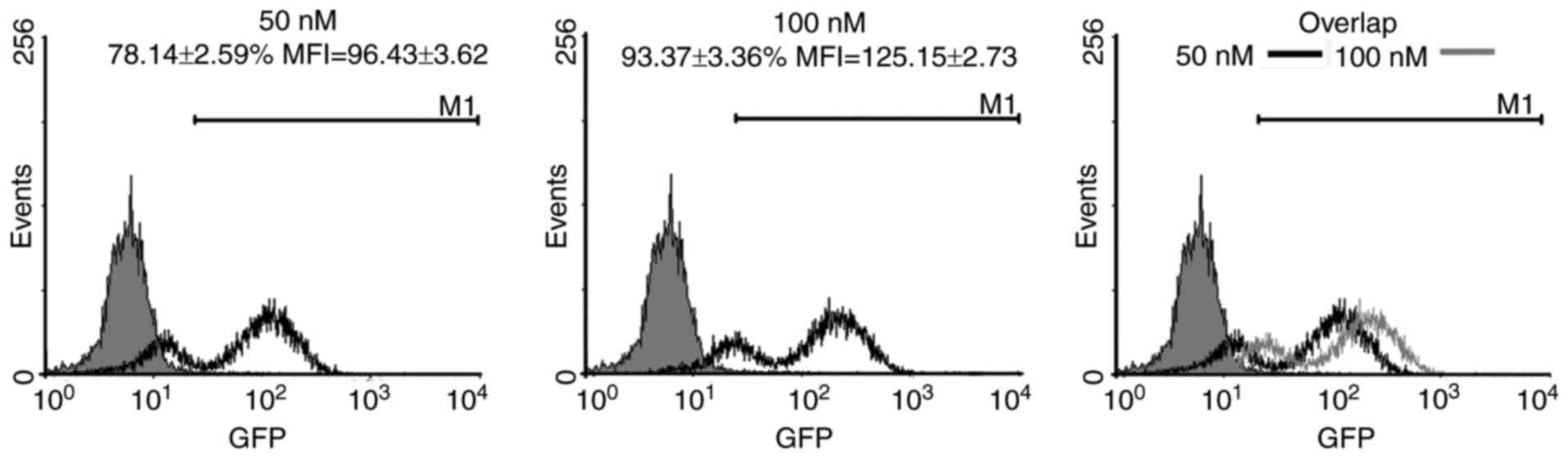
siRNA transfection efficiency
This study designed and synthesised scrambled siRNA
with green fluorescent protein (GFP) to investigate whether siRNA
can inhibit K562 cells in vitro. After the cells were
transfected with 50 and 100 nmol/l (nM) siRNA, the transfection
efficiency was detected through FCM. The inhibition rate of 50 nM
siRNA was 78.14±2.59%. By comparison, the inhibition rate of 100 nM
siRNA was 93.37±3.36%. The mean fluorescent intensities (MFI) of 50
and 100 nM siRNA were 96.43±3.62 and 125.15±2.73, respectively.
These results indicated that siRNA was transfected into the cells
and the transfection induced by 100 nM siRNA was higher than that
caused by 50 nM (Fig. 1).
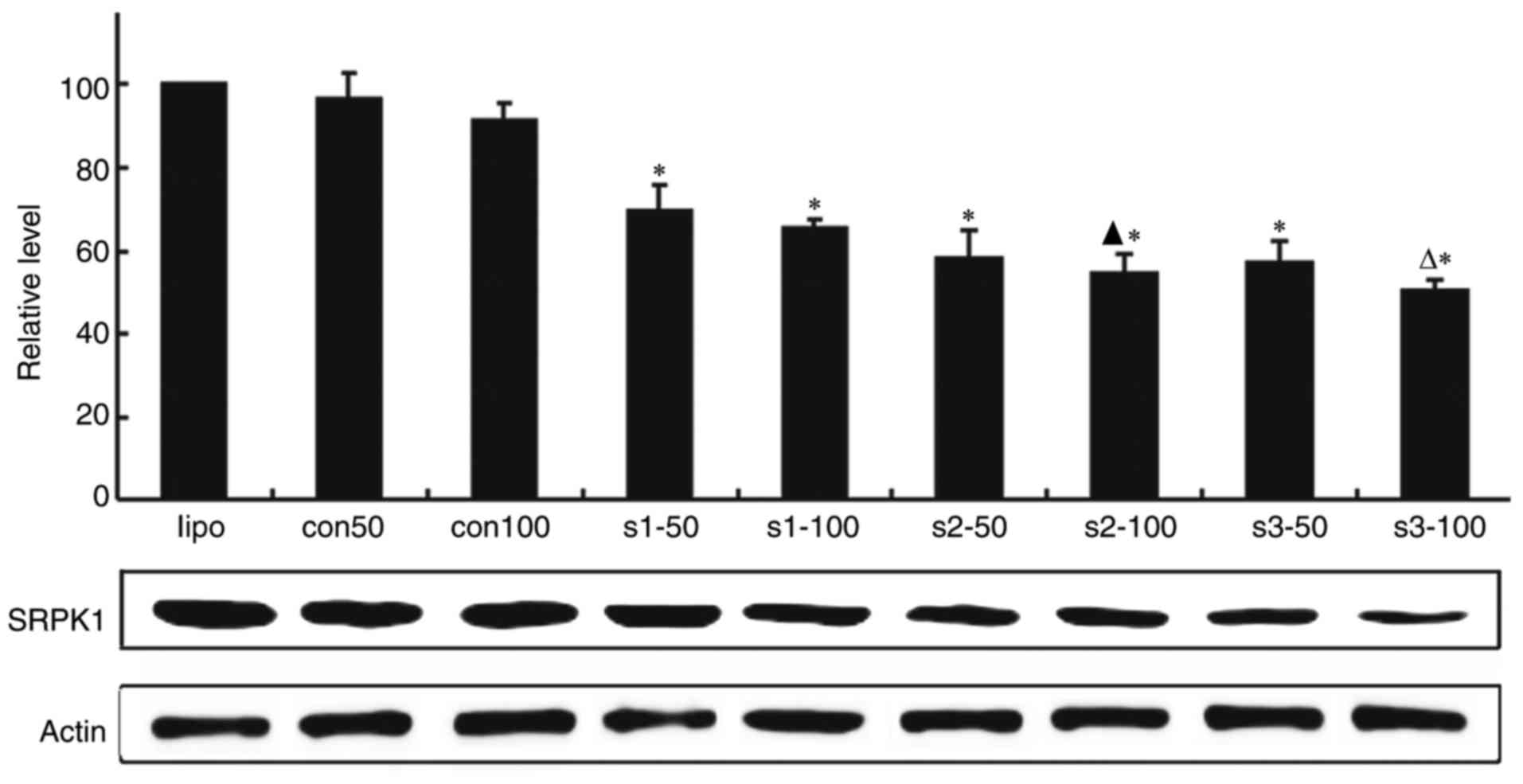
Effect of SRPK1-siRNA on SRPK1
expression in K562 cells
To explore the effect of SRPK1 silenced by siRNA on
K562 cells, we synthesised three SRPK1-siRNA (s1, s2 and s3) by
company. After K562 cells were transfected with two concentrations
of siRNA, the SRPK1 expression was detected through real-time
polymerase chain reaction (RT-PCR) analysis and western blot
analysis. The results showed that the mRNA levels of SRPK1 were
lower in the transfected cells groups than in the transfection
reagent and scrambled siRNA groups. SRPK1-siRNA at 100 nM was more
effective than 50 nM in silencing the SRPK1 gene. SRPK1 protein was
tested by western blot, the result was consistent with RT-PCR
method (Fig. 2).
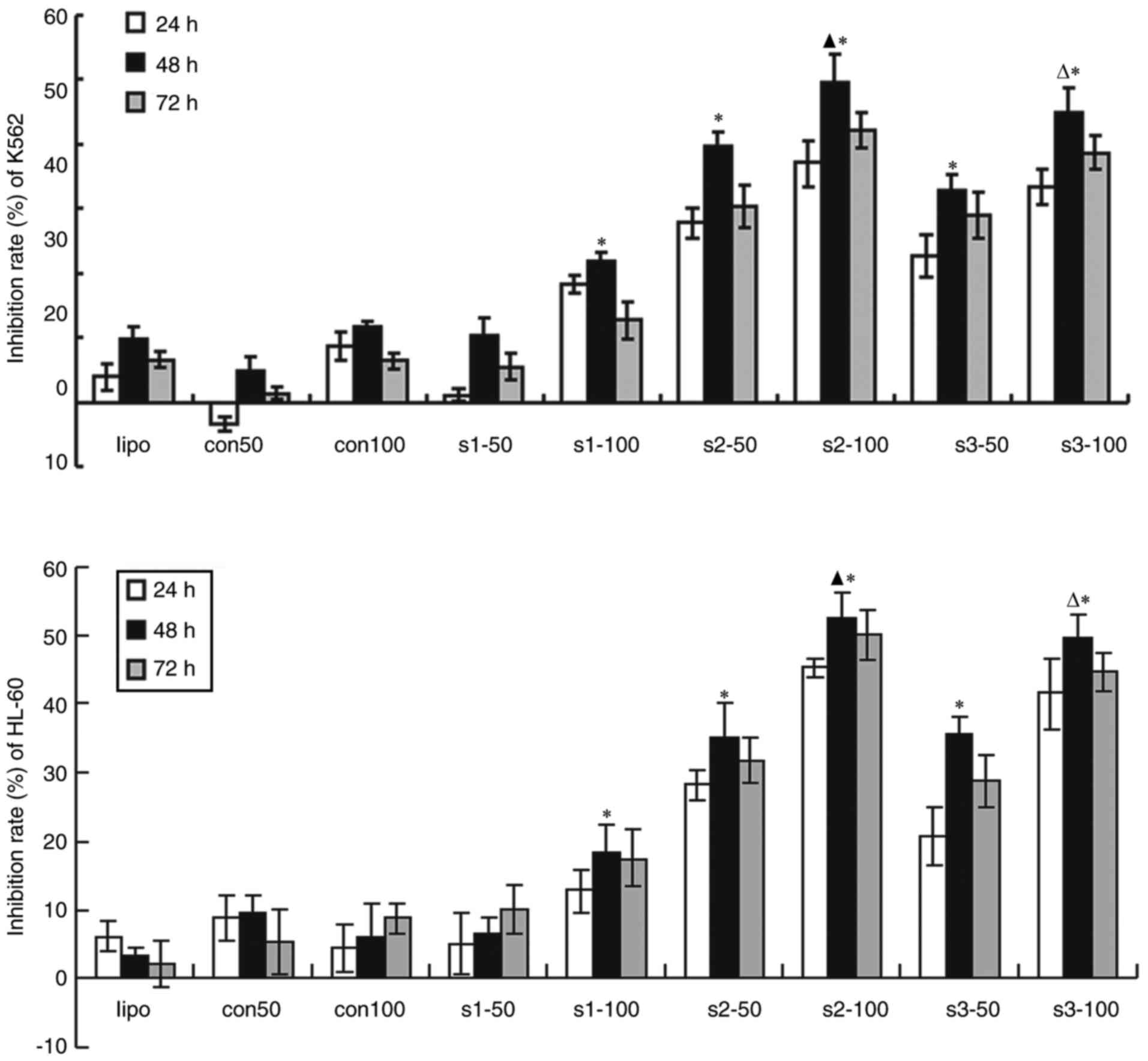
Suppression of tumor cells
proliferation by SRPK1
The tumor cells with knocked down SRPK1 grew slowly.
The transfected K562 and HL-60 cells were determined through MTT
test to evaluate whether SRPK1 inhibits cell proliferation. In this
study, 50 and 100 nM siRNA were transfected into K562 and HL-60
cells for 24, 48 and 72 h. The results revealed that the inhibition
rate of the transfected groups was higher than that of the SRPK1
nontransfected groups. No statistical difference was found between
the transfection reagent and siRNA scrambled groups. Compared with
that of the SRPK1 nontransfection groups, the maximum inhibition
rates of the transfection groups were s2 and s3 sequences at 100 nM
siRNA cultured for 48 h. Therefore, 100 nM siRNA was superior to 50
nM. The inhibition rate increased at 48 h but slightly decreased at
48–72 h. These results indicated that s2 sequence was more
effective than s3 in inhibiting cell proliferation in K562 and
HL-60 cells (Fig. 3).
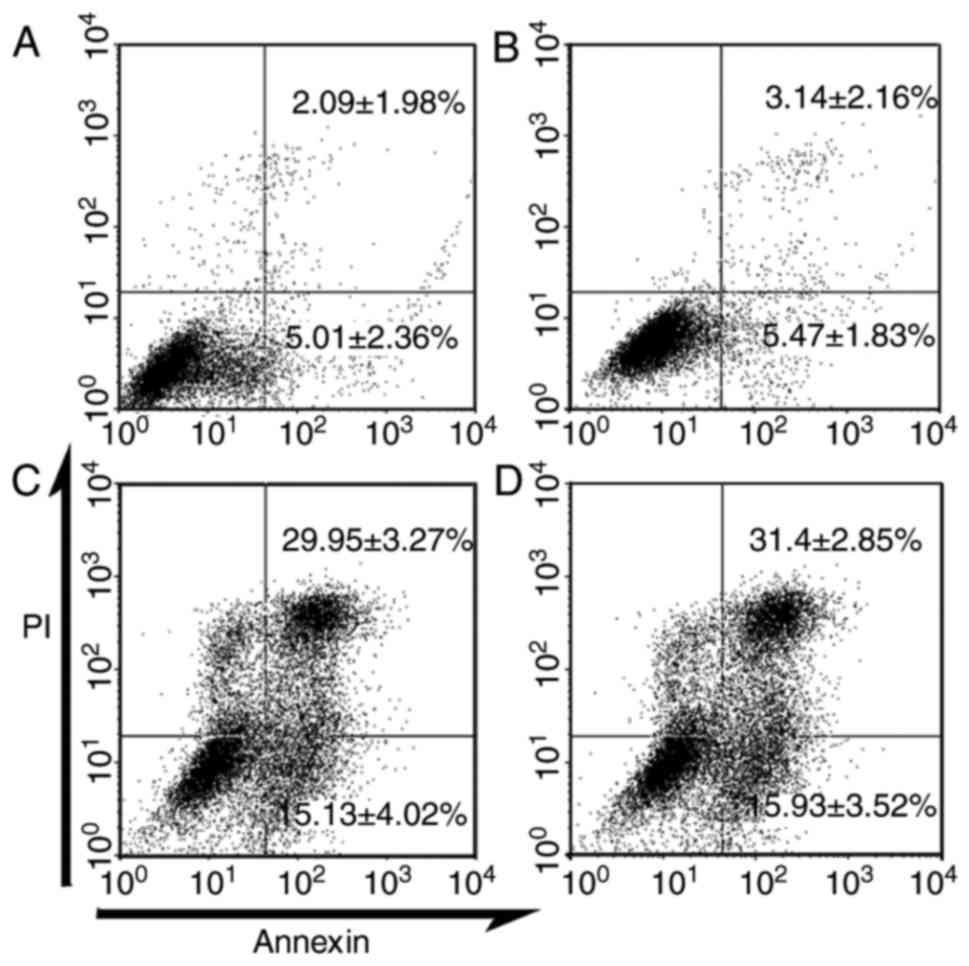
SRPK1-siRNA induced K562 cells
apoptosis
In previous experiments, SRPK1 silencing can inhibit
cell proliferation. The synthesised sequences s2 and s3 for 100 nM
siRNA are better than the others. To investigate whether SRPK1
silencing accelerates K562 cell apoptosis, we chose the s2 and s3
sequences as experimental groups, the transfection regent group as
control 1 and the scrambled siRNA group as control 2. Apoptosis was
analysed through FCM. Compared with those in the control groups,
the apoptotic cells in the SRPK1-silenced groups (s2 and s3)
increased by 29.95±3.27% and 31.4±2.85%. The early apoptotic cells
also increased by 15.13±4.02% and 15.93±3.52% (Fig. 4). No significant difference in
apoptotic cells was found between the control groups (2.09±1.98%
vs. 3.14±2.16%). These results indicated that silencing SRPK1
induced K562 cell apoptosis (Fig.
4).
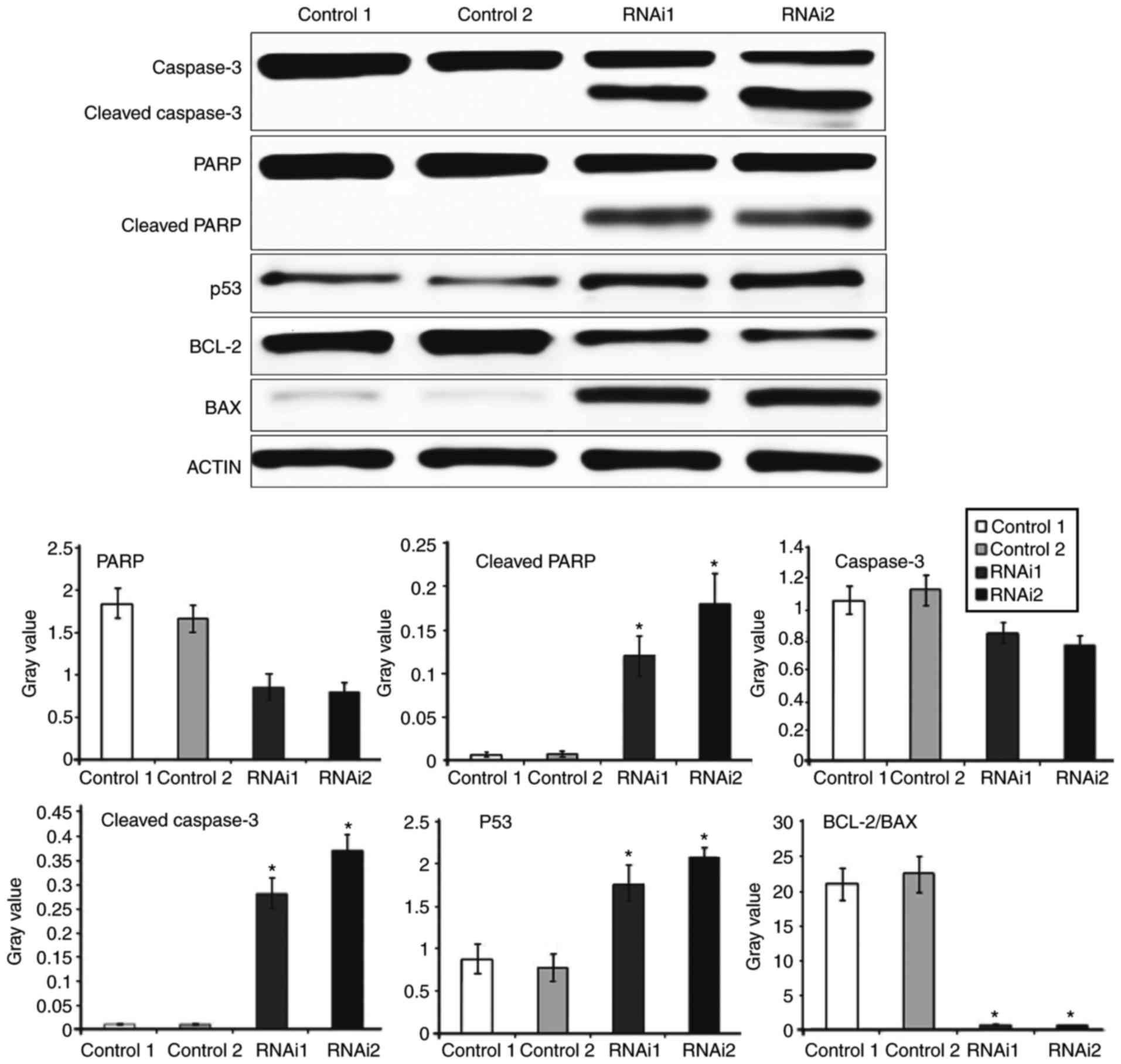
SRPK1-siRNA induced K562 cells
apoptosis via the PARP-caspase3 pathway
To determine the possible mechanism of SRPK1
contributing to K562 cells, we examined caspase-3, PARP, p53, Bcl-2
and Bax protein expression through western blot analysis. The
experiment was divided into four groups: Transfection regent group
as control 1, scrambled siRNA group as control 2, synthesised
sequence s2 for 100 nM siRNA as RNAi 1 and s3 100 nM siRNA as RNAi
2. The results revealed that the cleavage and activation of
caspase-3 and PARP were significantly higher in the RNAi groups
than in the control groups, whereas the expression levels of PARP
and caspase-3 in the four groups did not significantly differ. The
expression of RNAi 2 group was significantly higher than that of
the RNAi 1 group. We further examined whether SRPK1 inhibition can
modulate p53, pro-apoptotic Bax and anti-apoptotic Bcl-2 proteins
during induced apoptosis. The expression of p53 was significantly
higher in the RNAi groups than in the control groups, while
Bcl-2/Bax was significantly decreased. No significant difference
was observed between the control and RNAi groups in Bcl-2/Bax
(Fig. 5). These results suggested
that the inhibition of SRPK1 could induce the apoptosis of K562
cells via the PARP-caspase-3 pathway.
Discussion
Recent years, SRPKs were found as an important part
of serine-threonine protein kinase family, which played an
important role in specific phosphorylation of serine. SRPK1 is one
of SRPKs that studied most deeply. As a SR splicing factor
phosphorylation protein, SRPK1 plays an important role in
accumulating function signal and regulating the other members of
SRPKs family. SR protein kinase is a protein kinase family, all
SRPKs show the ability of phosphorylating SR protein, regulating
the alternative splicing of SR protein, influencing the
distribution and location of SR protein in the nuclear and playing
an important role in pre-mRNA splicing regulation. In addition,
SRPKs can be involved in intracellular iron homeostasis, polyamine
transportation, sperm development, paedomorphosis, cell cycle
regulation, cell apoptosis and other important life activities by
interacting with other proteins (15–17).
Current research has suggested that SRPKs could be
potential to be the target of tumor therapy because of high
expression in some tumor cells, such as prostate, breast, colon,
and lung cancers (18–21). After interfering with SRPK1 gene,
the proliferation of tumor cells was reduced, apoptosis potential
increased and the sensitivity to chemotherapy drugs improved. This
effect may be based on the mechanism of splicing. Sanidas et
al (22) suggested that SRPK1a
may play an important role in linking ribosomal assembly and/or
function to elytroid differentiation in human leukemic cells. Wu
et al (23) showed that
knockdown of SRPK1 can inhibit tumor cells growth, invasion and
migration in normoxic condition, but portion of the effect could be
reversed in hypoxia. SRPK1 may be a new molecular player
contributing to the early treatment of glioma. The study of van
Roosmalen et al (24)
provided comprehensive information on the molecular determinants of
tumor cell migration and suggested that SRPK1 had potential for
limiting breast cancer metastasis as a drug target. Aberrant SRPK1
expression in either direction can induce constitutive Akt
activation and provide a mechanistic basis for previous
observations that SRPK1 can be downregulated in some cancer types
but upregulated in others (25).
Ren et al (26) revealed
that SRPK1 mediated TGF-Î2−induced proliferation and
apoptosis by regulating AKT and JNK in ESCC. Thus, the
TGF-Î2−SRPK1 pathway may be a useful target to affect
the progression of ESCC. Sigala et al (27) had verified that the effect of SRPK1
knockdown on the viability of glioma cell lines was limited at
least in vitro, whereas the in vivo effects of this
process can be attributed to the modulation of angiogenesis by
SRPK1. Our study demonstrated that the mRNA levels of SRPK1 were
lower in the transfected cell groups than in the transfection
reagent and scrambled siRNA groups. The transfected K562 cells were
determined through MTT and FCM test to evaluate whether SRPK1 can
inhibit cell proliferation and induce apoptosis. The results showed
that the inhibition rate in the transfected groups was higher than
that in the SRPK1 nontransfected groups. HL-60 cells after
transfection were tested by MTT, it revealed SRPK1-siRNA can
inhibit HL-60 cells proliferation as well. Compared with that in
the control groups, the apoptotic cells in the SRPK1-silenced
groups (s2 and s3) increased, and early apoptotic cells also
increased.
Apoptosis is a complex process regulated by various
factors. For example, caspase-3 is generally considered as the main
executor of apoptosis. One of the essential substrates cleaved by
caspase-3 is PARP, an abundant DNA-binding enzyme that detects and
signals DNA strand breaks (28).
Caspase-3 and its substrate PARP are the key modulators of
apoptosis, especially through the cleavage of PARP and caspase-3.
p53 is a stress response protein, which is induced by DNA damage
and deregulated oncogene expression (29,30).
To confirm our findings, we analysed the protein levels of
caspase-3, PARP, p53 and Bcl-2/Bax by western blot analysis. The
results revealed that the cleavage and activation of caspase-3 and
PARP were significantly higher in the RNAi groups than in the
control groups, whereas the expression levels of PARP and caspase-3
in the four groups had no significant difference. The expression of
the RNAi 2 group was significantly higher than that of the RNAi 1
group. These observations could imply that the inhibitory effects
on the cyclic stretch-induced apoptosis of human K562 cells
occurred primarily at the post-cleavage level. The lever of p53
protein was significantly higher in the RNAi groups than that in
the control groups. The data suggested that p53 might mediate
cellular sensitivity to apoptosis. The expression of Bcl-2/Bax was
significantly decreased in the RNAi groups compared with that in
the control groups. No significant difference was observed between
the control and RNAi groups. p53 is implicated in the induction of
two distinct apoptotic signaling pathways: The intrinsic and
extrinsic pathways. The extrinsic pathway involves death receptors,
which including caspase-3. The two pathways can trigger the
activation of PARP and lead cells to apoptosis. This study showed
that the expression of p53, cleaved caspase-3 and cleaved PARP
increased while anti-apoptotic protein Bcl-2/Bax decreased in the
RNAi groups, may indicate the apoptosis of tumor cells.
This study provided evidence revealing a novel
function of SRPK1-siRNA in human K562 cells induced apoptosis by
the activation of the PARP-caspase3 pathway. The limitation of the
present study is the lack of a detailed molecular mechanism and
in vivo experiments. In future studies, we will study
further and related molecular mechanism will be identified to
elucidate the potential role in tumor therapy.
Acknowledgements
This study was supported by Shandong Provincial
Natural Science Foundation (no. ZR2013HQ059).
References
|
1
|
Ding JH, Zhong XY, Hagopian JC, Cruz MM,
Ghosh G, Feramisco J, Adams JA and Fu XD: Regulated cellular
partitioning of SR protein-specific kinases in mammalian cells. Mol
Biol Cell. 17:876–885. 2006. View Article : Google Scholar :
|
|
2
|
Ngo JC, Chakrabarti S, Ding JH,
Velazquez-Dones A, Nolen B, Aubol BE, Adams JA, Fu XD and Ghosh G:
Interplay between SRPK and Clk/Sty kinases in phosphorylation of
the splicing factor ASF/SF2 is regulated by a docking motif in
ASF/SF2. Mol Cell. 20:77–89. 2005. View Article : Google Scholar
|
|
3
|
Mavrou A, Brakspear K, Hamdollah-zadeh M,
Damodaran G, Babaei-Jadidi R, Oxley J, Gillatt DA, Ladomery MR,
Harper SJ, Bates DO and Oltean S: Serine-arginine protein kinase 1
(SRPK1) inhibition as a potential novel targeted therapeutic
strategy in prostate cancer. Oncogene. 34:4311–4319. 2015.
View Article : Google Scholar
|
|
4
|
Hayes GM, Carrigan PE, Beck AM and Miller
LJ: Targeting the RNA splicing machinery as a novel treatment
strategy for pancreatic carcinoma. Cancer Res. 66:3819–3827. 2006.
View Article : Google Scholar
|
|
5
|
Hayes GM, Carrigan PE and Miller LJ:
Serine-arginine protein kinase 1 overexpression is associated with
tumorigenic imbalance in mitogen-activated protein kinase pathways
in breast, colonic, and pancreatic carcinomas. Cancer Res.
67:2072–2080. 2007. View Article : Google Scholar
|
|
6
|
Lassus P, Opitz-Araya X and Lazebnik Y:
Requirement for caspase-2 in stress-induced apoptosis before
mitochondrial permeabilization. Science. 297:1352–1354. 2002.
View Article : Google Scholar
|
|
7
|
Wurzer WJ, Planz O, Ehrhardt C, Giner M,
Silberzahn T, Pleschka S and Ludwig S: Caspase 3 activation is
essential for efficient influenza virus propagation. EMBO J.
22:2717–2728. 2003. View Article : Google Scholar :
|
|
8
|
Chun HJ, Zheng L, Ahmad M, Wang J, Speirs
CK, Siegel RM, Dale JK, Puck J, Davis J, Hall CG, et al:
Pleiotropic defects in lymphocyte activation caused by caspase-8
mutations lead to human immunodeficiency. Nature. 419:395–399.
2002. View Article : Google Scholar
|
|
9
|
Song E, Lee SK, Wang J, Ince N, Ouyang N,
Min J, Chen J, Shankar P and Lieberman J: RNA interference
targeting Fas protects mice from fulminant hepatitis. Nat Med.
9:347–351. 2003. View
Article : Google Scholar
|
|
10
|
Liu J, Liu J, Mao J, Yuan X, Lin Z and Li
Y: Caspase-3-mediated cyclic stretch-induced myoblast apoptosis via
a Fas/FasL-independent signaling pathway during myogenesis. J Cell
Biochem. 107:834–844. 2009. View Article : Google Scholar
|
|
11
|
Jiang J, Qi YX, Zhang P, Gu WT, Yan ZQ,
Shen BR, Yao QP, Kong H, Chien S and Jiang ZL: Involvement of Rab28
in NF-κB nuclear transport in endothelial cells. PLoS One.
8:e560762013. View Article : Google Scholar :
|
|
12
|
Rennier K and Ji JY: Effect of shear
stress and substrate on endothelial DAPK expression, caspase
activity, and apoptosis. BMC Res Notes. 6:102013. View Article : Google Scholar :
|
|
13
|
Xu C, Hao Y, Wei B, Ma J, Li J, Huang Q
and Zhang F: Apoptotic gene expression by human periodontal
ligament cells following cyclic stretch. J Periodontal Res.
46:742–748. 2011. View Article : Google Scholar
|
|
14
|
Medina V, Edmonds B, Young GP, James R,
Appleton S and Zalewski PD: Induction of caspase-3 protease
activity and apoptosis by butyrate and trichostatin A (inhibitors
of histone deacetylase): Dependence on protein synthesis and
synergy with a mitochondrial/cytochrome c-dependent pathway.
Cancer Res. 57:3697–3707. 1997.
|
|
15
|
Forment J, Mulet JM, Vicente O and Serrano
R: The yeast SR protein kinase Sky1p modulates salt tolerance,
membrane potential and the Trk1,2 potassium transporter. Biochim
Biophys Acta. 1565:36–40. 2002. View Article : Google Scholar
|
|
16
|
Kondoh H, Yuasa T and Yanagida M: Mis3
with a conserved RNA binding motif is essential for ribosome
biogenesis and implicated in the start of cell growth and S phase
checkpoint. Genes Cells. 5:525–541. 2000. View Article : Google Scholar
|
|
17
|
Kamachi M, Le TM, Kim SJ, Geiger ME,
Anderson P and Utz PJ: Human autoimmune sera as molecular probes
for the identification of an autoantigen kinase signaling pathway.
J Exp Med. 196:1213–1225. 2002. View Article : Google Scholar :
|
|
18
|
Mavrou A, Brakspear K, Hamdollah-Zadeh M,
Damodaran G, Babaei-Jadidi R, Oxley J, Gillatt DA, Ladomery MR,
Harper SJ, Bates DO and Oltean S: Serine arginine protein kinase 1
(SRPK1) inhibition as a potential novel targeted therapeutic
strategy in prostate cancer. Oncogene. 34:4311–4319. 2015.
View Article : Google Scholar
|
|
19
|
Lin JC, Lin CY, Tarn WY and Li FY:
Elevated SRPK1 lessens apoptosis in breast cancer cells through
RBM4-regulated splicing events. RNA. 20:1621–1631. 2014. View Article : Google Scholar :
|
|
20
|
Hu ZY, Wang XY, Guo WB, Xie LY, Huang YQ,
Liu YP, Xiao LW, Li SN, Zhu HF, Li ZG and Kan H: Long non-coding
RNA MALAT1 increases AKAP-9 expression by promoting SRPK1-catalyzed
SRSF1 phosphorylation in colorectal cancer cells. Oncotarget.
7:11733–11743. 2016. View Article : Google Scholar :
|
|
21
|
Liu H, Hu X, Zhu Y, Jiang G and Chen S:
Up-regulation of SRPK1 in non-small cell lung cancer promotes the
growth and migration of cancer cells. Tumour Biol. 37:7287–7293.
2016. View Article : Google Scholar
|
|
22
|
Sanidas I, Kotoula V, Ritou E, Daans J,
Lenz C, Mairhofer M, Daniilidou M, Kolbus A, Kruft V, Ponsaerts P
and Nikolakaki E: The ratio of SRPK1/SRPK1a regulates erythroid
differentiation in K562 leukaemic cells. Biochim Biophys Acta.
1803:1319–1331. 2010. View Article : Google Scholar
|
|
23
|
Wu Q, Chang Y, Zhang L, Zhang Y, Tian T,
Feng G, Zhou S, Zheng Q, Han F and Huang F: SRPK1 dissimilarly
impacts on the growth, metastasis, chemosensitivity and
angiogenesis of glioma in normoxic and hypoxic conditions. J
Cancer. 4:727–735. 2013. View
Article : Google Scholar :
|
|
24
|
van Roosmalen W, Le Dévédec SE, Golani O,
Smid M, Pulyakhina I, Timmermans AM, Look MP, Zi D, Pont C, de
Graauw M, et al: Tumor cell migration screen identifies SRPK1 as
breast cancer metastasis determinant. J Clin Invest. 125:1648–1664.
2015. View
Article : Google Scholar :
|
|
25
|
Wang P, Zhou Z, Hu A, de Albuquerque Ponte
C, Zhou Y, Hong L, Sierecki E, Ajiro M, Kruhlak M, Harris C, et al:
Both decreased and increased SRPK1 levels promote cancer by
interfering with PHLPP-mediated dephosphorylation of Akt. Mol Cell.
54:378–391. 2014. View Article : Google Scholar :
|
|
26
|
Ren G, Sheng L, Liu H, Sun Y, An Y and Li
Y: The crucial role of SRPK1 in TGF-β-induced proliferation and
apoptosis in the esophageal squamous cell carcinomas. Med Oncol.
32:2092015. View Article : Google Scholar
|
|
27
|
Sigala I, Tsamis KI, Gousia A, Alexiou G,
Voulgaris S, Giannakouros T, Kyritsis AP and Nikolakaki E:
Expression of SRPK1 in gliomas and its role in glioma cell lines
viability. Tumour Biol. 37:8699–8707. 2016. View Article : Google Scholar
|
|
28
|
Decker P and Muller S: Modulating poly
(ADP-ribose) polymerase activity: Potential for the prevention and
therapy of pathogenic situations involving DNA damage and oxidative
stress. Curr Pharm Biotechnol. 3:275–283. 2002. View Article : Google Scholar
|
|
29
|
Bellodi C, Kopmar N and Ruggero D:
Deregulation of oncogene-induced senescence and p53 translational
control in X-linked dyskeratosis congenita. EMBO J. 29:1865–1876.
2010. View Article : Google Scholar :
|
|
30
|
Elkholi R and Chipuk JE: How do I kill
thee? Let me count the ways: p53 regulates PARP-1 dependent
necrosis. Bioessays. 36:46–51. 2014. View Article : Google Scholar
|