Best vitelliform macular dystrophy (BVMD), also
known as Best disease, is a hereditary retinal disease
characterized by the bilateral accumulation of large egg yolk-like
lesions in the sub-retinal and sub-retinal pigment epithelium (RPE)
spaces (1,2). This lesion may eventually break up
and spread throughout the macular area, leading to central vision
reduction and retinal detachment (3). The clinical manifestation of BVMD
varies substantially in different disease stages and among
individual patients (4). For
example, retinal lesions in BVMD typically occur bilaterally and
symmetrically; however, rarely, patients can exhibit unilateral
maculopathy (5,6). Patients with atypical BVMD
presentation can have multifocal macular and extramacular
involvement, including retinitis pigmentosa, microcornea, retinal
dystrophy, cataract, and posterior staphyloma syndrome (6–13).
Macular degeneration in BVMD can begin from childhood or adulthood,
and is classified as juvenile-onset BVMD or adult-onset BVMD,
respectively (14). The variation
in the age of onset is not clearly understood, and few studies have
compared the differences between these two classifications
(13,15). Currently, there is no effective
treatment for BVMD.
This study aimed to characterize the clinical
manifestations and investigate the underlying genetic variations of
a 16-year-old male and 43-year-old female with juvenile-onset and
adult-onset BVMD, respectively.
A 16-year-old male with juvenile-onset BVMD (Patient
1) and a 43-year-old female with adult-onset BVMD (Patient 2), both
from southern China, were diagnosed at Zhongshan Ophthalmic Center
(Guangzhou, P.R. China). Visual acuity was examined using the Early
Treatment Diabetic Retinopathy Study chart (Precision Vision, La
Salle, IL, USA) (28). Images of
the anterior segment were captured using a BX 900 Slit Lamp
(Haag-Streit, Bern, Switzerland). Measurements of the anterior
segment were recorded with Pentacam HR version 70700 (Oculus VR,
LLC, Wetzlar, Germany). Optical Coherence Tomography (OCT) was
performed by Cirrus HD-OCT (Carl Zeiss Meditec, Inc., Dublin, CA,
USA). Fundus photography and fundus fluorescein angiography (FFA)
imaging was performed using a Heidelberg Retina Angiograph
(Heidelberg Engineering, Heidelberg, Germany). Multifocal
electroretinography (mfERG) was performed to assess the amplitudes
of the rod and cone responses using the Espion electrophysiology
system (Diagnosys LLC, Littleton, MA, USA). Physical examinations
were performed to exclude systemic diseases.
Venous blood samples were collected from the study
subjects, their family members and 200 subjects without BVMD within
the same population (15–48 years old; sex ratio:
Male/female=108/92). Genomic DNA was extracted from peripheral
blood leukocytes using standard protocols. Briefly, a total amount
of 1 ml of blood sample was collected from each subject, lysed by
red blood cell lysis buffer (Sigma Aldrich, Merck KGaA, Darmstadt,
Germany), and centrifuged at 2,000 × g for 5 min at room
temperature. Genomic DNA was extracted from peripheral blood
leucocytes using a DNA extraction kit (Qiagen GmbH, Hilden,
Germany) (29,30). Exons of the BEST1 gene were
amplified by polymerase chain reaction (PCR) with primers as
previously described (31–34). The primer sequences are listed in
Table I. PCR was conducted in a 50
µl reaction system. using the PCR amplification kit (Takara Bio,
Inc., Otsu, Japan). The amplification included a single 5 min step
at 94°C, followed by 40 cycles of 94°C for 45 sec, 58–61°C for 45
sec, 72°C for 45 sec and a final 10 min step at 72°C. The PCR
products were sequenced from both directions with an ABI3730
Automated Sequencer (PE Biosystems Inc., Foster City, CA).
Sequenced products were analyzed using Seqman (version 2.3;
Technelysium Pty Ltd., Brisbane, Australia), and compared with
reference sequences in the database at the National Center for
Biotechnology Information (NC_000011.10).
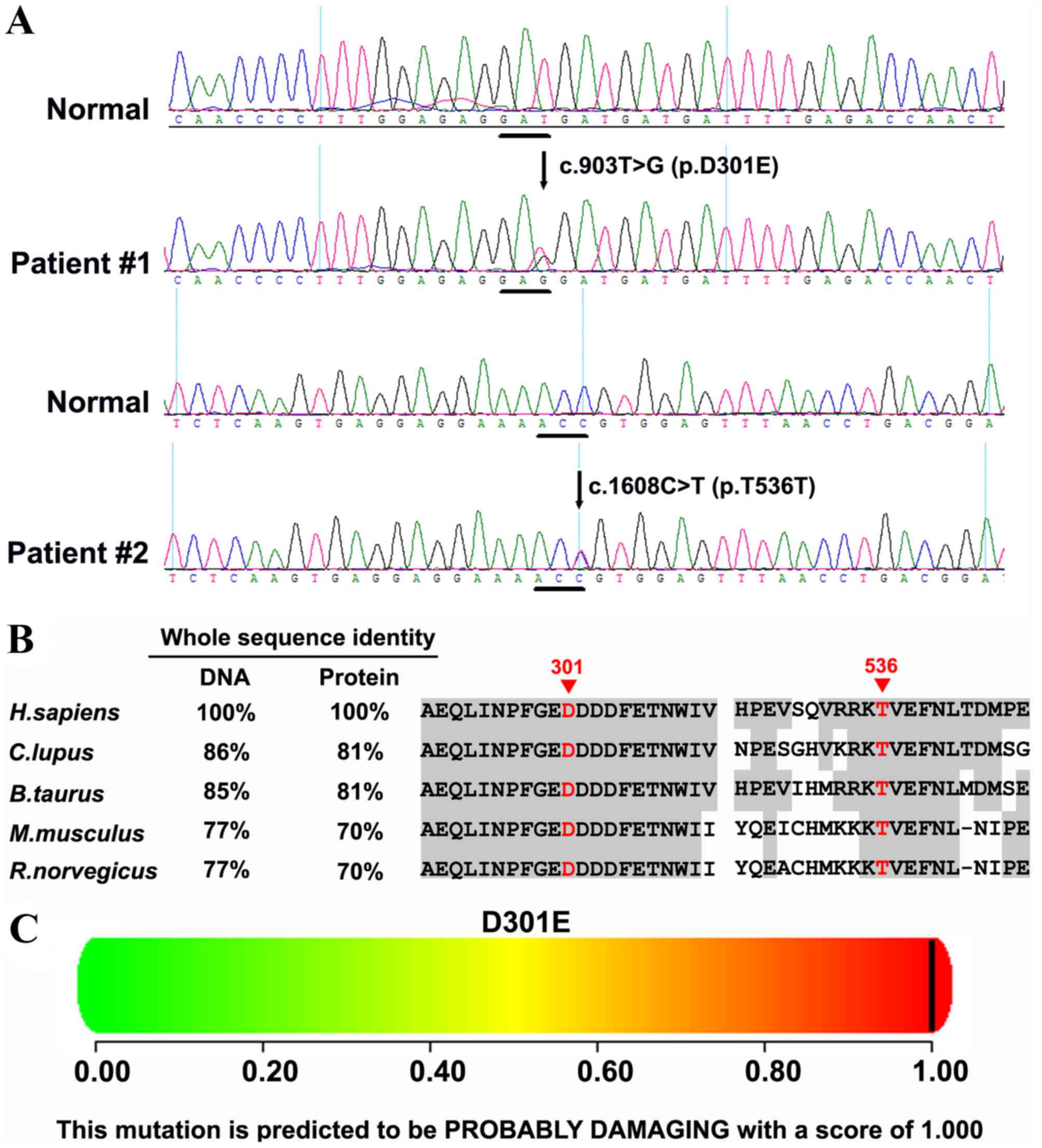
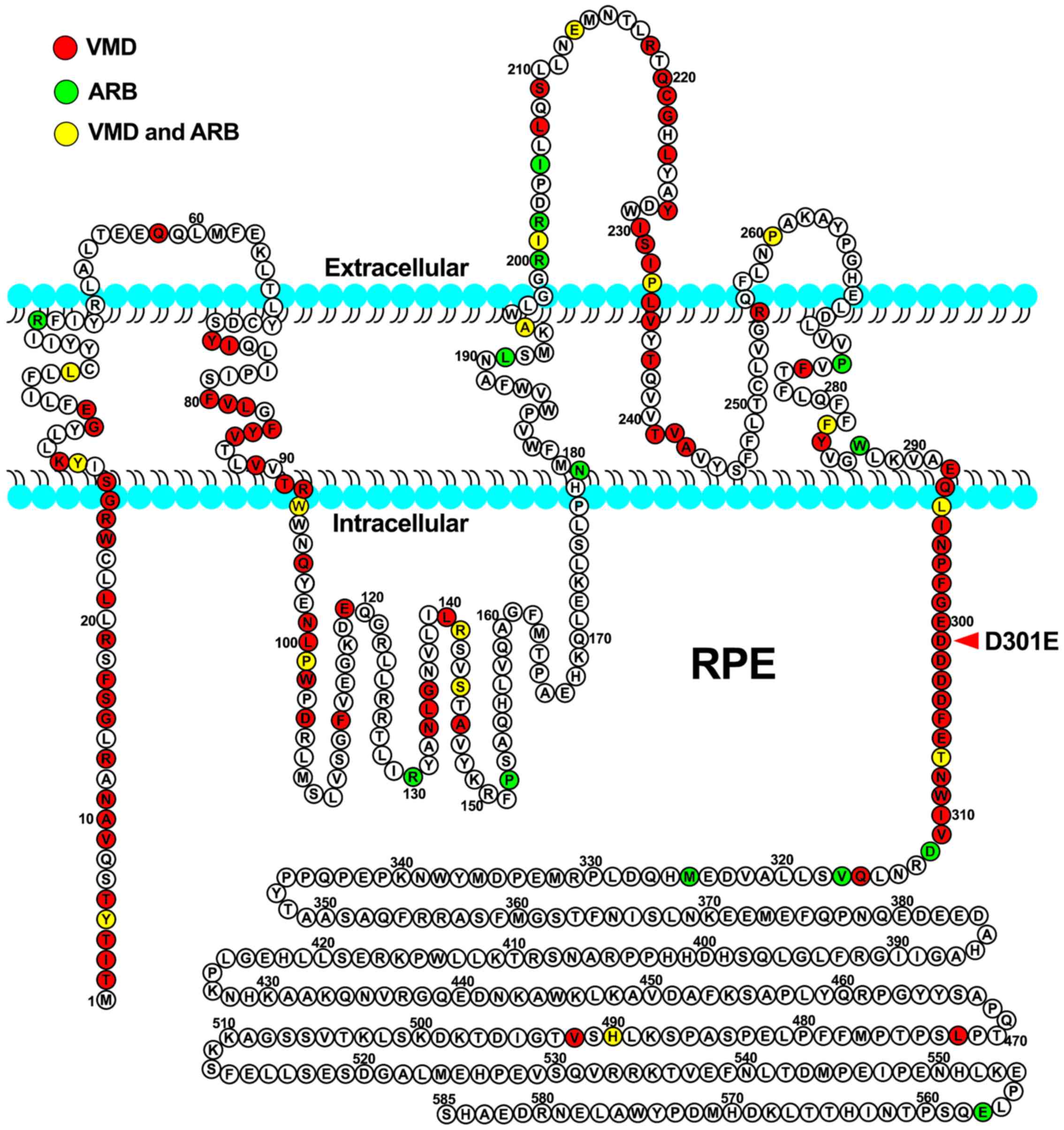
To analyze the effect of missense variants,
polymorphism phenotyping (PolyPhen)and the sorting intolerant from
tolerant (SIFT) algorithms were used to predict the possible impact
of an amino acid substitution on the protein structure and
function, using straightforward physical and comparative
considerations (35–39). Variants were considered to be
pathogenic when at least one of the two programs predicted a
deleterious effect of the amino acid substitution on the protein
structure and function. The Human Gene Mutation Database
(http://www.hgmd.cf.ac.uk/ac/index.php) was used to
screen mutations reported in published studies. HomoloGene
(https://www.ncbi.nlm.nih.gov/homologene) was used to
check whether the mutated amino acid residues were conserved across
different species.
All experimental protocols and the methods were
carried out in accordance with the guidelines approved by the
ethics committee of Zhongshan Ophthalmic Center of Sun Yat-sen
University (Guangzhou, P.R. China). Written informed consent was
obtained from each subject in accordance with The Declaration of
Helsinki. All participants provided informed consent for the
publication of their data, including images and examination
results.
Patient 1 had no known familial history of ocular
disease. Refractive error was +4.50 diopter sphere (DS) in both
eyes, with best corrected visual acuity (BVCA) at 1.0 in the right
eye and 0.1 in the left eye. The cornea and the lens were
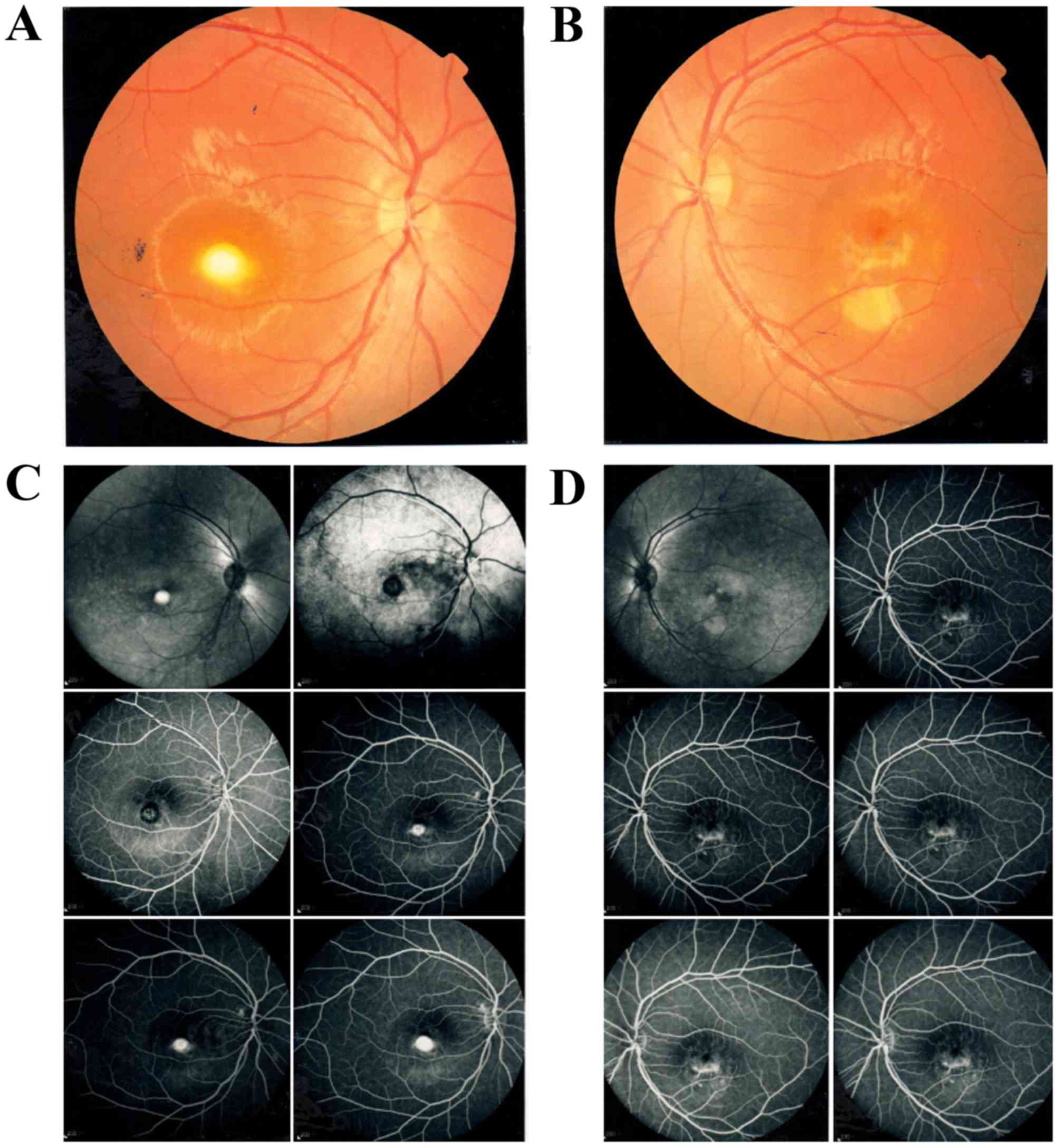
transparent. Fundus examination revealed that the right eye had
prominent yellow-white sub-retinal scarring with pigmented borders,
surrounded by a serous retinal detachment. The left eye exhibited
fragmented vitelliform lesions (Fig.
1A and B). FFA revealed a mild hyperfluorescence with moderate
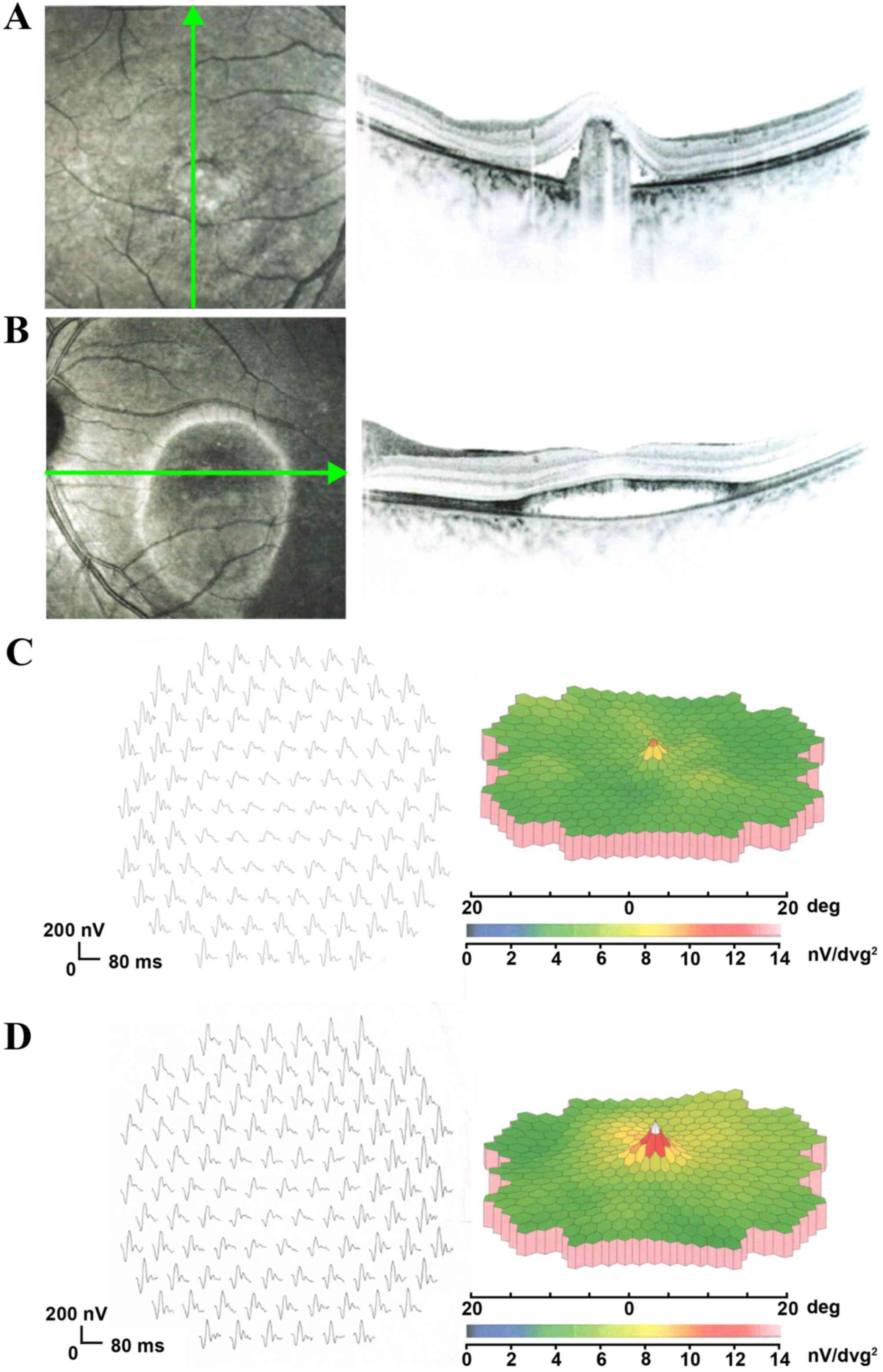
leakage in the fovea of the macula in both eyes (Fig. 1C and D). OCT scans revealed that
the foveal region in both eyes was abnormally thick, due to
neuroretinal detachment from the RPE. In the right eye, the
neuroretinal detachment was likely triggered by the abnormal
accumulation of hyperreflective materials beneath the retina
(Fig. 2A), whereas in the left
eye, it was likely due to the accumulation of sub-retinal fluid
(Fig. 2B). mfERGs (response of the
posterior fundus) revealed a mild decrease in the amplitude of the
foveal response in both eyes, although the peripheral mfERG
amplitudes were within normal limits (Fig. 2C and D).
Patient 2 had myopia and their decline in vision had
occurred over the last 3 years. Refractive error was −10.0 DS in
the right eye and −9.0 DS in the left eye. The BVCA was 0.4 in the
right eye and counting fingers at 50 cm away from the left eye. The
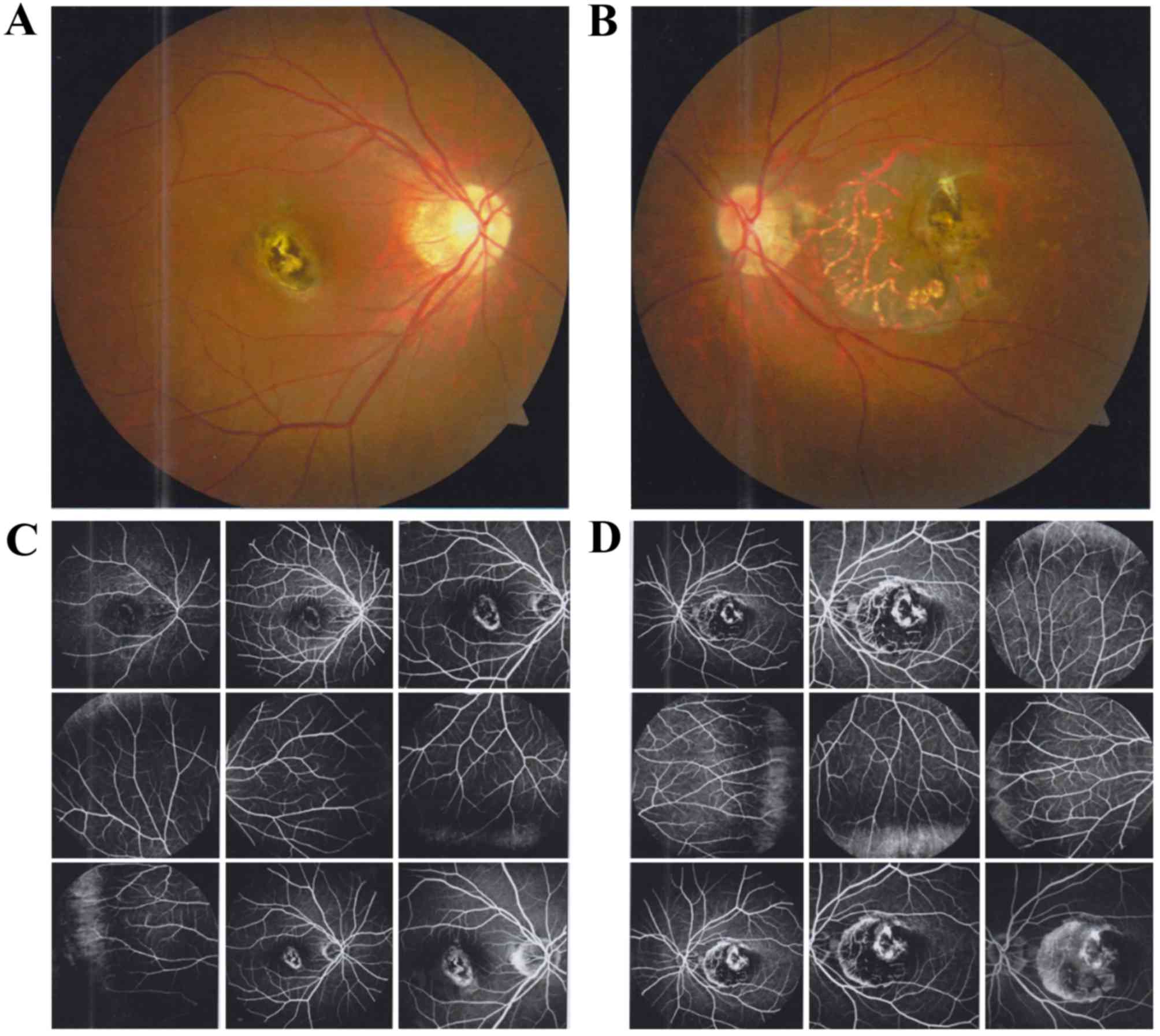
cornea and the lens were transparent. Fundus examination revealed
atrophic lesions in both eyes (Fig. 3A
and B). FFA revealed significant early hyperfluorescence that
had increased intensity at the late stage of the angiographic
sequence, with mild leakage in the right eye (Fig. 3C). The macular lesions exhibited a
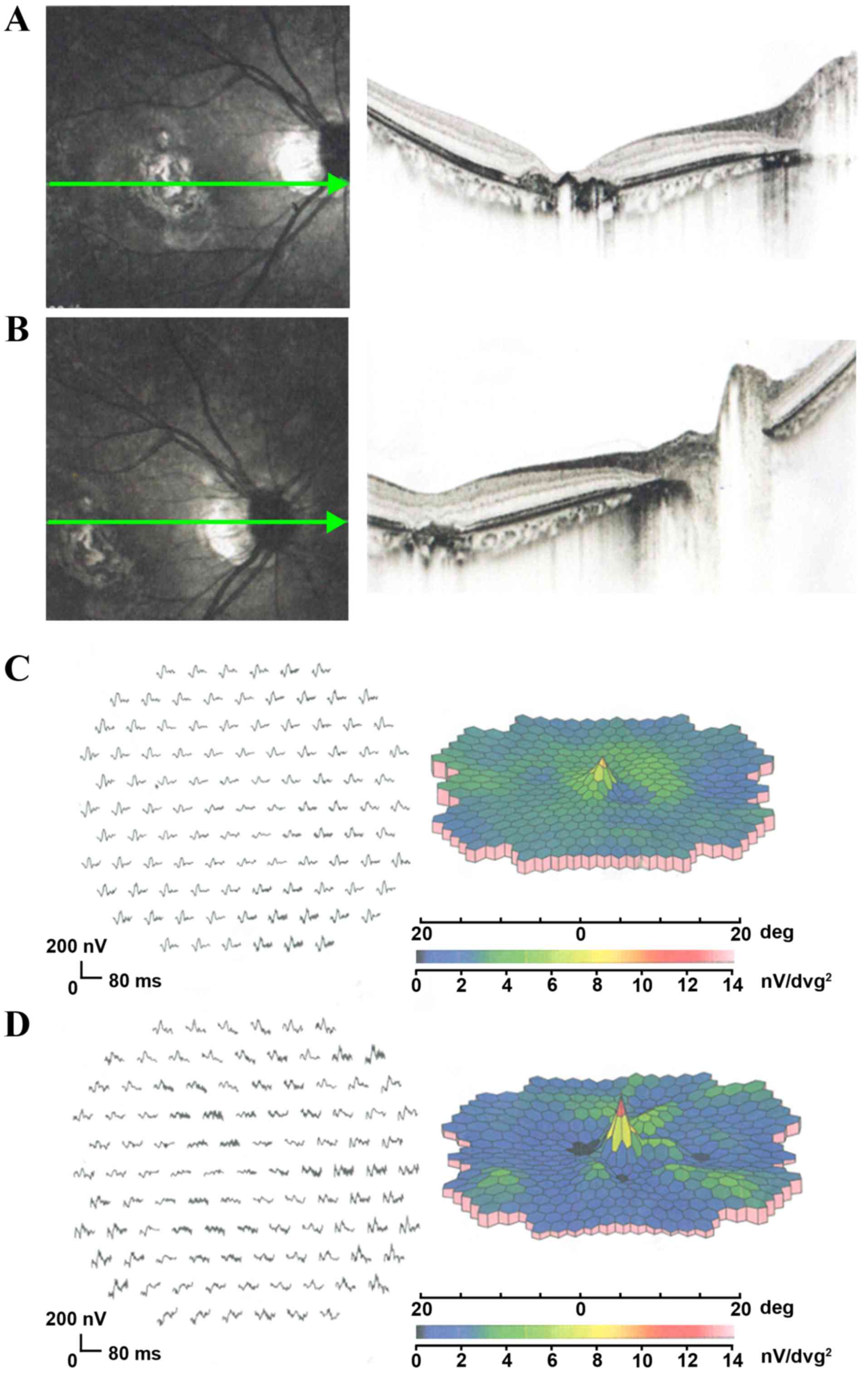
dystrophic pattern in the left eye (Fig. 3D). OCT revealed that the foveal
regions in both eyes were abnormally thin due to atrophy of the
retina and RPE (Fig. 4A and B).
Similar to Patient 1, mfERGs of Patient 2 revealed a significant
decrease in the amplitude of the foveal response in both eyes,
although most of the peripheral mfERG amplitudes were within normal
limits (Fig. 4C and D).
The present study was supported by the National
Natural Science Foundation of China (grant nos. 81500709, 81570862
and 81670872) and the State Scholarship Fund from the China
Scholarship Council.
|
1
|
Johnson AA, Guziewicz KE, Lee CJ, Kalathur
RC, Pulido JS, Marmorstein LY and Marmorstein AD: Bestrophin 1 and
retinal disease. Prog Retin Eye Res. 58:45–69. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xiao Q, Hartzell HC and Yu K: Bestrophins
and retinopathies. Pflugers Arch. 460:559–569. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Qian CX, Charran D, Strong CR, Steffens
TJ, Jayasundera T and Heckenlively JR: Optical coherence tomography
examination of the retinal pigment epithelium in best vitelliform
macular dystrophy. Ophthalmology. 124:456–463. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Guziewicz KE, Sinha D, Gómez NM, Zorych K,
Dutrow EV, Dhingra A, Mullins RF, Stone EM, Gamm DM,
Boesze-Battaglia K and Aguirre GD: Bestrophinopathy: An
RPE-photoreceptor interface disease. Prog Retin Eye Res. 58:70–88.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Arora R, Khan K, Kasilian ML, Strauss RW,
Holder GE, Robson AG, Thompson DA, Moore AT and Michaelides M:
Unilateral BEST1-associated retinopathy. Am J Ophthalmol.
169:24–32. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Subash M, Rotsos T, Wright GA, Devery S,
Holder GE, Robson AG, Pal B, Tufail A, Webster AR, Moore AT and
Michaelides M: Unilateral vitelliform maculopathy: A comprehensive
phenotype study with molecular screening of BEST1 and PRPH2. Br J
Ophthalmol. 96:719–722. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Liu J, Xuan Y, Zhang Y, Liu W and Xu G:
Bilateral macular holes and a new onset vitelliform lesion in Best
disease. Ophthalmic Genet. 38:79–82. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Elkhoyaali A, Chatoui S, Bercheq N,
Elouatassi N, Zerrouk R, Elasri F, Reda K and Oubaaz A: Choroidal
neovascularization complicating Best's vitelliform macular
dystrophy in a child. J Fr Ophtalmol. 39:69–73. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Peiretti E, Caminiti G, Forma G, Carboni
G, Dhaenens CM, Querques L, Souied E and Querques G: A Novel p.
Asp304Gly Mutation In Best1 gene associated with atypical best
vitelliform macular dystrophy phenotype and high intrafamilial
variability. Retina. 36:1733–1740. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Chacon-Camacho OF, Camarillo-Blancarte L
and Zenteno JC: OCT findings in young asymptomatic subjects
carrying familial BEST1 gene mutations. Ophthalmic Genet. 32:24–30.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Stanca HT and Nicolaescu M: Adult onset
foveomacular vitelliform dystrophy. Oftalmologia. 55:82–86.
2011.PubMed/NCBI
|
|
12
|
Brecher R and Bird AC: Adult vitelliform
macular dystrophy. Eye (Lond). 4:210–215. 1990. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Liu J, Zhang Y, Xuan Y, Liu W and Wang M:
Novel BEST1 Mutations and special clinical features of best
vitelliform macular dystrophy. Ophthalmic Res. 56:178–185. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lin Y, Li T, Gao H, Lian Y, Chen C, Zhu Y,
Li Y, Liu B, Zhou W, Jiang H, et al: Bestrophin 1 gene analysis and
associated clinical findings in a Chinese patient with Best
vitelliform macular dystrophy. Mol Med Rep. 16:4751–4755.
2017.PubMed/NCBI
|
|
15
|
Krämer F, White K, Pauleikhoff D, Gehrig
A, Passmore L, Rivera A, Rudolph G, Kellner U, Andrassi M, Lorenz
B, et al: Mutations in the VMD2 gene are associated with
juvenile-onset vitelliform macular dystrophy (Best disease) and
adult vitelliform macular dystrophy but not age-related macular
degeneration. Eur J Hum Genet. 8:286–292. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Glavač D, Jarc-Vidmar M, Vrabec K,
Ravnik-Glavač M, Fakin A and Hawlina M: Clinical and genetic
heterogeneity in Slovenian patients with BEST disease. Acta
Ophthalmol. 94:e786–e794. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Matson ME, Ly SV and Monarrez JL: Novel
Mutation in BEST1 Associated with Atypical Best Vitelliform
Dystrophy. Optom Vis Sci. 92:e180–e189. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Pasquay C, Wang LF, Lorenz B and Preising
MN: Bestrophin 1-phenotypes and functional aspects in
bestrophinopathies. Ophthalmic Genet. 36:193–212. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Seddon JM, Afshari MA, Sharma S, Bernstein
PS, Chong S, Hutchinson A, Petrukhin K and Allikmets R: Assessment
of mutations in the Best macular dystrophy (VMD2) gene in patients
with adult-onset foveomacular vitelliform dystrophy, age-related
maculopathy, and bull's-eye maculopathy. Ophthalmology.
108:2060–2067. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Marmorstein AD, Kinnick TR, Stanton JB,
Johnson AA, Lynch RM and Marmorstein LY: Bestrophin-1 influences
transepithelial electrical properties and Ca2+ signaling in human
retinal pigment epithelium. Mol Vis. 21:347–359. 2015.PubMed/NCBI
|
|
21
|
Tan X, Zhu Y, Chen C, Chen X, Qin Y, Qu B,
Luo L, Lin H, Wu M, Chen W and Liu Y: Sprouty2 Suppresses
Epithelial-Mesenchymal transition of human lens epithelial cells
through Blockade of Smad2 and ERK1/2 pathways. PLoS One.
11:e01592752016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zhang Y, Morgan R, Chen C, Cai Y, Clark E,
Khan WN, Shin SU, Cho HM, Al Bayati A, Pimentel A and Rosenblatt
JD: Mammary-tumor-educated B cells acquire LAP/TGF-β and PD-L1
expression and suppress anti-tumor immune responses. Int Immunol.
28:423–433. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Qin Y, Zhu Y, Luo F, Chen C, Chen X and Wu
M: Killing two birds with one stone: Dual blockade of integrin and
FGF signaling through targeting syndecan-4 in postoperative
capsular opacification. Cell Death Dis. 8:e29202017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Tan X, Chen C, Zhu Y, Deng J, Qiu X, Huang
S, Shang F, Cheng B and Liu Y: Proteotoxic stress desensitizes
TGF-beta signaling through receptor downregulation in retinal
pigment epithelial cells. Curr Mol Med. Jun 19–2017.(Epub ahead of
print). View Article : Google Scholar :
|
|
25
|
Tan X, Zhan J, Zhu Y, Cao J, Wang L, Liu
S, Wang Y, Liu Z, Qin Y, Wu M, et al: Improvement of uveal and
capsular biocompatibility of hydrophobic acrylic intraocular lens
by surface grafting with 2-Methacryloyloxyethyl
phosphorylcholine-methacrylic acid copolymer. Sci Rep. 7:404622017.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Aldehni F, Spitzner M, Martins JR,
Barro-Soria R, Schreiber R and Kunzelmann K: Bestrophin 1 promotes
epithelial-to-mesenchymal transition of renal collecting duct
cells. J Am Soc Nephrol. 20:1556–1564. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Oh SJ and Lee CJ: Distribution and
function of the bestrophin-1 (Best1) Channel in the Brain. Exp
Neurobiol. 26:113–121. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Xi L, Liu Y, Zhao F, Chen C and Cheng B:
Analysis of glistenings in hydrophobic acrylic intraocular lenses
on visual performance. Int J Ophthalmol. 7:446–451. 2014.PubMed/NCBI
|
|
29
|
Lin Y, Gao H, Ai S, Eswarakumar JVP, Chen
C, Zhu Y, Li T, Liu B, Liu X, Luo L, et al: C278F mutation in FGFR2
gene causes two different types of syndromic craniosynostosis in
two Chinese patients. Mol Med Rep. 16:5333–5337. 2017.PubMed/NCBI
|
|
30
|
Lin Y, Gao H, Ai S, Eswarakumar JV, Zhu Y,
Chen C, Li T, Liu B, Jiang H, Liu Y, et al: FGFR2 mutations and
associated clinical observations in two Chinese patients with
Crouzon syndrome. Mol Med Rep. Aug 29–2017.(Epub ahead of print).
View Article : Google Scholar
|
|
31
|
Lin Y, Gao H and Liu Y, Liang X, Liu X,
Wang Z, Zhang W, Chen J, Lin Z, Huang X and Liu Y: Two novel
mutations in the bestrophin-1 gene and associated clinical
observations in patients with best vitelliform macular dystrophy.
Mol Med Rep. 12:2584–2588. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Lin Y, Ai S, Chen C, Liu X, Luo L, Ye S,
Liang X, Zhu Y, Yang H and Liu Y: Ala344Pro mutation in the FGFR2
gene and related clinical findings in one Chinese family with
Crouzon syndrome. Mol Vis. 18:1278–1282. 2012.PubMed/NCBI
|
|
33
|
Lin Y, Liang X, Ai S, Chen C, Liu X, Luo
L, Ye S, Li B, Liu Y and Yang H: FGFR2 molecular analysis and
related clinical findings in one Chinese family with Crouzon
syndrome. Mol Vis. 18:449–454. 2012.PubMed/NCBI
|
|
34
|
Lin Y, Liu X, Yu S, Luo L, Liang X, Wang
Z, Chen C, Zhu Y, Ye S, Yan H and Liu Y: PAX6 analysis of two
sporadic patients from southern China with classic aniridia. Mol
Vis. 18:2190–2194. 2012.PubMed/NCBI
|
|
35
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Salvo J, Lyubasyuk V, Xu M, Wang H, Wang
F, Nguyen D, Wang K, Luo H, Wen C, Shi C, et al: Next-generation
sequencing and novel variant determination in a cohort of 92
familial exudative vitreoretinopathy Patients. Invest Ophthalmol
Vis Sci. 56:1937–1946. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Nikopoulos K, Gilissen C, Hoischen A, van
Nouhuys CE, Boonstra FN, Blokland EA, Arts P, Wieskamp N, Strom TM,
Ayuso C, et al: Next-generation sequencing of a 40 Mb linkage
interval reveals TSPAN12 mutations in patients with familial
exudative vitreoretinopathy. Am J Hum Genet. 86:240–247. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Kumar P, Henikoff S and Ng PC: Predicting
the effects of coding non-synonymous variants on protein function
using the SIFT algorithm. Nat Protoc. 4:1073–1081. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Avila-Fernandez A, Perez-Carro R, Corton
M, Lopez-Molina MI, Campello L, Garanto A, Fernandez-Sanchez L,
Duijkers L, Lopez-Martinez MA, Riveiro-Alvarez R, et al:
Whole-exome sequencing reveals ZNF408 as a new gene associated with
autosomal recessive retinitis pigmentosa with vitreal alterations.
Hum Mol Genet. 24:4037–4048. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Jansson Wivestad R, Berland S, Bredrup C,
Austeng D, Andréasson S and Wittström E: Biallelic mutations in the
BEST1 gene: Additional families with autosomal recessive
bestrophinopathy. Ophthalmic Genet. 37:183–193. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Wittström E, Ponjavic V, Bondeson ML and
Andréasson S: Anterior segment abnormalities and angle-closure
glaucoma in a family with a mutation in the BEST1 gene and Best
vitelliform macular dystrophy. Ophthalmic Genet. 32:217–227. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Gass JD: A clinicopathologic study of a
peculiar foveomacular dystrophy. Trans Am Ophthalmol Soc.
72:139–156. 1974.PubMed/NCBI
|
|
43
|
Chowers I, Tiosano L, Audo I, Grunin M and
Boon CJ: Adult-onset foveomacular vitelliform dystrophy: A fresh
perspective. Prog Retin Eye Res. 47:64–85. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Tiosano L, Grunin M, Hagbi-Levi S, Banin
E, Averbukh E and Chowers I: Characterising the phenotype and
progression of sporadic adult-onset foveomacular vitelliform
dystrophy. Br J Ophthalmol. 100:1476–1481. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Rishi E, Rishi P and Mahajan S:
Intravitreal bevacizumab for choroidal neovascular membrane
associated with Best's vitelliform dystrophy. Indian J Ophthalmol.
58:160–162. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Mimoun G, Caillaux V, Querques G,
Rothschild PR, Puche N and Souied EH: Ranibizumab for choroidal
neovascularization associated with adult-onset foveomacular
vitelliform dystrophy: One-year results. Retina. 33:513–521. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lee CS, Jun I, Choi SI, Lee JH, Lee MG,
Lee SC and Kim EK: A Novel BEST1 mutation in autosomal recessive
bestrophinopathy. Invest Ophthalmol Vis Sci. 56:8141–8150. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Caldwell GM, Kakuk LE, Griesinger IB,
Simpson SA, Nowak NJ, Small KW, Maumenee IH, Rosenfeld PJ, Sieving
PA, Shows TB and Ayyagari R: Bestrophin gene mutations in patients
with Best vitelliform macular dystrophy. Genomics. 58:98–101. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Allikmets R, Seddon JM, Bernstein PS,
Hutchinson A, Atkinson A, Sharma S, Gerrard B, Li W, Metzker ML,
Wadelius C, et al: Evaluation of the best disease gene in patients
with age-related macular degeneration and other maculopathies. Hum
Genet. 104:449–453. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Li T, Lin Y, Gao H, Chen C, Zhu Y, Liu B,
Lian Y, Li Y, Zhou W, Jiang H, et al: Two heterozygous mutations
identified in one Chinese patient with bilateral macular coloboma.
Mol Med Rep. 16:2505–2510. 2017.PubMed/NCBI
|
|
51
|
Carter DA, Smart MJ, Letton WV, Ramsden
CM, Nommiste B, Chen LL, Fynes K, Muthiah MN, Goh P, Lane A, et al:
Mislocalisation of BEST1 in iPSC-derived retinal pigment epithelial
cells from a family with autosomal dominant
vitreoretinochoroidopathy (ADVIRC). Sci Rep. 6:337922016.
View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Moshfegh Y, Velez G, Li Y, Bassuk AG,
Mahajan VB and Tsang SH: BESTROPHIN1 mutations cause defective
chloride conductance in patient stem cell-derived RPE. Hum Mol
Genet. 25:2672–2680. 2016.PubMed/NCBI
|
|
53
|
Qu Z, Cheng W, Cui Y, Cui Y and Zheng J:
Human disease-causing mutations disrupt an NC-terminal interaction
and channel function of bestrophin 1. J Biol Chem. 284:16473–16481.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Hartzell HC, Qu Z, Yu K, Xiao Q and Chien
LT: Molecular physiology of bestrophins: Multifunctional membrane
proteins linked to best disease and other retinopathies. Physiol
Rev. 88:639–672. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Tsunenari T, Sun H, Williams J, Cahill H,
Smallwood P, Yau KW and Nathans J: Structure-function analysis of
the bestrophin family of anion channels. J Biol Chem.
278:41114–41125. 2003. View Article : Google Scholar : PubMed/NCBI
|