Introduction
Due to the limited ability of cardiac myocytes to
proliferate, low levels of apoptosis can result in profound
structural and functional consequences in the myocardium, leading
to cardiac dysfunction and heart failure (HF). Therefore, novel
therapeutic targets are required to investigate the reversal of HF
pathogenesis.
Apelin (APL), isolated from the digestive juice of
cattle by Tatemoto et al (1) in 1998, is a protein hormone derived
from the adipose tissue family. APL-13 is a member of the APL
endogenous peptide family, with powerful inotropic and
cardio-protective properties (2).
APL exerts its effects by binding and activating APL receptor (APJ,
gene symbol APLNR), a member of the G-protein coupled receptor
(GPCR) super-family. GPCRs are central to many endocrine pathways
in the body, and represent a major therapeutic target class. Both
APL and APJ receptors are widely distributed in most tissues,
including the lung, heart, brain, skeletal muscle, kidney and
liver. APL can promote proliferation in various cell types, as
retinal Müller and retinal endothelial cells (3–7).
Furthermore, APL-13 exerts a cardio-protective effect of the
myocardium under pathological states, including myocardial
infarction (MI) (8,9). However, to the best of our knowledge,
there is currently no information about the effect of APL in H9c2
cells physiological conditions.
The signaling cascades of the mitogen activated
protein kinase (MAPK) family, and phosphoinositide 3-kinase
(PI3K)/protein kinase B (Akt), are believed to be involved in
cell-cycle regulation, apoptosis and inflammation (10,11).
A growing body of evidence suggests that the Akt and extracellular
signal-regulated (ERK) signaling pathway serve important roles in
the development of cardiac hypertrophy and progression to HF.
Therefore, it was hypothesized that APL-13 may exert proliferative
effects on H9c2 cells under physiological conditions, which may be
mediated by the Akt and ERK1/2 signaling pathways.
The present study investigated the potential
proliferative role of APL under physiological conditions in H9c2.
The effect of exogenous recombinant APL on cell proliferation and
phosphorylation of ERKs and Akt, and the underlying mechanisms,
were examined.
Materials and methods
Reagents
APL-13 trifluoroacetate salt and MTT were purchased
from Sigma-Aldrich; Merck KGaA (Darmstadt, Germany). Dulbecco's
modified Eagle's medium (DMEM) was purchased from Hyclone; GE
Healthcare Life Sciences (Logan, UT, USA). Fetal bovine serum (FBS)
was purchased from Gibco; Thermo Fisher Scientific, Inc. (Waltham,
MA, USA). LY294002 (Akt inhibitor) and U0126 (ERK1/2 inhibitor)
were purchased from Selleck Chemicals (Houston, TX, USA). An
anti-β-actin primary antibody (ab8226; 1:5,000) was purchased from
Abcam (Cambridge, MA, USA,). Antibodies for phosphorylated (p)-Akt
(Ser473; 4060S; 1:1,000), Akt (2920S; 1:1,000), p-ERK1/2 (4370S;
1:1,000) and ERK1/2 (9102S; 1:1,000) were purchased from Cell
Signaling Technology, Inc. (Danvers, MA, USA).
Cell culture
H9c2 cells were purchased from the Chinese Academy
of Sciences (Shanghai, China) and were cultured in DMEM
supplemented with FBS, 2 mM glutamine and 1%
penicillin/streptomycin (Hyclone; GE Healthcare Life Sciences,
Logan, UT, USA). Cells were allowed to reach 80% confluence in
complete DMEM and were incubated for an additional 24 h in
serum-free medium prior to experimental treatments. Following this,
H9c2 cells were treated with 0, 5, 25, 50, 100, 200, 400, 600, 800
or 1,000 nM APL-13 for 24 h (5).
In experiments involving kinase inhibitors, LY294002
and U0126 were dissolved in dimethyl sulfoxide (DMSO) and diluted
with DMEM. The final concentration of DMSO was 1%, which had no
effect on cell viability, and the final concentrations of LY294002
and U0126 were 10 µM (12). Drug
solutions were freshly prepared prior to each experiment.
A pathologist blinded to the study reviewed 10
sections per culture dish. All images were obtained using an
Olympus LCX100 Imaging system.
MTT assay of cell proliferation
Cell proliferation was evaluated by MTT assay
(13). After synchronization for
24 h by serum starvation, cells were treated with APL-13 (50, 100
or 200 nM) for 24 h, following which 5 mg/ml MTT was added and
cells were incubated for 4 h at 37°C. Subsequently, the supernatant
was removed from each well. The coloured formazan crystal produced
from MTT was dissolved in 150 µl DMSO and the absorbance was
measured at a wavelength of 490 nm using a microplate reader (Model
550; Bio-Rad Laboratories, Inc., Hercules, CA, USA). Percentage
viability was calculated as the optical density (OD) of
drug-treated sample/OD of control sample ×100%, assuming that the
absorbance of control sample was 100%.
Western blotting
Western blotting was used to determine the protein
expression levels of p-Akt and p-ERK, as described previously
(12). Briefly, H9c2 cells were
seeded into 6-well culture plates. After a 24-h drug incubation,
cells were collected and lysed for 30 min on ice in lysis buffer.
Radioimmunoprecipitation assay buffer (Beyotime Institute of
Biotechnology, Jiangsu, China) was used to extract total protein
from cultured cells. The quantity of protein extracted from the
cells was measured using a Bicinchoninic Acid protein assay reagent
kit (Pierce; Thermo Fisher Scientific, Inc.). An equal amount of
total protein (80 µg per lane) from each sample was separated by
5–10% SDS-PAGE and transferred onto a polyvinylidene difluoride
membrane. The membranes were blocked with 5% non-fat dry milk in
PBS with 0.05% Tween 20 and incubated overnight at 4°C with primary
antibodies followed by a 2 h incubation with HRP-conjugated goat
anti-rabbit IgG (7074S; 1:5,000; Cell Signaling Technology, Inc.,
Danvers, MA, USA). The blots were developed using an enhanced
chemiluminescence detection kit (EMD Millipore, Billerica, MA, USA)
and visualized using a FluroChem E Imager (ProteinSimple;
Bio-Techne, Minneapolis, MN, USA). Measurements to determine the
relative densities were normalized to that of β-actin using Image J
software (version 1.38x; National Institutes of Health, Bethesda,
MD, USA) (14).
Statistical analysis
The data are presented as the mean ± standard
deviation. Unpaired Student's t-test was used to compare values
between two groups, and one-way analysis of variance was used to
compare differences between more than two groups, followed by a
Newman-Keuls post hoc test. Analyses were performed using SPSS 17.0
software (SPSS, Inc., Chicago, IL, USA). P<0.05 was considered
to indicate a statistically significant difference.
Results
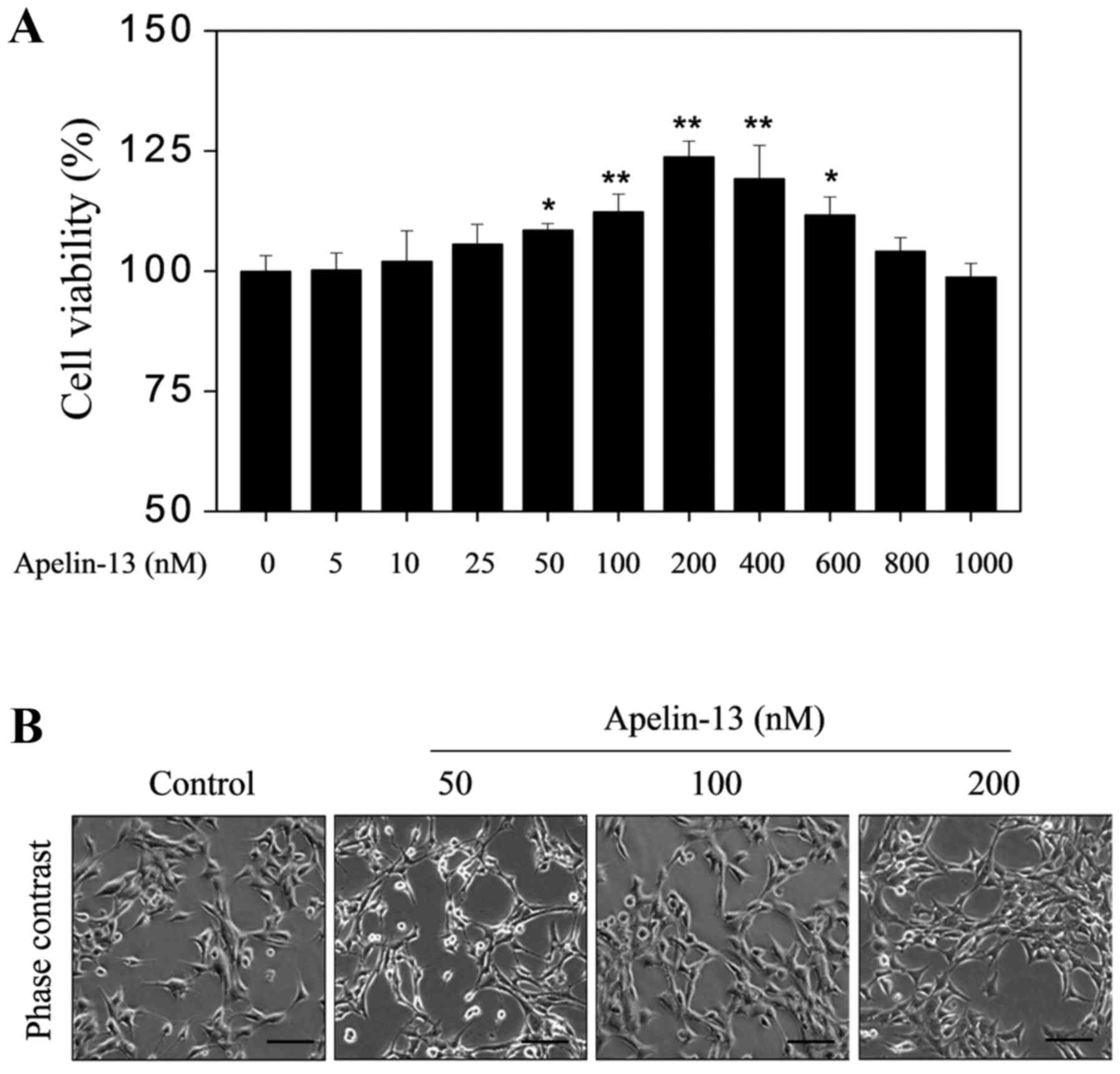
APL-13 alone can enhance H9c2
proliferation
To determine whether APL-13 itself promotes H9c2
proliferation in the absence of other stimuli, H9c2 growth was
assessed in response to various physiologically relevant
concentrations of APL-13 (0, 5, 25, 50, 100, 200. 400, 600, 800 and
1,000 nM) by MTT assay. APL-13 treatment alone increased the
percentage cell viability compared with the baseline constitutive
release over a range of physiological extracellular concentrations
from 50 to 600 nM, with a maximal effect at 200 nM (Fig. 1A). Microscopic examination
confirmed these results (Fig.
1B).
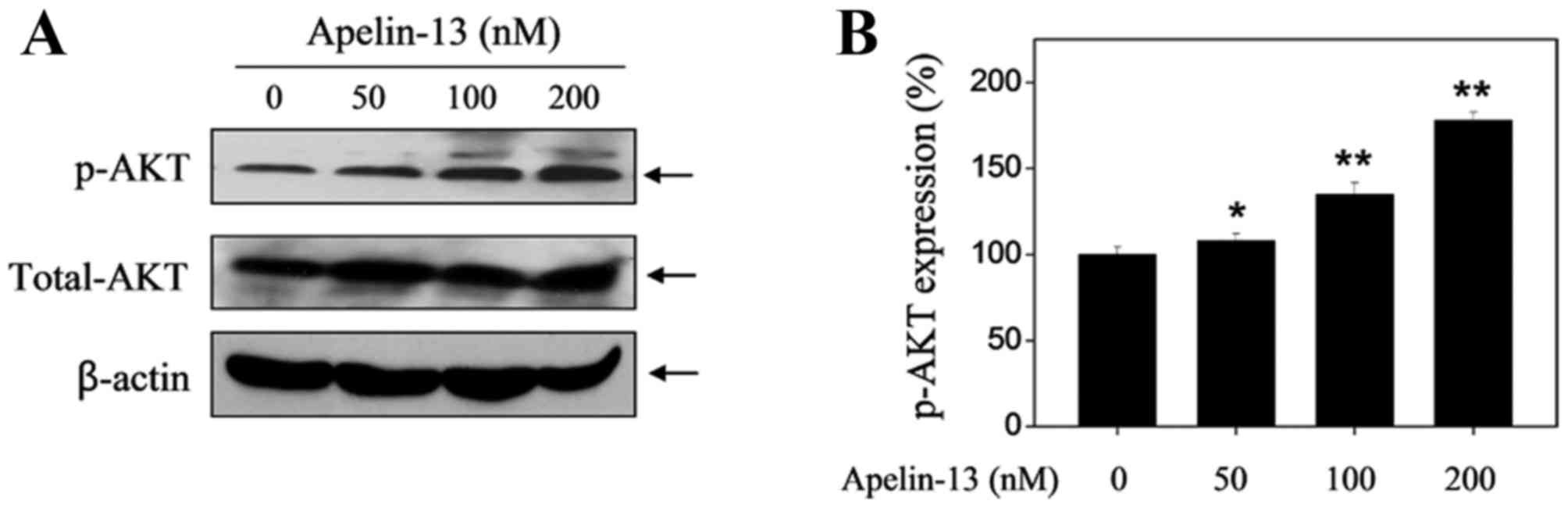
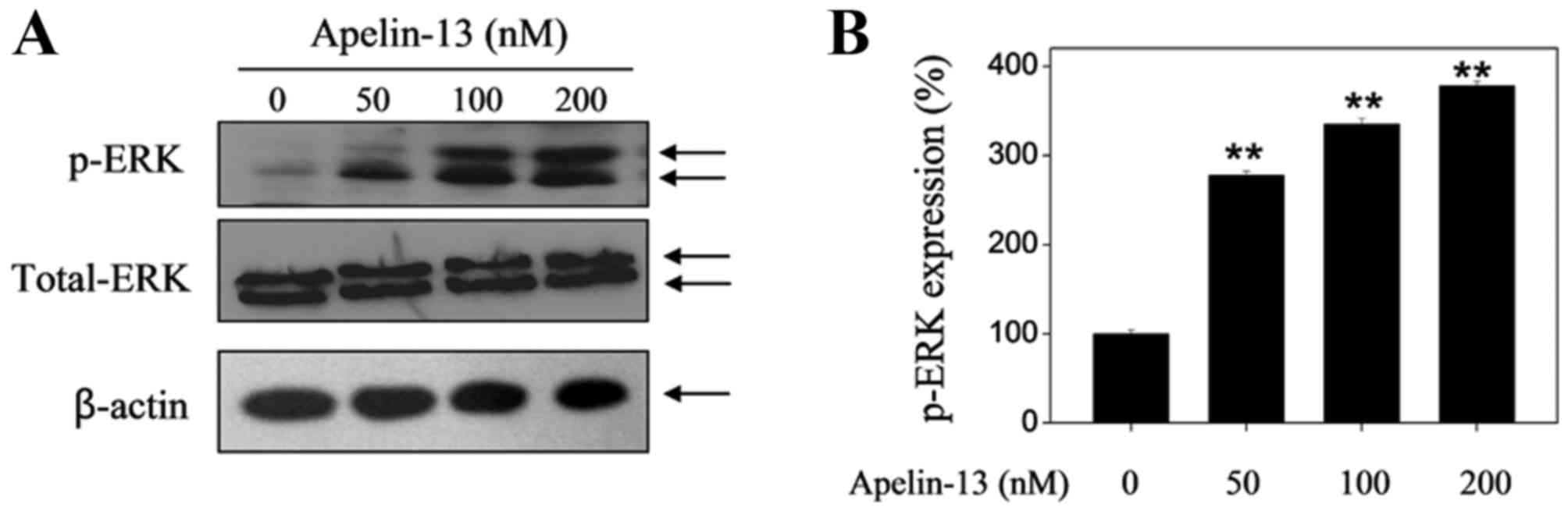
APL-13 upregulates the protein
expression levels of p-Akt and p-ERK
The Akt and ERK signaling pathways have been
suggested to serve an important role in cell proliferation,
differentiation, survival and apoptosis (15). To determine whether Akt and ERKs
are implicated in APL-13-mediated proliferative activity, changes
in the levels of p-Akt and p-ERK expression in H9c2 cells in the
presence or absence of APL-13 were examined. As presented in
Fig. 2, upregulation of p-Akt was
observed after APL-13 treatment alone (50, 100 and 200 nM) in a
dose-dependent manner when incubated for 24 h. In addition, APL-13
treatment increased the levels of p-ERK expression in a similar
manner (Fig. 3).
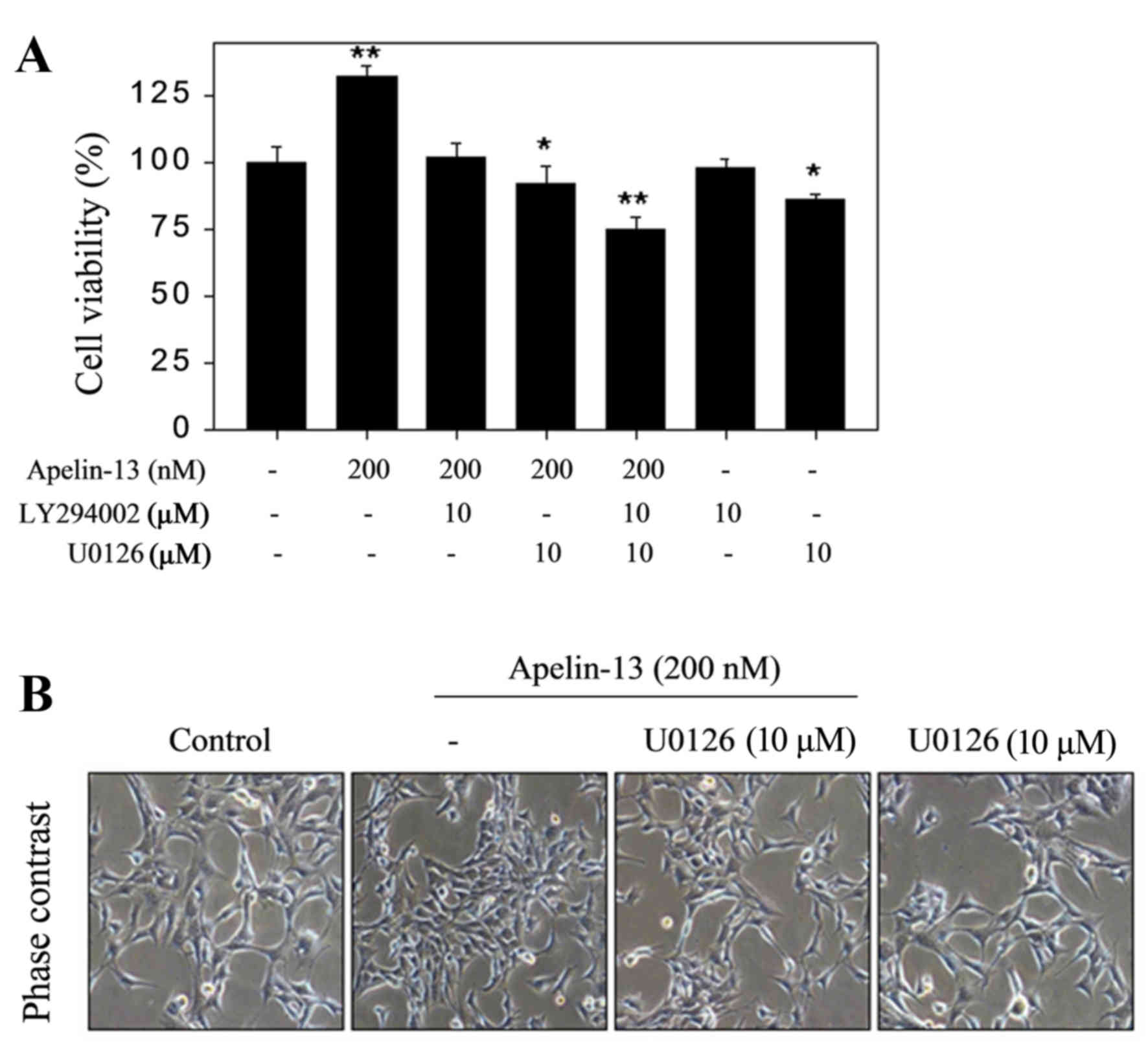
APL-13-mediated H9c2 proliferation
requires Akt and ERK signaling pathway activation
To further investigate the molecular mechanism by
which APL-13 induces H9c2 proliferation, H9c2 cells were pretreated
with inhibitors of Akt (LY294002; 10 µM) and ERK1/ERK2 (U0126; 10
µM) 2 h before stimulation with APL-13 (200 nM) to determine
whether ERK1/2 and Akt are involved in APL-13-induced cell
proliferation. LY294002 and U0126 co-treatment markedly abolished
APL-13 induced proliferation as determined by the MTT assay, while
LY294002 or U0126 alone had no effect on H9c2 proliferation in
cells not treated with APL-13. However, LY294002 reversed the
pro-proliferative H9c2 activity of APL-13. Treatment with U0126
reversed APL-13-induced cell proliferation (Fig. 4). Taken together, these results
suggest that the Akt and MAPK signaling pathways are involved in
the mechanisms underlying the proliferative effects of apelin-13.
Collectively, these data suggested that APL-13 mediates H9c2 growth
via activation of the Akt and ERK1/2 signaling pathways.
Discussion
To the best of our knowledge, the present study
demonstrated for the first time that APL-13 alone can enhance H9c2
cell proliferation, which is mediated by the ERK1/2 and Akt
signaling pathway. APL has been reported to activate multiple
protective mechanisms to prevent heart, brain, liver and kidney
injury, thus rising to be a promising therapeutic target for
ischemic and other associated diseases (16). APL-13 appears to be the predominant
isopeptide in the human myocardium and plasma (17), and it has greater biological
activity than APL-36 or −17, measured as the extracellular
acidification rate in cultured cells expressing the APJ receptor
(18). In particular, APL-13
serves a fundamental role in the occurrence and development of
cardiovascular diseases.
A biological rationale for the potential use of APL
as a therapeutic agent is confirmed by its low level in patients
with acute coronary syndromes and established coronary artery
disease. Thus, plasma APL concentration is considerably reduced in
patients with acute MI in comparison with the control group, and
this low level is maintained over time (19,20).
Consistent with this, upregulation of myocardial APL mRNA
expression during ischaemic insult is reverses in response to
reperfusion in a rat model of MI (21). Its promise is heightened further by
the observation that, unlike other and more established
cardio-protective pathways, it appears to be downregulated in heart
failure, suggesting that augmentation of this axis may
significantly impact HF (17).
Therefore, downregulation of APL may contribute to myocardial
apoptosis in MI and HF, and supports the use of exogenous APL
peptides for the treatment of HF. However, APL synthesis appears to
occur predominantly in the endothelium, despite the broad
distribution of APJ receptors (17). Therefore, it was hypothesized that
prolonged exogenous supplementation of APL may encourage myocardial
survival by combining with the APJ receptor in established HF.
Based on the protective mechanism of APL-13 in response to various
injury model and stimuli, the present study aimed to elucidate the
key role of exogenous APL-13 in H9c2 cells. It was demonstrated
that when H9c2 cells were incubated with various concentrations of
APL-13, cell proliferation was significantly increased at
concentrations from 50 to 600 nM, as determined by MTT assay, which
is supported by the fact that defects in APL in mice induces
age-dependent progressive cardiac dysfunction (22).
Subsequently, the cellular and molecular mechanisms
underlying APL-13-induced H9c2 proliferation were examined. The
PI3K/Akt and ERK1/2 signaling pathways serve an important role in
cell proliferation, differentiation, survival and apoptosis,
especially under various pathological states (12,23).
In addition, APL has been reported to promote the phosphorylation
of ERK and Akt in umbilical endothelial cells and vascular smooth
muscular cells (6,7). Consist with this, the present study
revealed that treatment of APL-13 alone stimulated ERK activation.
Furthermore, APL-13 upregulated p-Akt and p-ERK1/2, indicating a
potential involvement of Akt and ERK signaling pathways in
APL-13-mediated H9c2 proliferation. To strengthen the potential
conclusion, the present study further tested the effects of the
specific inhibitors LY294002 and U0126 on the proliferative effects
of APL-13. The results demonstrated that pretreatment of LY294002
or U0126 blocked APL-13-mediated H9c2 proliferation, further
indicating that APL-13 exerts a neuroprotective activity via
activating PI3K/Akt and MAPK signaling pathways.
Further studies are required to evaluate the
functional effect of these novel analogs in vivo, with a
Pressure-Volume curve device, and to further test their comparative
cardio-protective potential in established experimental HF model.
In addition, determining the therapeutic potential of augmenting
APL signaling in patients with heart failure.
In conclusion, APL-13 upregulated p-Akt and p-ERK1/2
levels and increased H9c2 cell proliferation under physiological
states, suggesting that Akt and ERK1/2 signals are involved in the
mechanism of the proliferative role of APL. The present study
expanded on current knowledge of APL-13 as an endogenous surviving
signal, and implicate it as a potential treatment option for
cardiovascular disease.
References
|
1
|
Tatemoto K, Hosoya M, Habata Y, Fujii R,
Kakegawa T, Zou MX, Kawamata Y, Fukusumi S, Hinuma S, Kitada C, et
al: Isolation and characterization of a novel endogenous peptide
ligand for the human APJ receptor. Biochem Biophys Res Commun.
251:471–476. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lesur O: Myocardial impact and
cardioprotective effects of apelin-13 and a c-terminal-modified
analog during lps and clp experimental sepsis. Intensive Care Med
Exp. 3:A4362015. View Article : Google Scholar :
|
|
3
|
Paine SK, Basu A, Mondal LK, Sen A,
Choudhuri S, Chowdhury IH, Saha A, Bhadhuri G, Mukherjee A and
Bhattacharya B: Association of vascular endothelial growth factor,
transforming growth factor beta, and interferon gamma gene
polymorphisms with proliferative diabetic retinopathy in patients
with type 2 diabetes. Mol Vis. 18:2749–2757. 2012.PubMed/NCBI
|
|
4
|
Lu Q, Jiang YR, Qian J and Tao Y:
Apelin-13 regulates proliferation, migration and survival of
retinal Müller cells under hypoxia. Diabetes Res Clin Pract.
99:158–167. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bai B, Cai X, Jiang Y, Karteris E and Chen
J: Heterodimerization of apelin receptor and neurotensin receptor 1
induces phosphorylation of ERK(1/2) and cell proliferation via
Galphaq-mediated mechanism. J Cell Mol Med. 18:2071–2081. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Liu QF, Yu HW, Sun LL, You L, Tao GZ and
Qu BZ: Apelin-13 upregulates Egr-1 expression in rat vascular
smooth muscle cells through the PI3K/Akt and PKC signaling
pathways. Biochem Biophys Res Commun. 468:617–621. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Qin D, Zheng XX and Jiang YR: Apelin-13
induces proliferation, migration, and collagen I mRNA expression in
human RPE cells via PI3K/Akt and MEK/Erk signaling pathways. Mol
Vis. 19:2227–2236. 2013.PubMed/NCBI
|
|
8
|
Tao J, Zhu W, Li Y, Xin P, Li J, Liu M,
Redington AN and Wei M: Apelin-13 protects the heart against
ischemia-reperfusion injury through inhibition of ER-dependent
apoptotic pathways in a time-dependent fashion. Am J Physiol Heart
Circ Physiol. 301:H1471–H1486. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang S, Li H, Tang L, Ge G, Ma J, Qiao Z,
Liu H and Fang W: Apelin-13 protects the heart against
ischemia-reperfusion injury through the RISK-GSK-3β-mPTP
pathway. Arch Med Sci. 11:1065–1073. 2015.PubMed/NCBI
|
|
10
|
Liou SF, Hsu JH, Chen YT, Chen IJ and Yeh
JL: KMUP-1 attenuates endothelin-1-induced cardiomyocyte
hypertrophy through activation of heme oxygenase-1 and suppression
of the Akt/GSK-3β, calcineurin/NFATc4 and RhoA/ROCK
pathways. Molecules. 20:10435–10449. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Dong WQ, Chao M, Lu QH, Chai WL, Zhang W,
Chen XY, Liang ES, Wang LB, Tian HL, Chen YG and Zhang MX:
Prohibitin overexpression improves myocardial function in diabetic
cardiomyopathy. Oncotarget. 7:66–80. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zou Y, Wang B, Fu W, Zhou S, Nie Y and
Tian S: Apelin-13 protects PC12 cells from corticosterone-induced
apoptosis through PI3K and ERKs activation. Neurochem Res.
41:1635–1644. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Li YG, Han BB, Li F, Yu JW, Dong ZF, Niu
GM, Qing YW, Li JB, Wei M and Zhu W: High glucose induces
down-regulated GRIM-19 expression to activate STAT3 signaling and
promote cell proliferation in cell culture. PLoS One.
11:e01536592016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Yin J, Hu H, Li X, Xue M, Cheng W, Wang Y,
Xuan Y, Yang N, Shi Y and Yan S: Inhibition of Notch signaling
pathway attenuates sympathetic hyperinnervation together with the
augmentation of M2 macrophages in rats post-myocardial infarction.
Am J Physiol Cell Physiol. 310:C41–C53. 2016.PubMed/NCBI
|
|
15
|
Khan M, Maryam A, Qazi JI and Ma T:
Targeting apoptosis and multiple signaling pathways with icariside
II in cancer cells. Int J Biol Sci. 11:1100–1102. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Bircan B, Cakir M, Kırbağ S and Gül HF:
Effect of apelin hormone on renal ischemia/reperfusion induced
oxidative damage in rats. Ren Fail. 38:1122–1128. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Dalzell JR, Rocchiccioli JP, Weir RA,
Jackson CE, Padmanabhan N, Gardner RS, Petrie MC and McMurray JJ:
The emerging potential of the apelin-APJ system in heart failure. J
Card Fail. 21:489–498. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Boal F, Timotin A, Roumegoux J, Alfarano
C, Calise D, Anesia R, Parini A, Valet P, Tronchere H and Kunduzova
O: Apelin-13 administration protects against
ischaemia/reperfusion-mediated apoptosis through the FoxO1 pathway
in high-fat diet-induced obesity. Br J Pharmacol. 173:1850–1863.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Wang W, McKinnie SM, Patel VB, Haddad G,
Wang Z, Zhabyeyev P, Das SK, Basu R, McLean B, Kandalam V, et al:
Loss of Apelin exacerbates myocardial infarction adverse remodeling
and ischemia-reperfusion injury: Therapeutic potential of synthetic
Apelin analogues. J Am Heart Assoc. 2:e0002492013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tycinska AM, Sobkowicz B, Mroczko B,
Sawicki R, Musial WJ, Dobrzycki S, Waszkiewicz E, Knapp MA and
Szmitkowski M: The value of apelin-36 and brain natriuretic peptide
measurements in patients with first ST-elevation myocardial
infarction. Clin Chim Acta. 411:2014–2018. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Kleinz MJ and Baxter GF: Apelin reduces
myocardial reperfusion injury independently of PI3K/Akt and P70S6
kinase. Regul Pept. 146:271–277. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Helske S, Kovanen PT, Lommi J, Turto H and
Kupari M: Transcardiac gradients of circulating apelin: Extraction
by normal hearts vs. release by hearts failing due to pressure
overload. J Appl Physiol (1985). 109:1744–1748. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Yang Y, Zhang XJ, Li LT, Cui HY, Zhang C,
Zhu CH and Miao JY: Apelin-13 protects against apoptosis by
activating AMP-activated protein kinase pathway in ischemia stroke.
Peptides. 75:96–100. 2016. View Article : Google Scholar : PubMed/NCBI
|