Introduction
Cardiac gap junctions serve a crucial role in
maintaining heart tissue homeostasis. There is abundant evidence
that alterations of gap junctional intercellular communication
(GJIC) is strongly associated with the development of heart
diseases, including hypertension (1), arrhythmias (2,3),
atherosclerosis and restenosis (4). It has been demonstrated that
connexin43 (Cx43) is predominantly located in ventricular muscles
(5). Cx43 forms gap junction
channels and is additionally involved in the modification of
cell-cycle (6). Accumulating
evidence has indicated that Cx43 expression levels decrease in
diabetes (7,8), myocardial infraction (2,3) and
heart failure (9). In heterozygous
Cx43-deficient mice, reduced Cx43 expression levels greatly
increased the risk of ventricular arrhythmias (10). Downregulation of Cx43 may activate
endothelial cells to a pathological status (11). Taken together, these results
suggest that the reduction of cardiac Cx43 expression levels is a
potential indicator of heart dysfunction.
Hypoglycemia is the most common side effect of
exogenous insulin or insulin secretagogue administration in
patients with type 1 and 2 diabetes. Hypoglycemia has been
demonstrated to be strongly associated with adverse cardiovascular
events and mortality (12).
Numerous efforts have been made to identify potential underlying
mechanisms between hypoglycemia and cardiovascular events. An
increase in sympathetic system activity may lead to destabilization
of atherosclerotic plaques during hypoglycemic episodes (13). In addition, previous studies have
indicated that hypoglycemic episodes may induce cardiac and
cerebral ischemia (14),
arrhythmia (15), thrombosis and
inflammation (16), all of which
are strongly associated with the pathogenesis of cardiovascular
diseases. However, establishing a direct causal link is difficult.
Cardiac Cx43 acts as a potential biomarker of heart function;
however, the association between cardiac Cx43 and hypoglycemia
remains unclear.
Based on previous observations, the present study
aimed to investigate the effect of glucose deprivation (GD) on Cx43
expression levels in H9c2 cells and to examine the underlying
mechanisms.
Materials and methods
Cell culture and reagents
H9c2 cells were obtained from the Cell Bank of
Chinese Academy of Science (Shanghai, China). The cells were
cultured in Dulbecco's modified Eagle's medium supplemented with
10% fetal bovine serum (Gibco; Thermo Fisher Scientific, Waltham,
MA, USA) at 37°C in a humidified incubator in 5% CO2 and
95% air.
U0126 and diphenyleneiodonium (DAPI) were purchased
from Sigma-Aldrich; Merck Millipore (Darmstadt, Germany).
Anti-extracellular signal-regulated kinase (ERK; cat no. 9102;
dilution, 1:1,000), anti-phosphorylated (p)-ERK (cat no. 9101;
dilution, 1:1,000), anti-Cx43 (cat no. 3512; dilution, 1:1,000),
anti-Beclin-1 (cat no. 3738; dilution, 1:1,000), anti-p62 (cat no.
5114; dilution, 1:1,000), anti-microtubule-associated protein
1A/1B-light chain 3 (LC3; cat no. 2775; dilution, 1:1,000),
anti-B-cell lymphoma 2 (Bcl-2; cat no. 2870; dilution, 1:1,000) and
anti-Bcl-2-associated X protein (Bax; cat no. 2772; dilution,
1:1,000) primary antibodies were obtained from Cell Signaling
Technology, Inc. (Danvers, MA, USA).
GD treatment
H9c2 cells were exposed to media containing no
glucose (NG) for 2 h. The U0126 group were treated with 15 µM U0126
1 h prior to and during GD. The high glucose (HG) group was exposed
to medium containing HG for 2 h.
Cell viability assay
Cell viability was measured by MTT assay. H9c2 cells
were seeded into 96-well plates at a density of 2×104
cells/well. After 24 h culture, the cells were subjected to GD
treatment in the presence or absence of U0126. MTT solution (0.5
mg/ml; Beyotime Institute of Biotechnology, Shanghai, China) was
added to each well and incubated for 4 h at 37°C. Following this,
the medium was removed and dimethyl sulfoxide was added to dissolve
the blue-colored formazan product. Absorbance was measured at a
wavelength of 490 nm using a microplate reader. Cell survival rates
were expressed as the percentage of the absorbance of treated cells
compared with the sham group.
Lactate dehydrogenase (LDH)
release
Cell death was assessed using an LDH Activity Assay
kit (Beyotime Institute of Biotechnology) according to the
manufacturer's protocol. Cell medium was collected and mixed with
LDH reaction buffer for 30 min at room temperature. Following this,
the absorbance was read at a wavelength of 450 nm using a
microplate reader.
Transfection of small interfering RNA
(siRNA)
siRNA-ERK was synthesized by Shanghai GenePharma
Co., Ltd. (Shanghai, China). Primer sequences were as follows:
Forward, 5′-GCUAUUACCGAGCAGACUUTT-3′ and reverse,
5′-AAGUCUGCUCGGUAAUAGCTT-3′ for ERK. H9c2 cells were seeded into
6-well plates to ensure 50–60% confluence and subsequently
transfected with siRNA. Briefly, 1 µl siRNA-ERK was mixed with 3 µl
transfection reagent, diluted with 250 µl transfection buffer (both
from Qiagen GmbH, Hilden, Germany) and subsequently incubated for
25 min at 37°C. Cells were incubated with the media for 24 h at
37°C, following which the medium was replaced and cells were
incubated for a further 48 h. The efficacy of transfection was
measured by western blot analysis.
Immunofluorescence
Confluent cells were seeded onto coverslips and
fixed with 4% paraformaldehyde. Following fixation, 0.1% Triton
X-100 and 10% goat serum were used to permeabilize cells and
saturate the non-specific binding sites, respectively. Cells were
subsequently incubated with anti-Cx43 (4°C overnight), followed by
incubation with an Alexa Fluor 568 goat secondary antibody (cat no.
175471; Abcam, Cambridge, MA, USA; 25°C for 2 h; dilution,
1:2,000). Finally, cell nuclei were stained with 0.5 µg/ml DAPI.
The cells were examined using an inverted fluorescent
microscope.
Western blot analysis
Following treatment, H9c2 cells were harvested and
lysed with radioimmunoprecipitation assay lysis buffer (50 mM
Tris-HCl, pH 7.4, 150 mM NaCl, 0.1% SDS, 1% sodium deoxycholate)
supplemented with protease and phosphatase inhibitor cocktails
(Roche Applied Science, Penzburg, Germany). The supernatant
fractions were collected by centrifugation (10,000 × g for 15 min
at 4°C) and protein concentration was determined using a
Bicinchoninic Acid kit (Beyotime Institute of Biotechnology).
Lysate protein was separated by 10–12% SDS-PAGE and transferred
onto polyvinylidene difluoride membranes. The membranes were
incubated with primary antibodies against ERK, p-ERK, Cx43,
Beclin-1, p62, LC3, Bcl-2 and Bax at 4°C overnight. Subsequently
incubated for 2 h with a horseradish peroxidase-conjugated goat
anti-rabbit secondary antibody at room temperature. Proteins were
detected using Enhanced Chemiluminescence reagents (EMD Millipore,
Billerica, MA, USA). The bands were imaged using the LAS-4500
system and subsequently quantified using Gel-Pro Analyzer software
(version 4.0; Media Cybernetics, Inc., Rockville, MD, USA).
Statistical analysis
Data are expressed as mean ± standard deviation.
Student's t-test was performed to compare the differences between
the HG and NG groups, and between NG and U0126 or siRNA groups.
Statistical analysis was performed using SPSS software (version
13.0; SPSS Inc., Chicago, IL, USA). P<0.05 was considered to
indicate a statistically significant difference.
Results
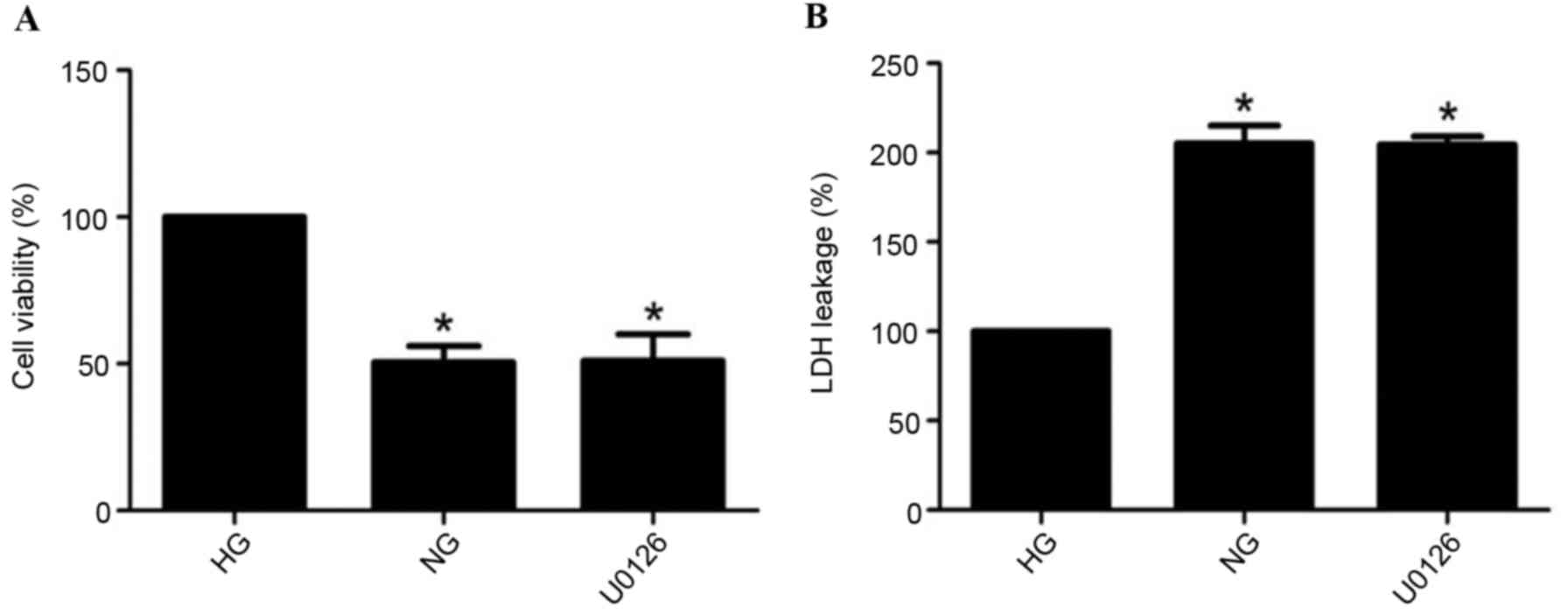
Cell viability and LDH release
Compared with the HG group, cell survival rates
significantly decreased and LDH release markedly increased during
GD for 2 h in the NG group (P<0.05). Compared with the NG group,
U0126 administration had no effect on cell viability (Fig. 1A) and LDH release (Fig. 1B; P>0.05).
Cx43 expression levels
Cx43 protein expression levels were significantly
elevated in H9c2 cells exposed to GD (P<0.05; Fig. 2A). Notably, the effect of GD on
Cx43 expression levels in H9c2 cells was attenuated by U0126
treatment (P<0.05; Fig. 2A),
whereas U0126 treatment alone demonstrated no significant effect on
Cx43 expression levels without GD (data not shown).
 | Figure 2.Effect of GD on Cx43, ERK, p-ERK, Bax,
Bcl-2 protein expression and autophagy indicator levels in H9c2
cells. (A) Compared with the HG group, Cx43 expression levels were
significantly increased in the NG group. U0126 (ERK inhibitor)
treatment abolished the effect of GD on Cx43 expression levels. (B)
Compared with the HG group, p-ERK expression levels were
significantly increased in the NG group; however, p-ERK protein
expression levels were inhibited following U0126 treatment under GD
conditions. Total ERK expression levels remained unaltered.
Compared with the HG group, (C) Bax expression levels were
significantly increased and (D) Bcl-2 expression levels were
markedly decreased in the NG group. U0126 treatment marginally
altered these protein expression levels; however, not
significantly. (E-G) Compared with HG group, (E) Beclin-1, (F) p62
and (G) LC3 expression levels were significantly increased in the
NG group, and U0126 treatment abolished the effect of GD on these
protein expression levels. Data are expressed as the mean ±
standard deviation. *P<0.05 vs. HG group; #P<0.05
vs. NG group. GD, glucose deprivation; Cx43, connexin43; ERK,
extracellular signal-regulated kinase; p-, phosphorylated; Bax,
B-cell lymphoma-2-associated X protein; Bcl-2, B-cell lymphoma 2;
HG, high glucose; NG, no glucose; LC3, microtubule-associated
protein 1A/1B-light chain 3. |
Activation of the ERK/MAPK signaling
pathway
GD treatment activated the phosphorylation levels of
ERK, as measured by western blotting (P<0.05), whereas total ERK
expression levels remained unaltered. As presented in Fig. 2B, the ERK inhibitor totally
abolished GD-induced Cx43 upregulation. These results suggested
that the ERK/mitogen-activated protein kinase (MAPK) signaling
pathway may be involved in the effect of GD on Cx43 expression
levels.
Cell apoptosis
Compared with the HG group, protein expression
levels of Bax were increased (Fig.
2C), whereas Bcl-2 protein expression levels were decreased
(Fig. 2D) in GD-stimulated cells
(P<0.05). However, no significant differences were observed in
Bax and Bcl-2 expression levels between the NG and U0126
groups.
Autophagy-associated protein
expression levels
Protein expression levels of Beclin-1 (Fig. 2E), p62 (Fig. 2F) and LC3 (Fig. 2G) were significantly increased in
the NG group compared with the HG group (P<0.05). However,
inhibition of the ERK/MAPK signaling pathway (U0126 treatment)
completely abolished the effect of GD on the protein expression
levels of Beclin-1, p62 and LC3 (P<0.05).
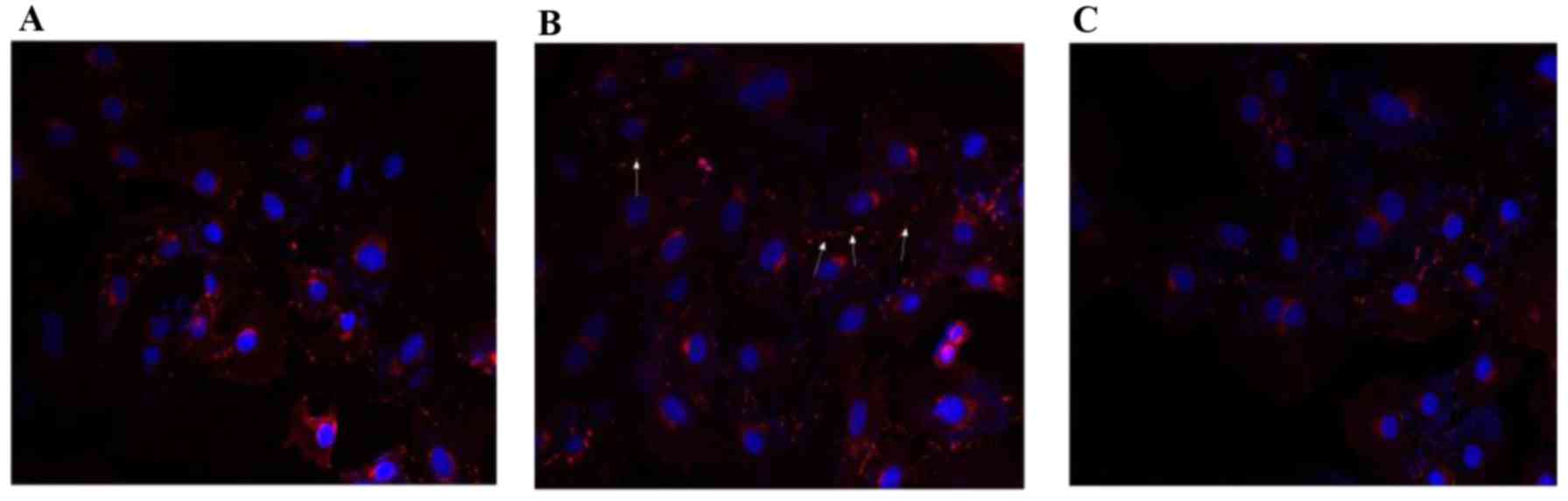
Immunofluorescence of the effect of GD
on Cx43 expression and localization
Consistent with the western blotting results,
immunofluorescence microscopy of HG group cells demonstrated
reduced levels of Cx43 localizing to cell borders in H9c2 cells
(Fig. 3A). Increased
immunoreactivity of Cx43 was observed between adjacent cells
following GD for 2 h (Fig. 3B).
U0126 administration significantly decreased levels of Cx43
compared with GD treatment (Fig.
3C).
Silencing of the ERK/MAPK signaling
pathway
As presented in Fig.
4A, total ERK and p-ERK protein expression levels were
inhibited by siRNA-ERK (P<0.05). Furthermore, Cx43 expression
levels were significantly decreased in H9c2 cells transfected with
siRNA-ERK under GD conditions (P<0.05; Fig. 4B). Protein expression levels of
Beclin-1, p62 and LC3 were markedly decreased in the siRNA group
compared with the NG group (P<0.05; Fig. 4C).
 | Figure 4.Effectof ERK-specific siRNA on Cx43
expression and autophagy indicator levels in H9c2 cells under GD
conditions. (A) Compared with the control and siRNA-con groups,
total ERK and p-ERK protein expression levels were significantly
reduced in the siRNA group. (B) Compared with the HG group, Cx43
expression levels were significantly increased in the NG group;
however, Cx43 protein expression levels were inhibited by siRNA-ERK
treatment under GD conditions. (C) Compared with the HG group,
Beclin-1, p62 and LC3 protein expression levels were significantly
increased in the NG group. siRNA-ERK abolished the effect of GD on
the expression levels of these proteins. Data are expressed as the
mean ± standard deviation. *P<0.05 vs. HG group;
#P<0.05 vs. NG group. ERK, extracellular
signal-regulated kinase; siRNA, small interfering RNA; Cx43,
connexin43; GD, glucose deprivation; p-, phosphorylated; HG, high
glucose; NG, no glucose; LC3, microtubule-associated protein
1A/1B-light chain 3; siRNA-con, non-silencing ERK. |
Discussion
The present study demonstrated that Cx43 protein
expression levels markedly increased under GD conditions,
accompanied by an increase in p-ERK expression levels.
Immunocytochemistry indicated that increased Cx43 localization at
the membrane between neighboring cells, consistent with the results
of the western blot analysis. Furthermore, Cx43 expression levels
in H9c2 cells under GD conditions were decreased following
inhibition of p-ERK, indicating that p-ERK may be involved in the
regulation of cardiac Cx43 expression levels during GD. Similarly,
decreased Cx43 expression levels were observed by silencing ERK
under GD conditions. Notably, upregulation of Beclin-1, p62 and LC3
protein expression levels following GD treatment was abrogated by
inhibitor or siRNA. These observations suggested that increased
Cx43 expression levels may be mediated by the ERK-autophagy
signaling pathway during GD.
Previous studies have demonstrated that
hyperglycemia causes downregulation of Cx43 expression levels in
endothelial cells and cardiomyocytes (7,8).
Hypoglycemic episodes are frequent in diabetic patients undergoing
glucose-lowering therapy. However, there is limited information
regarding the effect of hypoglycemia on Cx43 expression levels in
H9c2 cells. The present study demonstrated that Cx43 expression
levels were significantly increased following GD treatment for 2 h
in H9c2 cells. The exchange rates of injury insult via gap junction
channels may increase during GD, which may increase cell damage and
death. In the current study, Bax protein expression levels were
increased and Bcl-2 protein expression levels were decreased in the
NG group compared with the control group, indicating that increased
Cx43 may increase GJIC activity. Furthermore, cell viability was
significantly decreased and LDH release was markedly increased
under GD conditions, which was in parallel with the results of
western blot analysis.
It is understood that the ERK/MAPK signaling pathway
serves an essential role in regulating Cx43 expression levels
(17–20). Yu et al (20) reported that advanced glycation end
products upregulated Cx43 expression levels in rat cardiomyocytes
via protein kinase C and ERK/MAPK signaling pathways. Activation of
ERK/MAPK by epigallocatechingallate (EGCG) attenuated Cx43
downregulation by serum deprivation in H9c2 cells. Furthermore,
inhibition of ERK by pretreatment with PD98059 may reverse the
increase of Cx43 protein expression levels induced by EGCG in H9c2
cells (19). Inhibition of ERK by
siRNA may suppress Cx43 expression levels induced by angiotensin II
in smooth muscle cells (21). Wang
et al (17) reported that
Cx43 expression levels were suppressed in human aortic endothelial
cells via activation of the ERK signaling pathway. The present
study demonstrated that increased Cx43 protein expression levels
following GD were blocked by ERK/MAPK inhibitors or siRNA-ERK.
Thus, these data suggested that the ERK/MAPK signaling pathway was
involved in the regulation of Cx43 expression levels during GD.
Previous studies have provided evidence that Cx43
may be regulated by autophagy (9,22).
Autophagy is a proteolytic pathway that provides cells with
nutrients to adjust to environmental changes (23). Lichtenstein et al (24) observed that during starvation, Cx43
was enclosed by membrane structures containing the
autophagy-associated proteins LC3 and p62. LC3 was involved in the
formation of auto phagosomes and persisted for the lifespan of auto
phagosome. p62 may serve as a cargo receptor for clearance of
protein aggregates (25). In
starved mice, Cx43 expression levels were significantly decreased
at membranes, whereas they increased intracellularly (26). Furthermore, Hesketh et al
(9) demonstrated that Cx43 was
incorporated into the autophagosome during heart failure. The
results of the present study demonstrated that protein expression
levels of Beclin-1, p62 and LC3 were significantly increased during
GD. It is unclear whether this increase results from an increase of
autophagy flow, or damage to the degradation pathway. Notably,
these protein expression levels were significantly decreased
following pretreatment of U0126 or siRNA, suggesting that
autophagic flow was decreased. Therefore, increased Cx43 protein
expression levels may be a response to GD via activation of the
autophagy pathway. Furthermore, the marked effect of U0126 on
reduction of Cx43 expression levels in H9c2 cells suggested the
involvement of compromised autophagosome formation. Further studies
are required to confirm these interpretations.
Downregulation of Cx43 protein expression levels in
the heart may limit the spread of injury insult by intercellular
transmission. In a previous study, Kanno et al (10) demonstrated that Cx43-deficient mice
developed smaller infracts compared with wild-type mice following
coronary ligation. This may be due to decreased intercellular
exchange of deleterious mediums induced by ischemia. This
hypothesis was supported by Garcia-Dorado et al (27); they additionally reported that
heptanol treatment may improve contractile function and decrease
necrosis. These studies suggested that decreased Cx43 expression
levels may limit injury by decreasing intercellular transfer of
molecules under certain conditions. The results of the present
study additionally indicated that increased Cx43 expression levels
may be responsible for decreased cell viability and increased LDH
release.
In conclusion, the present study provided evidence
that GD induced upregulated Cx43 expression levels in H9c2 cells
via the ERK/MAPK pathway.
Acknowledgements
The present study was supported by the Shanghai
Committee of Science and Technology, Shanghai, China (grant no.
13ZR1431500).
Glossary
Abbreviations
Abbreviations:
|
Cx43
|
connexin43
|
|
HG
|
high glucose
|
|
NG
|
no glucose
|
|
GD
|
glucose deprivation
|
|
siRNA
|
small interfering RNA
|
|
LDH
|
lactate dehydrogenase
|
References
|
1
|
Yeh HI, Lee PY, Su CH, Tian TY, Ko YS and
Tsai CH: Reduced expression of endothelial connexins 43 and 37 in
hypertensive rats is rectified after 7-day carvedilol treatment. Am
J Hypertens. 19:129–135. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Rutledge CA, Ng FS, Sulkin MS, Greener ID,
Sergeyenko AM, Liu H, Gemel J, Beyer EC, Sovari AA, Efimov IR and
Dudley SC: c-Src kinase inhibition reduces arrhythmia inducibility
and connexin43 dysregulation after myocardial infarction. J Am Coll
Cardiol. 63:928–934. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Greener ID, Sasano T, Wan X, Igarashi T,
Strom M, Rosenbaum DS and Donahue JK: Connexin43 gene transfer
reduces ventricular tachycardia susceptibility after myocardial
infarction. J Am Coll Cardiol. 60:1103–1110. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Brisset AC, Isakson BE and Kwak BR:
Connexins in vascular physiology and pathology. Antioxid Redox
Signal. 11:267–282. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Vozzi C, Dupont E, Coppen SR, Yeh HI and
Severs NJ: Chamber-related differences in connexin expression in
the human heart. J Mol Cell Cardiol. 31:991–1003. 1999. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Giepmans BN: Gap junctions and
connexin-interacting proteins. Cardiovasc Res. 62:233–245. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Fernandes R, Girão H and Pereira P: High
glucose down-regulates intercellular communication in retinal
endothelial cells by enhancing degradation of connexin 43 by a
proteasome-dependent mechanism. J Biol Chem. 279:27219–27224. 2004.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lin H, Ogawa K, Imanaga I and Tribulova N:
Remodeling of connexin 43 in the diabetic rat heart. Mol Cell
Biochem. 290:69–78. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Hesketh GG, Shah MH, Halperin VL, Cooke
CA, Akar FG, Yen TE, Kass DA, Machamer CE, Van Eyk JE and Tomaselli
GF: Ultrastructure and regulation of lateralized connexin43 in the
failing heart. Circ Res. 106:1153–1163. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Kanno S, Kovacs A, Yamada KA and Saffitz
JE: Connexin43 as a determinant of myocardial infarct size
following coronary occlusion in mice. J Am Coll Cardiol.
41:681–686. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang HH, Kung CI, Tseng YY, Lin YC, Chen
CH, Tsai CH and Yeh HI: Activation of endothelial cells to
pathological status by down-regulation of connexin43. Cardiovasc
Res. 79:509–518. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zoungas S, Patel A, Chalmers J, de Galan
BE, Li Q, Billot L, Woodward M, Ninomiya T, Neal B, MacMahon S, et
al: Severe hypoglycemia and risks of vascular events and death. N
Engl J Med. 363:1410–1418. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Hilsted J, Bonde-Petersen F, Nørgaard MB,
Greniman M, Christensen NJ, Parving HH and Suzuki M: Haemodynamic
changes in insulin-induced hypoglycaemia in normal man.
Diabetologia. 26:328–332. 1984. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Dave KR, Tamariz J, Desai KM, Brand FJ,
Liu A, Saul I, Bhattacharya SK and Pileggi A: Recurrent
hypoglycemia exacerbates cerebral ischemic damage in
streptozotocin-induced diabetic rats. Stroke. 42:1404–1411. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Robinson RT, Harris ND, Ireland RH, Lee S,
Newman C and Heller SR: Mechanisms of abnormal cardiac
repolarization during insulin-induced hypoglycemia. Diabetes.
52:1469–1474. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Dandona P, Chaudhuri A and Dhindsa S:
Proinflammatory and prothrombotic effects of hypoglycemia. Diabetes
Care. 33:1686–1687. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang CY, Liu HJ, Chen HJ, Lin YC, Wang HH,
Hung TC and Yeh HI: AGE-BSA down-regulates endothelial connexin43
gap junctions. BMC Cell Biol. 12:192011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Lee KM, Kwon JY, Lee KW and Lee HJ:
Ascorbic acid 6-palmitate suppresses gap-junctional intercellular
communication through phosphorylation of connexin 43 via activation
of the MEK-ERK pathway. Mutat Res. 660:51–56. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhao Y, Yu L, Xu S, Qiu F, Fan Y and Fu G:
Down-regulation of connexin43 gap junction by serum deprivation in
human endothelial cells was improved by (−)-Epigallocatechin
gallate via ERK MAP kinase pathway. Biochem Biophys Res Commun.
404:217–222. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Yu L, Zhao Y, Xu S, Ding F, Jin C, Fu G
and Weng S: Advanced glycation end product (AGE)-AGE receptor
(RAGE) system upregulated connexin43 expression in rat
cardiomyocytes via PKC and Erk MAPK pathways. Int J Mol Sci.
14:2242–2257. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Jia G, Cheng G, Gangahar DM and Agrawal
DK: Involvement of connexin 43 in angiotensin II-induced migration
and proliferation of saphenous vein smooth muscle cells via the
MAPK-AP-1 signaling pathway. J Mol Cell Cardiol. 44:882–890. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Bejarano E, Girao H, Yuste A, Patel B,
Marques C, Spray DC, Pereira P and Cuervo AM: Autophagy modulates
dynamics of connexins at the plasma membrane in a
ubiquitin-dependent manner. Mol Biol Cell. 23:2156–2169. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Mitchener JS, Shelburne JD, Bradford WD
and Hawkins HK: Cellular autophagocytosis induced by deprivation of
serum and amino acids in HeLa cells. Am J Pathol. 83:485–492.
1976.PubMed/NCBI
|
|
24
|
Lichtenstein A, Minogue PJ, Beyer EC and
Berthoud VM: Autophagy: A pathway that contributes to connexin
degradation. J Cell Sci. 124:910–920. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Bjorkoy G, Lamark T, Brech A, Outzen H,
Perander M, Overvatn A, Stenmark H and Johansen T: p62/SQSTM1 forms
protein aggregates degraded by autophagy and has a protective
effect on huntingtin-induced cell death. J Cell Biol. 171:603–614.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
McLachlan CS, Almsherqi ZA, Mossop P,
Suzuki J, Leong ST and Deng Y: Down regulation of immuno-detectable
cardiac connexin-43 in BALB/c mice following acute fasting. Int J
Cardiol. 136:99–102. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Garcia-Dorado D, Inserte J, Ruiz-Meana M,
González MA, Solares J, Juliá M, Barrabés JA and Soler-Soler J: Gap
junction uncoupler heptanol prevents cell-to-cell progression of
hypercontracture and limits necrosis during myocardial reperfusion.
Circulation. 96:3579–3586. 1997. View Article : Google Scholar : PubMed/NCBI
|