Introduction
Previously, it was believed that antiangiogenic
drugs inhibit tumors by promoting degradation of tumor blood
vessels, causing ischemia and hypoxia in the tumor, leading to
tumor cell death (1). According to
this theory, anti-angiogenic drugs would antagonize the effect of
other cancer treatments, such as chemotherapy, because the lack of
oxygen and blood supply impedes the delivery of other drugs.
However, the truth is that the antiangiogenic drugs actually
enhance the efficacy of radiotherapy and chemotherapy. Therefore,
antiangiogenic drugs may inhibit tumors by some other mechanisms.
In 2004, Lin and Sessa (2) first
proposed the ‘time window’ concept for tumor angiogenesis. They
believed that there is a certain period, during which
antiangiogenic drugs have the greatest anti-tumor effect. In this
time window, the newly formed tumor vessels tended to be normal in
vascular endothelial maturation, morphological regulation and tight
junctions, and thus, anticancer drugs can easily reach the tumor
cells, directly damaging tumor cell DNA and inhibiting vascular
endothelial cell proliferation.
The time window of tumor vessel normalization is
specifically marked by improved tumor hypoxic status and change in
pericyte coverage (1,3). Generally, pericytes in the tumor
vascular bed display obviously abnormal structure and the pericyte
coverage is low (4). Pericyte
maturation and blood vessel normalization take place following the
depletion of regulator of G-protein signaling 5 (RGS5), one of the
proteins that reflects pericyte coverage (5,6).
Hypoxia, a common phenomenon in human tumors (7), can cause tumors to become resistant
to therapy and lead to tumor progression (8). Carbonic anhydrase 9 (CA9), a
transmembrane zinc metalloenzyme, can be induced by hypoxia and
promoting aggressive behaviors of tumors by helping maintain normal
intracellular pH and preventing apoptosis in a hypoxic environment
(9).
Endostatin is the 20 kDa c-terminal fragment of
collagen XVIII that is primarily located in the basement membrane
of blood vessels. It was first described by O'Reilly et al
(10) in 1997 and was licensed by
EntreMed. In spite of the fact that preclinical and clinical
studies on tumor suppression were very positive, EntreMed abandoned
phase III clinical trials due to an insufficient supply of
endostatin. Nevertheless, endostatin development was continued by
scientists in China and recombinant human endostatin (rh-ES, with
the trade name Endostar) has been approved by the China Food and
Drug Administration. The use of it in combination with chemotherapy
has been suggested by the National Comprehensive Cancer Network
(NCCN) Guidelines to treat non-small cell lung cancer (NSCLC).
However, the optimal administration time of rh-ES and its molecular
mechanism still remain unclear. In the current study, by using a
Lewis lung cancer (LLC) tumor model, the authors aimed to identify
the optimal administration time of rh-ES in cancer treatment and
its possible molecular mechanism.
Materials and methods
Ethical statement
The present study was approved by the Ethics
Committee of North Sichuan Medical College (Nanchang, China).
Cell culture and animal model
Mouse LLC tumor cells were purchased from the State
Key Laboratory of Cancer Biotherapy at Sichuan University (Chengdu,
China). The 6–8 week-old female mice with a body weight of 15–20 g
were purchased from the Laboratory Animal Center of North Sichuan
Medical College (Nanchong, China). LLC cells at logarithmic growth
phase were collected as a 1×106 cells/ml single cell
suspension. Cells (0.2 ml) were injected subcutaneously into the
left armpit of each C57/BL6 mouse. The growth and tumor formation
in the mice was observed and recorded.
Animal treatment and tumor sample
collection
Treatment started when the LLC tumors reached a
diameter of 6 mm. A total of 40 mice were randomly split into two
equal groups: NS and rh-ES. Mice in the NS group were
intraperitoneally injected with 0.2 ml 0.9% normal saline daily for
up to 9 days, while mice in rh-ES group were intraperitoneally
injected with 5 mg/kg rh-ES (Shandong Simcere-Medgenn
Bio-Pharmaceutical Co., Ltd., Yantai, China) daily for up to 9
days. A total of 5 mice from each group were sacrificed at days 2,
4, 6 and 9 following treatment. Each tumor sample was split into
two parts. One part was snap-frozen within 30 min following animal
sacrifice and stored at −80°C for further RNA and protein analyses.
The other part was fixed with 4% paraformaldehyde and dehydrated,
and embedded in paraffin for immunohistochemical analysis.
Immunohistochemistry (IHC)
Sections (5 µm) were prepared from
paraformaldehyde-fixed, paraffin-embedded tissues and were used for
immunocytochemistry. The CD31 antibody (cat no. ab28364; dilution,
1:500) was obtained from Abcam (Cambridge, MA, USA) and incubated
at 37°C for 2 h. RGS5 (cat no. ab138019; Abcam, Shanghai, China)
and CA9 (cat no. NB100-417; Novus Biologicals, LLC, Littleton, CO,
USA) antibodies were used at a 1:100 dilution at 37°C for 2 h. The
bound antibody was visualized with SABC IHC kit (Santa Cruz
Biotechnology, Inc., Dallas, TX, USA). The positive RGS5 signal was
located in the cytoplasm and had a color of light yellow to brown,
while the CA9 signal was located on the cell membrane and had a
color of brown. The authors followed the criteria used by Rahman
et al (11) and used the
gray color intensity of positive cells as the indicator of signal
strength: 0, negative; 1, weak positive; 2, positive; 3, strongly
positive. The staining range (% of positive cells) was scored from
0–4: 0, negative; 1, 1–25% positive cells; 2, 26–50%; 3, 51–75%; 4,
76–100%. Five non-overlapping fields (magnification, ×400) were
randomly selected and calculated the percentage of positive cells
in 1,000 tumor cells.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
Total RNA samples were extracted from tissues using
Total RNA extraction kit (Omega Bio-Tek, Inc., Norcross, GA, USA).
cDNA samples were synthesized using a cDNA synthesis kit (Thermo
Fisher Scientific, Inc., Waltham, MA, USA) (42°C, 60 min; 70°C, 5
min) and the cDNA used for RT-qPCR (Stage 1: 95°C, 30 sec; Stage 2:
95°C, 5 sec; 60°C, 34 sec; repeated 40 times. Stage 3: 95°C, 15
sec; 60°C, 60 sec and 95°C, 15 sec). Primers were purchased from
Shanghai Generay Biotech Co., Ltd. (Shanghai, China). Sequences are
as follows: GAPDH forward, 5′-AGAAGGTGGTGAAGCAGGCATC-3′ and
reverse, 5′-CGAAGGTGGAAGAGTGCGAGTTG-3′; RGS5 forward,
5′-ATCAAAATGGCGGAGAAGGCAAA-3′ and reverse,
5′-CACAAAGCGGGGCAGAGAATC-3′; CA9 forward,
5′-TGTGGGGACCTCGTGATTCTCG-3′ and reverse,
5′-TGGACTGGCTCAGGGCTGCTAT-3′. The relative levels of gene
expression were quantified by using the comparative Cq method
(12).
ELISA
Tumor tissues were homogenized using 1X cell lysis
buffer (cat no. 9803; Cell Signaling Technology, Inc., Danvers, MA,
USA) and centrifuged at −4°C and 12,000 × g for 15 min. The
supernatant was harvested and tested using ELISA kits (cat nos.
MBS2604739 and MBS939749; MyBiosource, Inc., San Diego, CA, USA)
for RGS5 and CA9 following manufacturer's protocols. A total volume
of 100 µl blank, standards, and tumor samples were added into
wells. Optical density was measured at 450 nm. Results are
expressed as ng/ml of homogenizing buffer.
Statistics
Data are presented as the mean ± standard deviation.
The significance between two groups was tested using Students
t-test (two tailed) and P<0.05 was considered to indicate a
statistically significant difference. SPSS software (version, 13.0;
SPSS, Inc., Chicago, IL, USA) was used for statistical
analysis.
Results
LLC tumor formation
The tumor formation rate in C57/BL6 female mice was
100%. On day 10, following subcutaneous injection of LLC cells,
tumor xenografts grew to a size of ~6 mm in diameter. The tumors
were hard and spherical, and displayed poor activity and expansive
growth.
Rh-ES causes transient LLC tumor
vascular normalization and improves tumor tissue hypoxia
To examine the changes in tumor vascularization, the
authors performed immunohistochemical staining for CD31, a
microvessel marker, on LLC tumors treated with rh-ES for 2, 4, 6
and 9 days. It was reported that LLC tumors treated with rh-ES for
4 or 6 days presented lower CD31 expression than those treated for
2 or 9 days. The authors calculated microvessel density as a ratio
of microvessel number to the area of the image to measure CD31
immunoreactivity. The data indicated a significant reduction in the
vascularity of the LLC tumor xenografts following treatment with
rh-ES for 4 or 6 days. In contrast, no significant change was
observed for LLC tumors treated with rh-ES for 2 days or 9 days
(Fig. 1 and Table I).
 | Table I.Microvessel density value in tumor
tissues. |
Table I.
Microvessel density value in tumor
tissues.
| Days following
injection (n) | n | Microvessel density
(cells/mm2) |
|---|
| 2 | 5 |
23.10±3.26 |
| 4a | 5 |
10.51±3.12 |
| 6a | 5 |
9.39±1.94 |
| 9 | 5 |
21.86±3.25 |
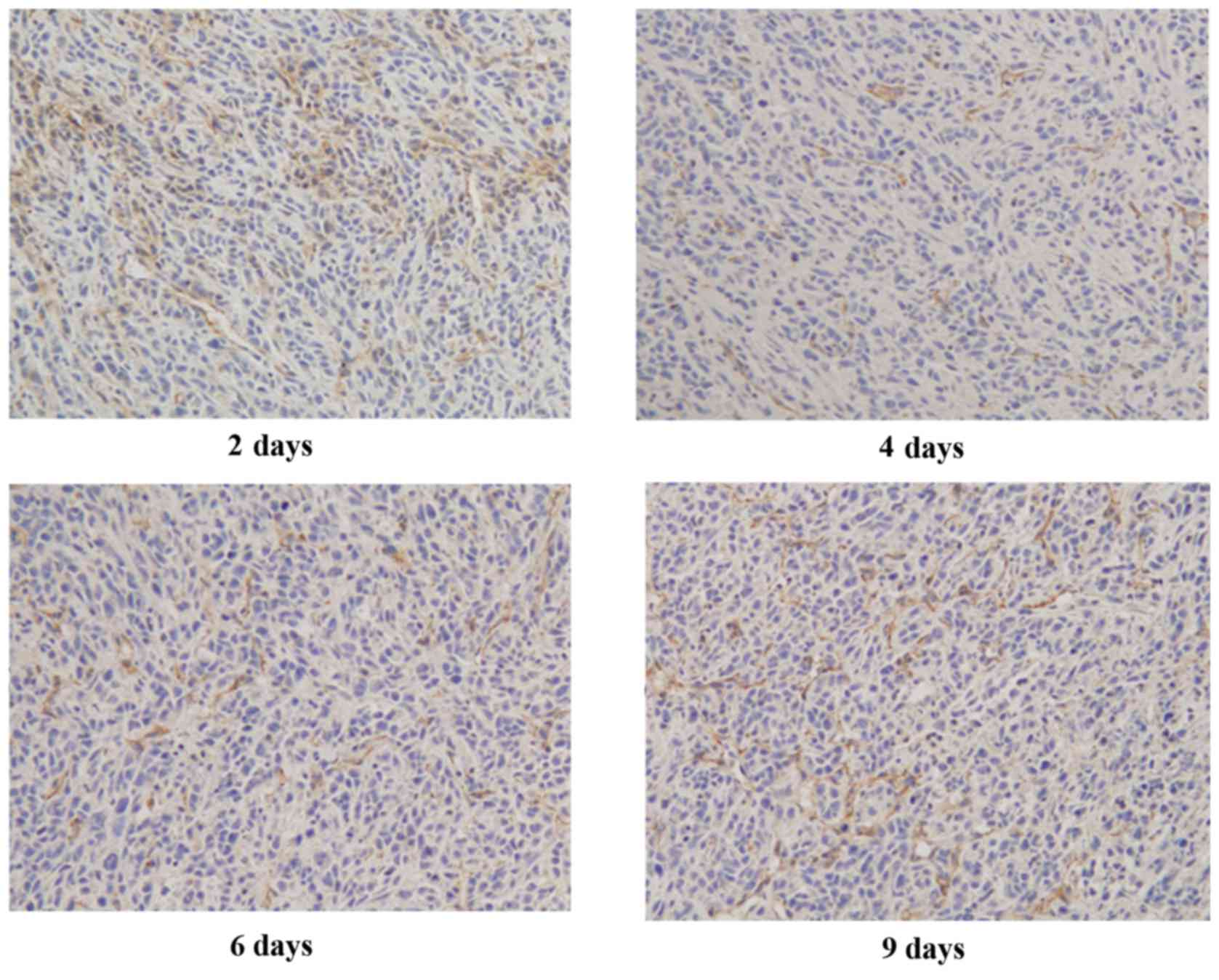
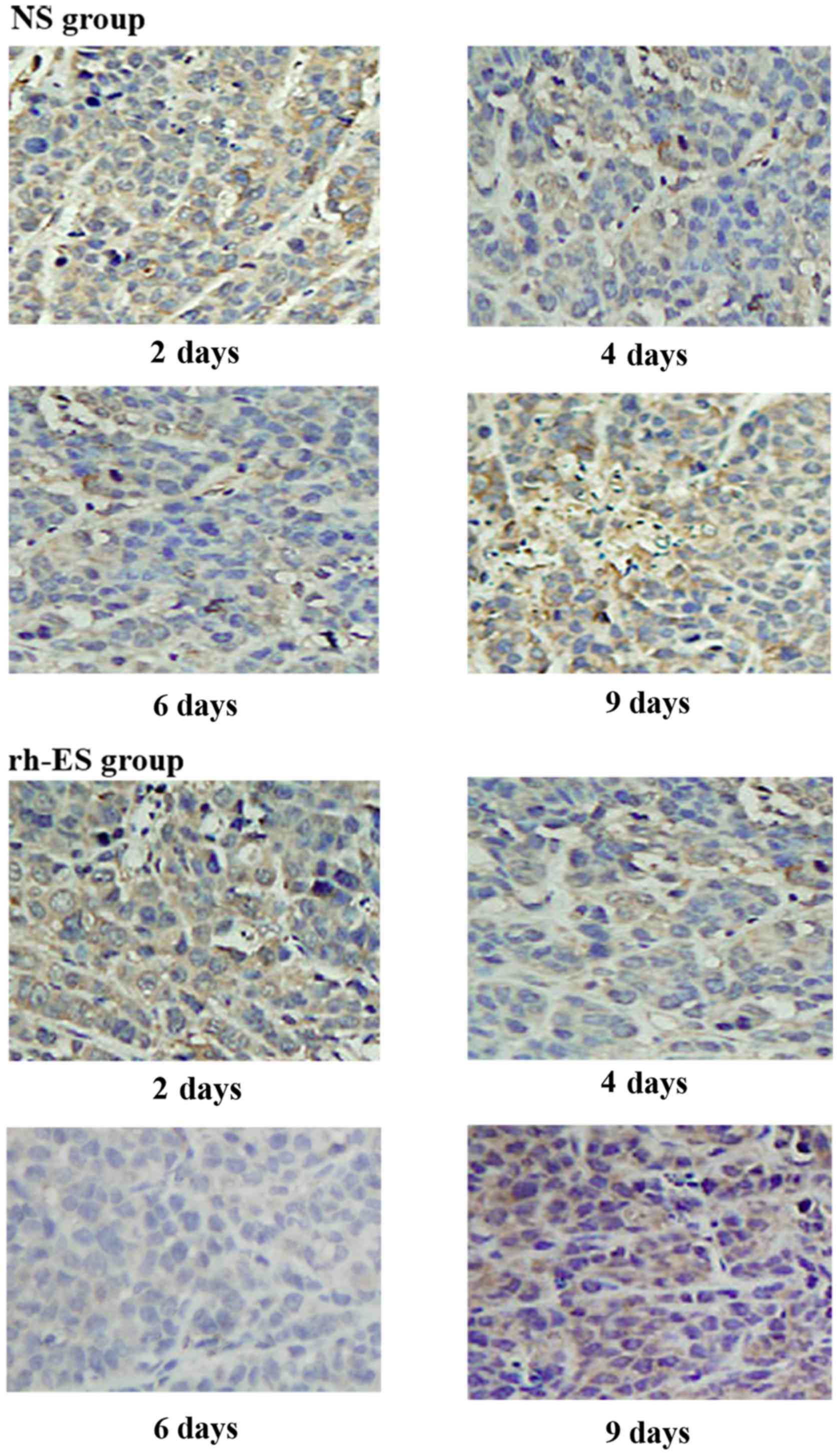
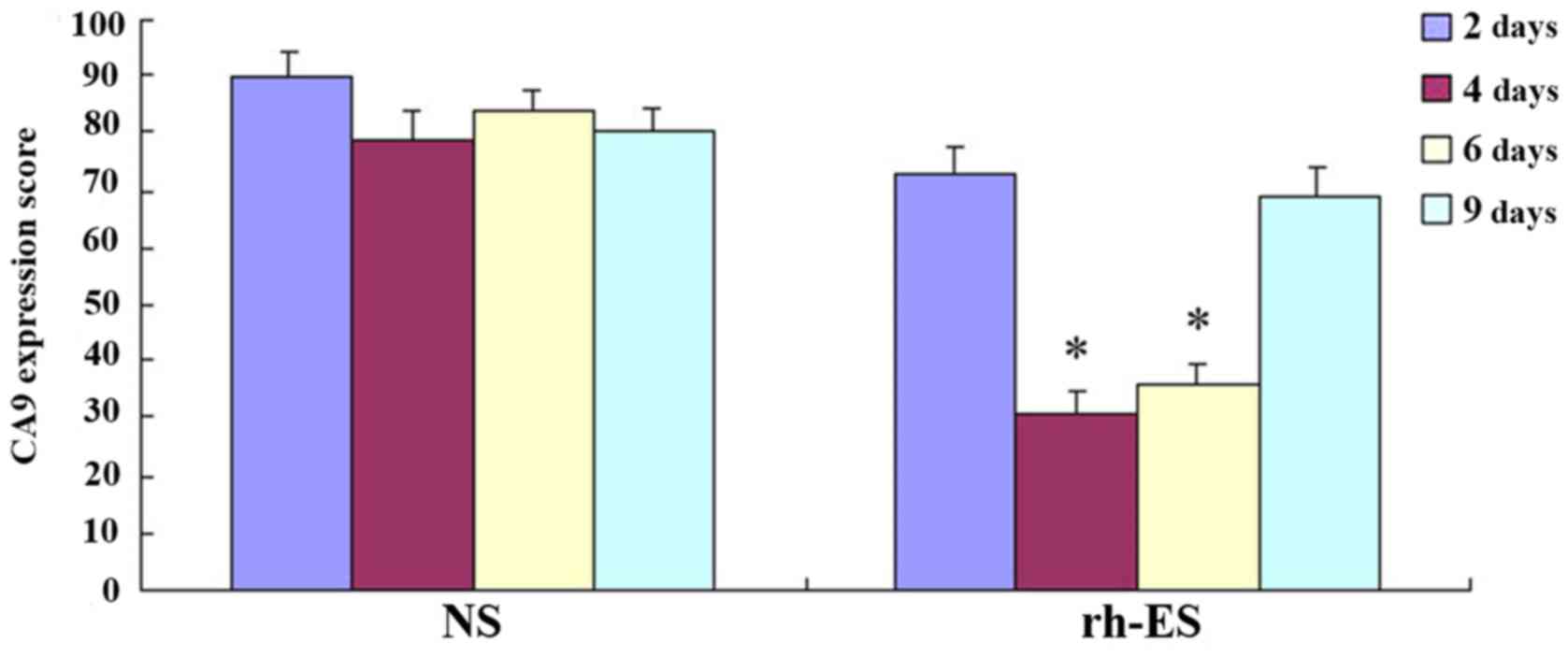
Immunohistochemical analysis
demonstrates the reduction of RGS5 and CA9 protein expression in
tumor cells at d4 and d6 of treatment, but not at d2 and d9
RGS5 is a master regulator for tumor vascular
normalization. CA9 is a biomarker of tumor hypoxia. To test whether
rh-ES can cause a change in the expression of these two proteins,
using immunohistochemical analysis, the authors examined the RGS5
and CA9 protein expression levels in LLC tumors treated with rh-ES
or saline for 2, 4, 6 and 9 days. It was determined that, compared
with the NS group, both RGS5 and CA9 levels in rh-ES group were
significantly lower at days 4 and 6 (P<0.05), while there was no
significant change between two groups at days 2 and 9 (Figs. 2–5
and Tables II and III).
| Table II.Expression of regulator of G-protein
signaling 5 in Lewis lunch cancer at different time-points. |
Table II.
Expression of regulator of G-protein
signaling 5 in Lewis lunch cancer at different time-points.
| Group | Time | Observed fields
(n) | Labeling index
(%) |
|---|
| NS | 2 | 5 |
76.13±4.13 |
|
| 4 | 5 |
70.24±4.08 |
|
| 6 | 5 |
68.66±3.76 |
|
| 9 | 5 |
79.60±3.99 |
| rh-ES | 2 | 5 |
70.02±4.03 |
|
| 4a | 5 |
44.77±3.41 |
|
| 6a | 5 |
40.49±3.38 |
|
| 9 | 5 |
77.04±4.26 |
| Table III.Expression of carbonic anhydrase 9 in
Lewis lung cancer at different time points. |
Table III.
Expression of carbonic anhydrase 9 in
Lewis lung cancer at different time points.
| Group | Time | Observed fields
(n) | Labeling index
(%) |
|---|
| NS | 2 | 5 |
90.10±3.85 |
|
| 4 | 5 |
78.98±4.98 |
|
| 6 | 5 |
83.62±3.68 |
|
| 9 | 5 |
80.15±3.96 |
| rh-ES | 2 | 5 |
73.09±4.19 |
|
| 4a | 5 |
30.68±3.95 |
|
| 6a | 5 |
35.87±3.70 |
|
| 9 | 5 |
68.81±4.92 |
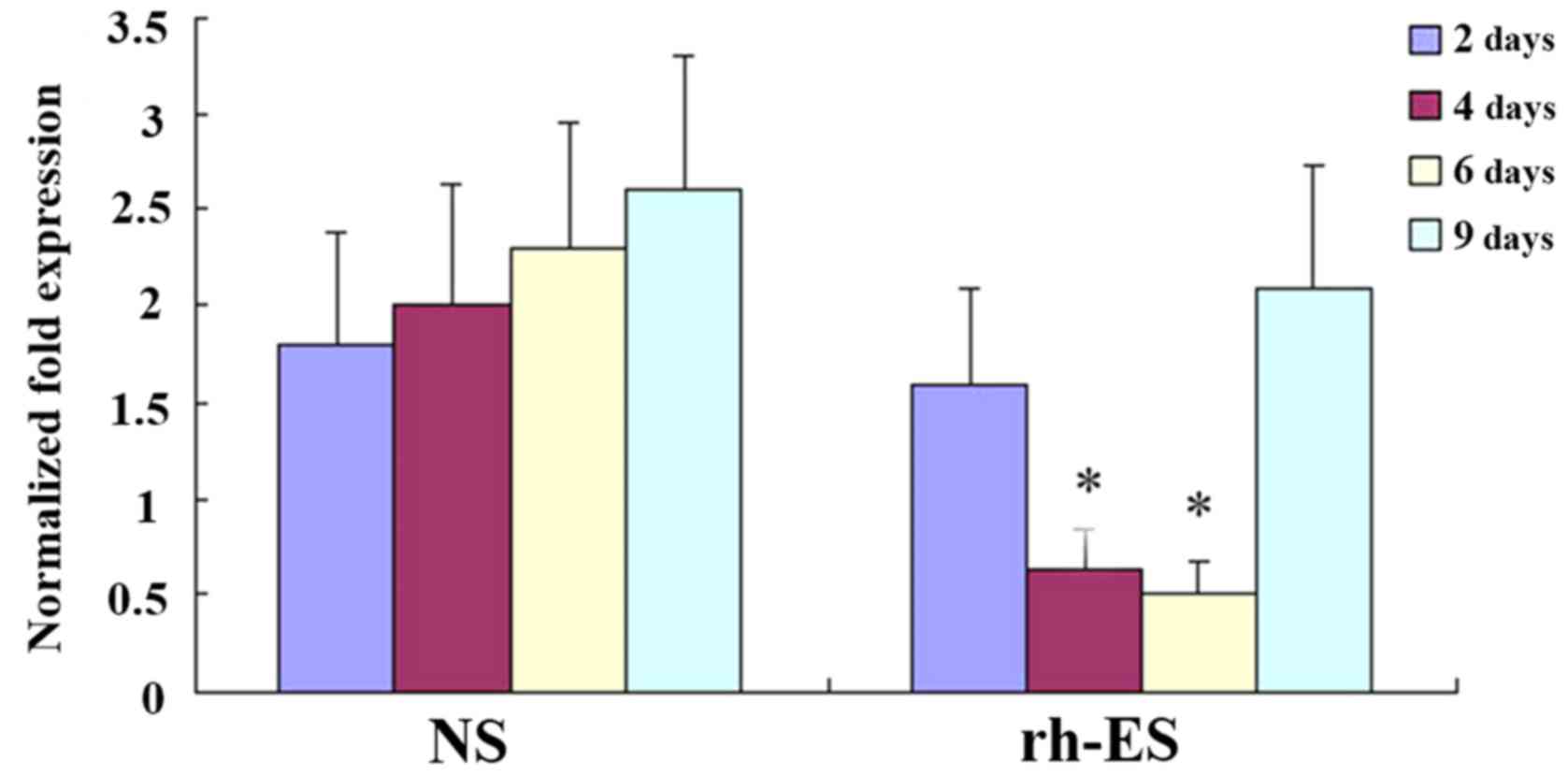
RT-qPCR analysis demonstrates a
reduction of RGS5 and CA9 mRNA levels in tumor cells at days 4 and
6 of treatment, but not at days 2 and 9
To confirm that rh-ES can cause a reduction in the
expression of RGS5 and CA9, the authors performed RT-qPCR on LLC
tumors of NS and rh-ES groups. Compared with the NS group, both
RGS5 and CA9mRNA levels in the rh-ES group were significantly lower
at days 4 and 6 (P<0.05), while there was no significant change
between two groups at days 2 and 9 (Figs. 6 and 7).
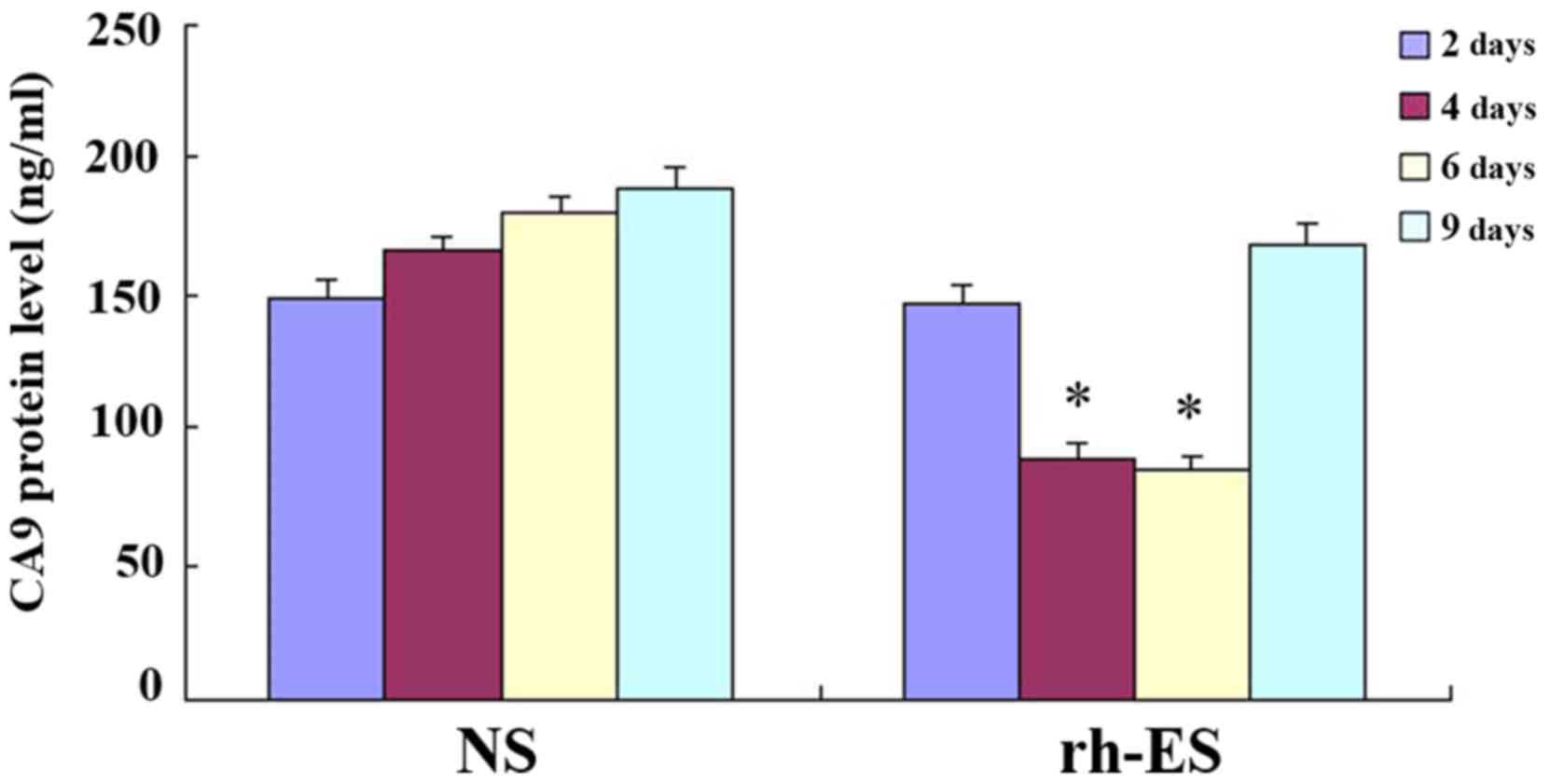
ELISA further confirms the reduction
of RGS5 and CA9 protein levels in tumor cells at days 4 and 6 of
treatment, but not at days 2 and 9
To further confirm the effect of rh-ES, ELISA was
performed on LLC tumors of NS and rh-ES groups. Compared with the
NS group, both RGS5 and CA9 protein levels in rh-ES group were
significantly lower at days 4 and 6 (P<0.05), while there was no
significant change between two groups at days 2 and 9 (Figs. 8 and 9 and Tables
IV and V).
| Table IV.Expression of regulator of G-protein
signaling 5 in Lewis lung cancer by ELISA (ng/ml). |
Table IV.
Expression of regulator of G-protein
signaling 5 in Lewis lung cancer by ELISA (ng/ml).
| Days following
injection (n) | 2 | 4 | 6 | 9 |
|---|
| NS |
7.88±0.72 |
8.16±0.88 |
10.51±1.44 |
11.08±1.54 |
| Rh-ES |
8.45±0.94 |
4.02±0.68a |
2.98±0.46a |
10.28±0.96 |
| Table V.Expression of carbonic anhydrase 9 in
Lewis lung cancer by ELISA (ng/ml). |
Table V.
Expression of carbonic anhydrase 9 in
Lewis lung cancer by ELISA (ng/ml).
| Days following
injection (n) | 2 | 4 | 6 | 9 |
|---|
| NS |
148.12±7.22 |
166.56±5.12 |
180.51±5.65 |
189.28±7.60 |
| Rh-ES |
146.45±6.28 |
88.63±6.16a |
83.98±5.76a |
168.20±8.02 |
Discussion
The use of rh-ES as an antiangiogenic drug in
advanced NSCLC has been suggested by the Chinese version of the
NCCN guidelines (13). Besides
lung cancer (14), studies and
clinical trials have demonstrated the anticancer effect of rh-ES in
other cancers such as melanoma, nasopharyngeal and esophageal
cancer (15–17). These studies have reported that
rh-ES can cause transient normalization of tumor vasculature.
Consistent with previous studies, using an LLC tumor model, the
authors demonstrated that the reduction of microvessel marker CD31
caused by rh-ES occurs between days 4 and 6 following the start of
rh-ES treatment, and is reversed at day 9 or even earlier,
demonstrating that the vascular normalization of LLC tumors caused
by rh-ES is transient and reversible. Based on the ‘vascular
normalization window’ hypothesis proposed by Jain (3), the time window ranging from day 4 to
day 6 would be the best scheduling time for a combination therapy
using rh-ES and chemo- or radio-therapy.
In the past decade, vast evidence has emerged in
support of the ‘normalization window’ hypothesis for tumor
angiogenesis. Molecular mechanisms have been investigated to
understand this process. Among these mechanisms, VEGF is
demonstrated to be a key regulator for angiogenesis. Although there
is no doubt concerning the anti-angiogenic effect of rh-ES, the
molecular mechanism for this effect still remains unclear.
Different research groups have demonstrated that rh-ES can reduce
the expression levels of vascular endothelial growth factor (VEGF),
indicating that VEGF may play an important role in rh-ES-induced
vascular normalization (14,16).
Aside from this, the general understanding of the effect of rh-ES
has been limited. The current findings reported that RGS5 and CA9
were reduced by rh-ES during the ‘vascular normalization window’
are striking. To the best of the authors' knowledge, this is the
first direct evidence to present the roles of RGS5 and CA9 in the
antiangiogenic effect of rh-ES.
Rgs5 has been identified as a master gene
responsible for the abnormal vasculature in mouse tumors. Loss of
Rgs5 can cause vascular normalization and a consequent reduction in
tumor hypoxia and vessel leakiness, leading to enhanced influx of
immune effector cells into the tumor (18). However, the relationship between
RGS5 and VEGF is not completely understood. On one hand, RGS5 can
be triggered by tumor vasculature and sustained by VEGF-rich
proangiogenic microenvironment (19). One the other hand, RGS5 can
antagonize the angiogenic effect of VEGF through the p38 signaling
pathway (20). Whether the
reduction of RGS5 by rh-ES is mediated by VEGF or is independent of
VEGF needs to be further investigated. The presented tumor model
provides the best in vivo system for such a follow-up study
to better the understanding of the molecular mechanism involved in
the anti-angiogenic effect of rh-ES.
Different to RGS5, the relationship between CA9 and
VEGF is clearer. The regulation of CA9 is differential from the
regulation of VEGF in hypoxic conditions, which has been
demonstrated in bladder cancer (21). By using this system, the authors
will be able to examine whether the rh-ES-induced CA9 reduction is
independently of VEGF in future studies.
In conclusion, the authors have provided evidence
that rh-ES can cause transient and reversible tumor vascular
normalization, optimizing the schedule of combination therapy in
human tumors. Most importantly, the novel findings that the
expression of RGS5 and CA9 is reduced during the ‘vascular
normalization window’ suggested that RGS5 and CA9 may be used as
biomarkers for defining the ‘vascular normalization window’.
Acknowledgements
The present study was supported by technology
support program of Science and Technology Department of Sichuan
Province (grant no. 2014SZ0020-7), the Department of Science and
Technology Innovation Talent Engineering Projects in Sichuan
Province (grant no. 20133007), Sichuan Province Health Department
of Scientific Research Projects (grant no. 130466), Nanchong
Technology Bureau Application Technology and Development of Capital
Projects (grant no. 13A0061) and doctoral startup fund of North
Sichuan Medical College (grant no. CBY15-QD09).
References
|
1
|
Winkler F, Kozin SV, Tong RT, Chae SS,
Booth MF, Garkavtsev I, Xu L, Hicklin DJ, Fukumura D, di Tomaso E,
et al: Kinetics of vascular normalization by VEGFR2 blockade
governs brain tumor response to radiation: Role of oxygenation,
angiopoietin-1, and matrix metalloproteinases. Cancer Cell.
6:553–563. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Lin MI and Sessa WC: Antiangiogenic
therapy: Creating a unique ‘window’ of opportunity. Cancer Cell.
6:529–531. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Jain RK: Normalizing tumor vasculature
with anti-angiogenic therapy: A new paradigm for combination
therapy. Nat Med. 7:987–989. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Welén K, Jennbacken K, Tesan T and Damber
JE: Pericyte coverage decreases invasion of tumour cells into blood
vessels in prostate cancer xenografts. Prostate Cancer Prostatic
Dis. 12:41–46. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Faraone D, Aguzzi MS, Toietta G, Facchiano
AM, Facchiano F, Magenta A, Martelli F, Truffa S, Cesareo E,
Ribatti D, et al: Platelet-derived growth factor-receptor alpha
strongly inhibits melanoma growth in vitro and in vivo. Neoplasia.
11:732–742. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Manzur M and Ganss R: Regulator of G
protein signaling 5: A new player in vascular remodeling. Trends
Cardiovasc Med. 19:26–30. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Park SY, Kim YJ, Gao AC, Mohler JL, Onate
SA, Hidalgo AA, Ip C, Park EM, Yoon SY and Park YM: Hypoxia
increases androgen receptor activity in prostate cancer cells.
Cancer Res. 66:5121–5129. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Yasuda H: Solid tumor physiology and
hypoxia-induced chemo/radio-resistance: Novel strategy for cancer
therapy: Nitric oxide donor as a therapeutic enhancer. Nitric
Oxide. 19:205–216. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Bussink J, Kaanders JH and van der Kogel
AJ: Tumor hypoxia at the micro-regional level: Clinical relevance
and predictive value of exogenous and endogenous hypoxic cell
markers. Radiother Oncol. 67:3–15. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
O'Reilly MS, Boehm T, Shing Y, Fukai N,
Vasios G, Lane WS, Flynn E, Birkhead JR, Olsen BR and Folkman J:
Endostatin: An endogenous inhibitor of angiogenesis and tumor
growth. Cell. 88:277–285. 1997. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Rahman MA, Dhar DK, Yamaguchi E, Maruyama
S, Sato T, Hayashi H, Ono T, Yamanoi A, Kohno H and Nagasue N:
Coexpression of inducible nitric oxide synthase and COX-2 in
hepatocellular carcinoma and surrounding liver: Possible
involvement of COX-2 in the angiogenesis of hepatitis C
virus-positive cases. Clin Cancer Res. 7:1325–1332. 2001.PubMed/NCBI
|
|
12
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Shi Y, Sun Y, Yu J, Ding C, Wang Z, Wang
C, Wang D, Wang C, Wang Z, Wang M, et al: China experts consensus
on the diagnosis and treatment of advanced stage primary lung
cancer (2016 version). Asia Pac J Clin Oncol. 13:87–103. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Li N, Zheng D, Wei X, Jin Z, Zhang C and
Li K: Effects of recombinant human endostatin and its synergy with
cisplatin on circulating endothelial cells and tumor vascular
normalization in A549 xenograft murine model. J Cancer Res Clin
Oncol. 138:1131–1144. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cui C, Mao L, Chi Z, Si L, Sheng X, Kong
Y, Li S, Lian B, Gu K, Tao M, et al: A phase II, randomized,
double-blind, placebo-controlled multicenter trial of Endostar in
patients with metastatic melanoma. Mol Ther. 21:1456–1463. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Zhu H, Yang X, Ding Y, Liu J, Lu J, Zhan
L, Qin Q, Zhang H, Chen X, Yang Y, et al: Recombinant human
endostatin enhances the radioresponse in esophageal squamous cell
carcinoma by normalizing tumor vasculature and reducing hypoxia.
Sci Rep. 5:145032015. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Peng F, Xu Z, Wang J, Chen Y, Li Q, Zuo Y,
Chen J, Hu X, Zhou Q, Wang Y, et al: Recombinant human endostatin
normalizes tumor vasculature and enhances radiation response in
xenografted human nasopharyngeal carcinoma models. PLoS One.
7:e346462012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Hamzah J, Jugold M, Kiessling F, Rigby P,
Manzur M, Marti HH, Rabie T, Kaden S, Gröne HJ, Hämmerling GJ, et
al: Vascular normalization in Rgs5-deficient tumours promotes
immune destruction. Nature. 453:410–414. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Silini A, Ghilardi C, Figini S, Sangalli
F, Fruscio R, Dahse R, Pedley RB, Giavazzi R and Bani M: Regulator
of G-protein signaling 5 (RGS5) protein: A novel marker of cancer
vasculature elicited and sustained by the tumor's proangiogenic
microenvironment. Cell Mol Life Sci. 69:1167–1178. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Jin Y, An X, Ye Z, Cully B, Wu J and Li J:
RGS5, a hypoxia-inducible apoptotic stimulator in endothelial
cells. J Biol Chem. 284:23436–23443. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Turner KJ, Crew JP, Wykoff CC, Watson PH,
Poulsom R, Pastorek J, Ratcliffe PJ, Cranston D and Harris AL: The
hypoxia-inducible genes VEGF and CA9 are differentially regulated
in superficial vs invasive bladder cancer. Br J Cancer.
86:1276–1282. 2002. View Article : Google Scholar : PubMed/NCBI
|