Introduction
Myocardial ischemia is a common cause of morbidity
and mortality in the world (1). It
is well known that well-differentiated tissues including heart
require large amounts of oxygen to support their specialized
functions. When oxygen is in short supply, the oxidative
phosphorylation of mitochondria stops rapidly, which results in a
resultant loss of the major source of ATP production for energy
metabolism and subsequently ischemia (2,3).
Myocardial ischemia can cause a characteristic pattern of
ultrastructural and metabolic changes, leading to irreversible
damage to the myocardium (4,5).
Presently, the mechanisms of myocardial ischemic injury are still
needed to be explored.
Long non-coding RNAs (lncRNAs) are a set of RNAs
longer than 200 nt, which involve in lots of cellular processes,
such as genomic imprinting, chromatin modification and RNA
alternative splice (6). In
addition, lncRNAs are associated with many human diseases (7–9).
Recently, a growing number of studies focus on the role of lncRNAs
in cardiac diseases. Several lncRNAs have been detected in
cardiomyocytes and are suggested to be involved in heart
development (10,11). LncRNA TUG1 is highly conserved in
mammals but it is not reported in other vertebrates (12). Previous studies have shown that
TUG1 is implicated in many cancers, affecting apoptosis and
proliferation of tumor cells (13,14).
However, to our best knowledge, there is no study concerning the
function of TUG1 in regulating myocardial ischemic injury.
Therefore, to explore the role and regulatory
mechanism of TUG1 in regulating myocardial ischemic injury, the
present study established a cell model of myocardial injury through
treating cardiomyocytes with hypoxia. Then the expression level of
TUG1 in hypoxia-induced myocardial injury model was detected.
Furthermore, the relationship between dysregulated expression of
TUG1 and myocardial injury were explored, as well as the potential
molecular mechanisms of TUG1 in regulating myocardial injury. This
study aimed to provide new theoretical explanation for the
mechanism of myocardial injury.
Materials and methods
Cell culture and treatment
The cardiomyocytes cell line H9c2 was purchased from
Sigma-Aldrich (Merck KGaA, Darmstadt, Germany), and cultured in
Dulbecco's modified Eagle's medium (Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) at 37°C in an incubator with 5% CO2.
The culture medium was supplemented with 10% fetal bovine serum
(FBS), 1% Penicillin/Streptomycin (100 U/ml:100 mg/ml) and 1%
GlutaMAX (Thermo Fisher Scientific, Inc.), and was changed every
other day. The H9c2 cells were cultured under the hypoxia (3%
O2) and normoxia (21% O2) conditions,
respectively.
Cell transfection
Short-hairpin (sh)RNA directed against TUG1 was
ligated into the plasmid of U6/GFP/Neo (GenePharma, Shanghai,
China), which was called sh-TUG1. TUG1 was ligated into the
pcDNA3.1, which was referred as to pc-TUG1. To analyze the
functions of Bcl2/adenovirus E1B 19 kDa-interacting protein 3
(Bnip3), the full-length Bnip3 sequences and shRNA directed against
Bnip3 were respectively ligated into plasmids of pEX-2 and
U6/GFP/Neo (GenePharma), referring as to pEX-Bnip3 and si-Bnip3.
Cells transfection was then performed using Lipofectamine 3000
reagent (Thermo Fisher Scientific, Inc.) according to the
manufacturer's instructions. The plasmid that carried a
non-targeting sequence was used as negative control (NC) of sh-TUG1
and si-Bnip3. The stably transfected cells were selected through
the culture medium containing 0.5 mg/ml G418 (Sigma-Aldrich; Merck
KGaA), and G418-resistant cell clones were established after about
4 weeks. miR-145-5p mimics, miR-145-5p inhibitors, and NC were
synthesized (Thermo Fisher Scientific, Inc.) and then transfected
into cells. Cells were harvested after 72 h of transfection.
RT-qPCR
Total RNA was extracted from cells using Trizol
reagent (Thermo Fisher Scientific, Inc.). Real-Time PCR analysis
was performed to detect the expression level of TUG1 using One Step
SYBR® PrimeScript®PLUS RT-RNA PCR Kit (TaKaRa
Biotechnology). Bnip3 expression was detected with RNA PCR kit
(AMV) Ver.3.0 (Takara Biotechnology Co., Ltd., Dalian, China).
GAPDH was used as the internal control. The expression level of
miR-145-5p was determined using the Taqman MicroRNA Reverse
Transcription Kit and Taqman Universal Master Mix II with the
TaqMan MicroRNA Assay of miR-145-5p and U6 (Applied Biosystems;
Thermo Fisher Scientific, Inc.). U6 was used for normalizing the
expression of miR-145-5p. Fold-changes were calculated according to
cycle quantitation (Cq) values with 2−ΔΔCq method.
Cell viability assay
Total 1×105 cells were seeded into 60-mm
dishes in duplicate. At the indicated time periods, Cells were
washed and the living cells were determined using trypan blue
exclusion.
Apoptosis assay
Cells were washed with phosphate-buffer saline (PBS)
and then fixed in 70% ethanol. Afterwards, cells were stained with
propidium iodide (PI)/fluorescein isothiocyanate (FITC)-Annexin V
in the presence of 50 µg/ml RNase A (Sigma-Aldrich; Merck KGaA).
Then cells were incubated in the dark for 1 h at 25°C. Flow
cytometry analysis was performed using a FACS can (Beckman Coulter,
Inc., Brea, CA, USA). The data were analyzed using the FlowJo
software.
Cell migration and invasion
assays
Cell migration was detected using a modified
two-chamber migration assay (pore size, 8 mm). Cells suspended in
200 ml serum-free medium were seeded on the upper compartment of
24-well Transwell culture chamber, and 600 ml complete medium was
added to the lower compartment. After incubation at 37°C, cells
were then fixed with methanol. On the upper surface of the chamber,
the non-traversed cells were removed with a cotton swab. The
traversed cells were stained with crystal violet and counted
microscopically.
The invasion behavior was detected with 24-well
Millicell Hanging Cell Culture inserts with 8 mm PET membranes (EMD
Millipore, Billerica, MA, USA). Total 5.0×104 cells
suspended in 200 µl serum-free dulbecco's modified eagle medium
were seeded onto BD BioCoat™ Matrigel TM Invasion Chambers (BD
Biosciences, Franklin Lakes, NJ, USA). Complete medium containing
10% FBS was added to the lower chamber. The invasion chambers were
incubated at 37°C for 48 h with 5% CO2. After removing
the non-invading cells, the invading cells were fixed with 100%
methanol and stained with crystal violet solution. Finally, cells
were counted microscopically.
Luciferase reporter assay
Fragment from Bnip3 that contained the predicted
miR-145-5p binding site was amplified via PCR, which were then
cloned into a pmirGlO Dual-luciferase miRNA Target Expression
Vector (Promega Corporation, Madison, WI, USA) to construct the
reporter vector Bnip3-wild-type (Bnip3-wt). Subsequently, the
reporter vectors and miR-145-5p mimics were co-transfected into HEK
293T cells. The luciferase activity was determined based on the
Dual-Luciferase Reporter Assay System (Promega Corporation).
Western blot analysis
Protein was extracted using RIPA lysis buffer
(Beyotime Biotechnology, Shanghai, China) that was supplemented
with protease inhibitors (Roche, Guangzhou, China). The protein
extracts were quantified with the BCA™ Protein Assay kit (Pierce;
Thermo Fisher Scientific, Inc.). According to the manufacturer's
instructions, the western blot system was established using a
Bio-Rad Bis-Tris Gel system. Primary antibodies (at a dilution of
1:1,000) were prepared in 5% blocking buffer. After incubation with
primary antibodies at 4°C overnight, secondary antibodies marked by
horseradish peroxidase were used to incubate the polyvinylidene
difluoride (PVDF) membrane at room temperature (approximately 25°C)
for 1 h. The membranes carried blots and antibodies were then
transferred into the Bio-Rad ChemiDoc™ XRS system, and 200 µl
Immobilon Western Chemiluminescent HRP Substrate (EMD Millipore)
was added to cover the membrane surface. The intensity of the bands
was quantified with Image Lab™ Software (Bio-Rad, Shanghai,
China).
Statistical analysis
The results were presented as mean ± standard
deviation. Statistical analyses were performed using Graphpad 6.0.
The P-values were calculated using a one-way analysis of variance
(ANOVA). P<0.05 was considered to indicate a statistically
significant difference.
Results
Hypoxia induces hypoxia injury in H9c2
cells
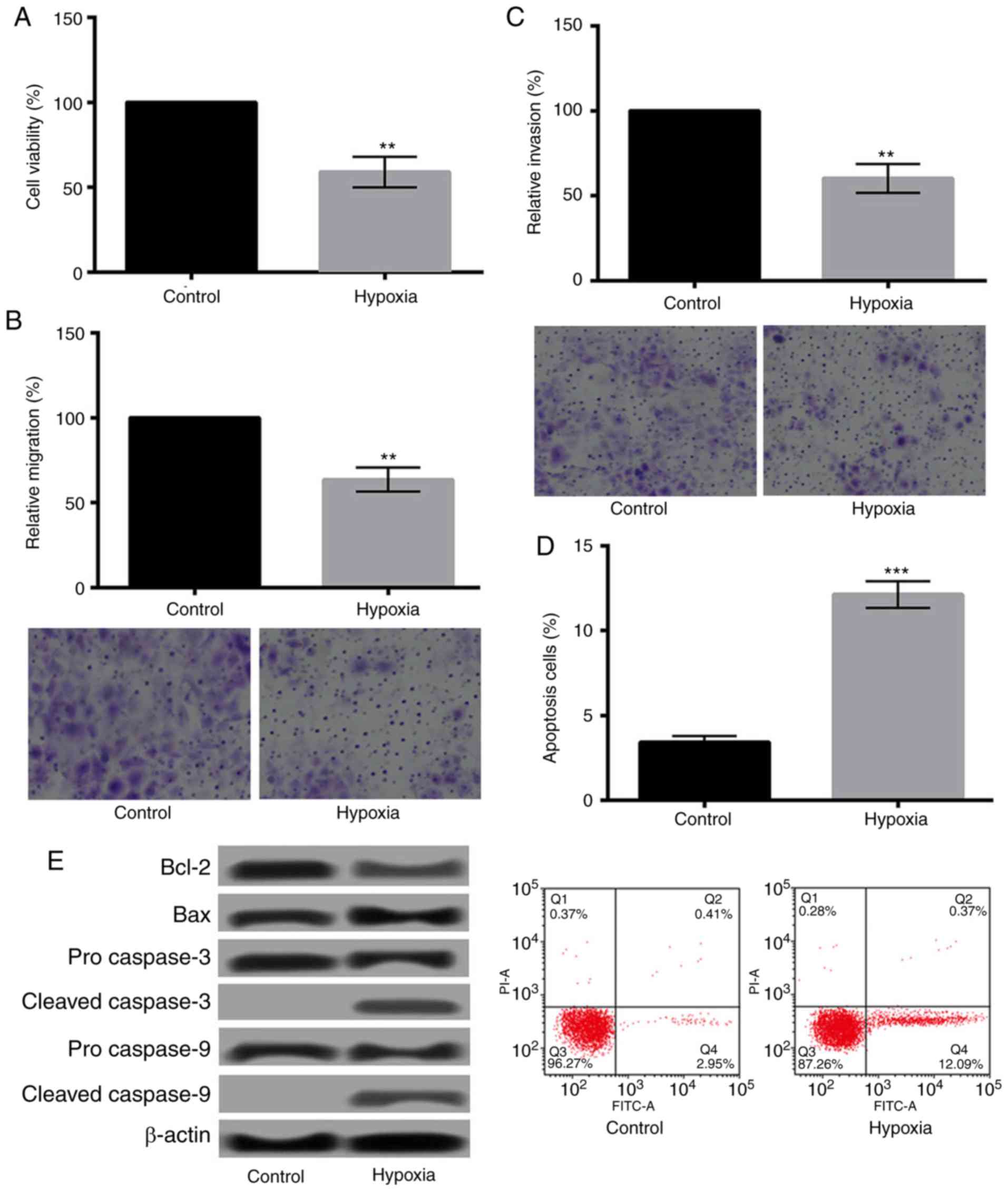
The effects of hypoxia on H9c2 cells were evaluated
by determination of the changes of cell viability, migration,
invasion, and apoptosis. As presented in Fig. 1A-C, hypoxia could significantly
decrease the viability, migration, and invasion of H9c2 cells
(P<0.01). Additionally, Fig. 1D
showed that hypoxia significantly increased apoptosis of H9c2 cells
(P<0.001). The relative expression levels of apoptosis-related
proteins changed obviously as well. As shown in Fig. 1E, the expression of Bcl-2 was
downregulated, while Bax was upregulated. Moreover, cleaved
caspase-3/9 were detected after hypoxia treatment.
Hypoxia promotes the expression of
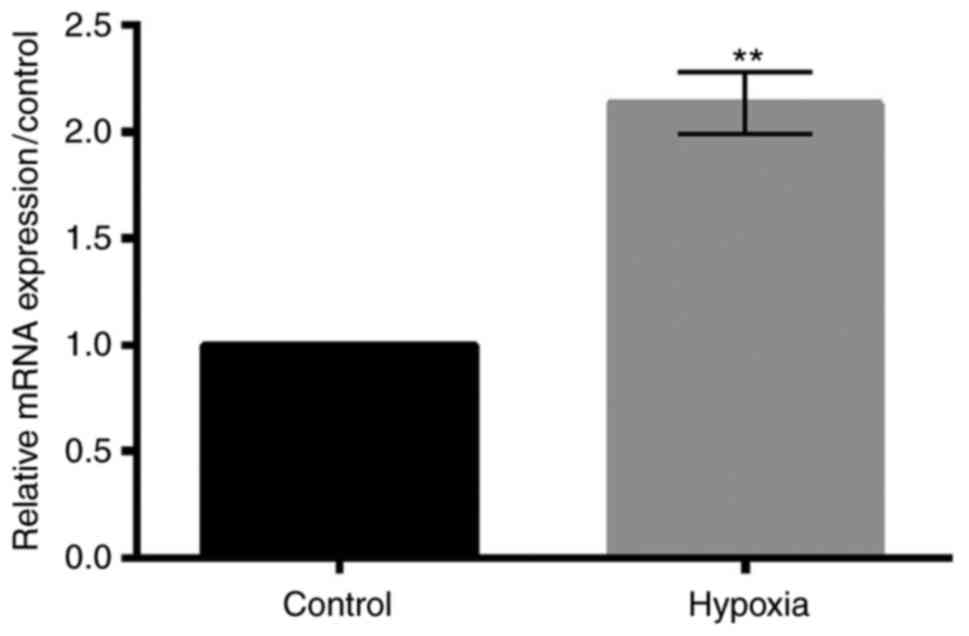
TUG1
The relative expression level of TUG1 under hypoxic
condition was detected using RT-qPCR. As shown in Fig. 2, hypoxia treatment significantly
increased the expression level of TUG1 (P<0.01).
Overexpression of TUG1 aggravates
hypoxia-induced injury in H9c2 cells, while suppression of TUG1
relieves the injury
To study whether abnormal expression of TUG1 could
influence hypoxia-induced injury in H9c2 cells, TUG1 was
overexpressed and suppressed in H9c2 cells. The overexpression or
suppression of TUG1 was confirmed by qRT-PCR (P<0.01) (Fig. 3A). After cell transfection, the
effects of hypoxia on cell viability, migration, invasion, and
apoptosis of H9c2 cells were further evaluated. The results showed
that compared with pcDNA3.1, overexpression of TUG1 (pc-TUG1)
significantly decreased the viability, migration, and invasion
(Fig. 3B-D), while increased the
apoptosis of H9c2 cells (Fig. 3E)
(P<0.05). Additionally, the expression of Bcl-2 was further
decreased after TUG1 overexpression, while of Bax was further
increased. Moreover, the expression levels of cleaved caspase-3 and
caspase-9 were higher in hypoxia+pc-TUG1 than that in
hypoxia+pcDNA3.1 (Fig. 3F). On the
contrary, reverse results were obtained when the TUG1 expression
was suppressed (Fig. 3B-F).
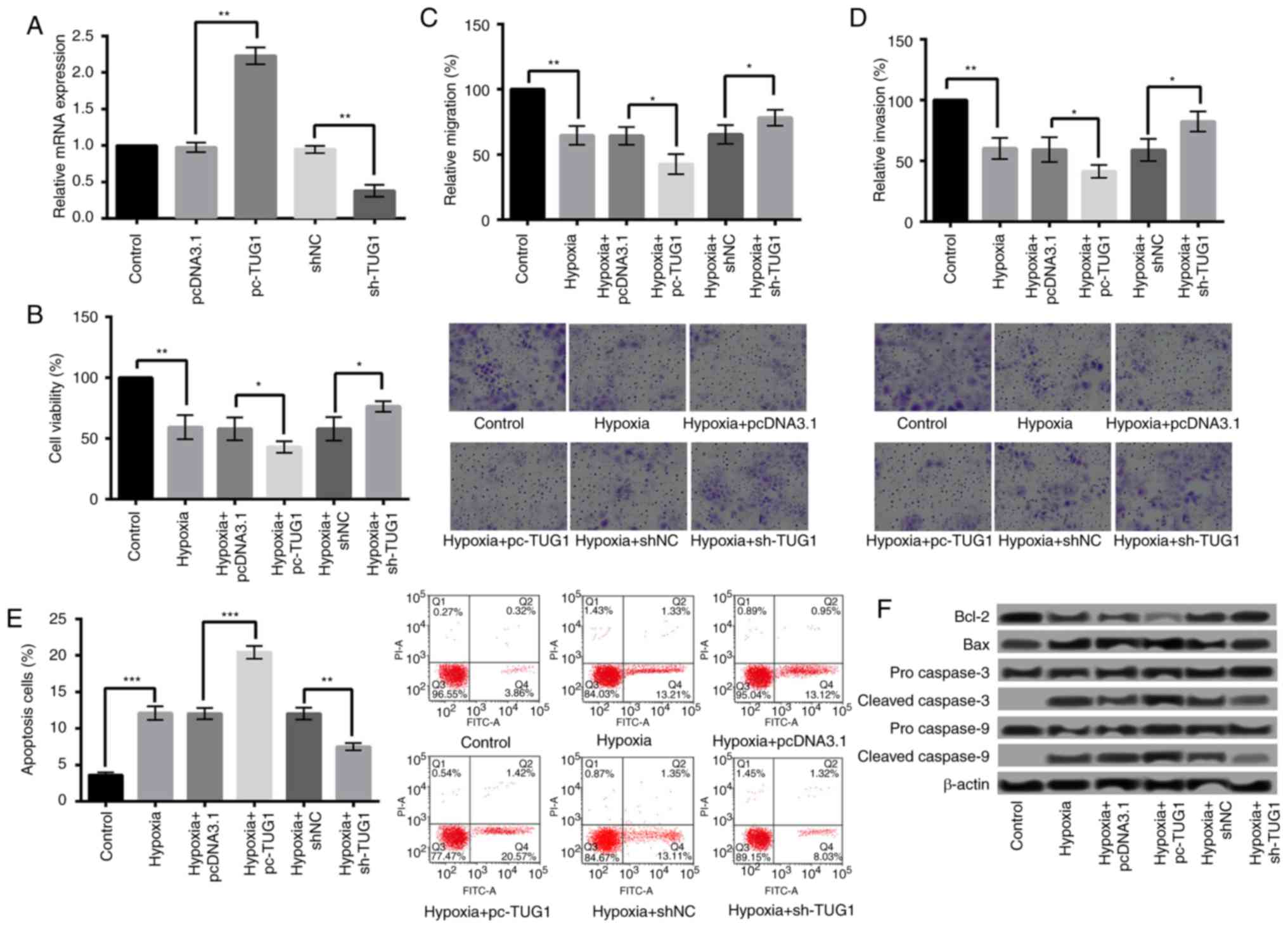 | Figure 3.Overexpression of TUG1 aggravated
hypoxia-induced injury. (A) TUG1 expression following H9c2 cell
transfection with sh-TUG1 and pc-TUG1. Overexpression of TUG1
significantly decreased the (B) viability, (C) migration and (D)
invasion, and (E) increased the apoptosis of H9c2 cells when
compared with control. Magnification, ×400. (F) The expression
levels of apoptosis-associated proteins following TUG1
overexpression. *P<0.05, **P<0.01 and ***P<0.001, as
indicated. TUG1, taurine upregulated 1 (non-protein coding); sh-,
small hairpin RNA; pcDNA3.1, plasmid cytomegalovirus promoter
DNA3.1; pc-TUG1, TUG1 overexpression group; Bcl-2, B-cell lymphoma
2; Bax, Bcl-2-associated X protein. |
TUG1 negatively regulates the
expression of miR-145-5p and overexpression of TUG1 aggravates
hypoxia injury by downregulation of miR-145-5p
Further study found that TUG1 could negatively
regulate the expression of miR-145-5p. The relative expression of
TUG1 after cell transfection was shown in Fig. 4A. Fig.
4B presented the expression level of miR-145-5p after H9c2
cells were transfected with miR-145-5p mimic or inhibitor.
Additionally, the effect of miR-145-5p overexpression on
hypoxia-induced cardiomyocyte injury was detected. As shown in
Fig. 4C-F, compared with
hypoxia+pc-TUG1+mimic NC group, hypoxia+pc-TUG1+miR-145-5p mimic
could relieve hypoxia injury by significantly increasing cell
viability, migration, and invasion (P<0.05), and decreasing
apoptosis (P<0.001). In addition, compared with
hypoxia+pc-TUG1+mimic NC group, miR-145-5p overexpression increased
Bcl-2 expression, and decreased the expression levels of Bax,
cleaved caspase-3 and caspase-9 (Fig.
4G).
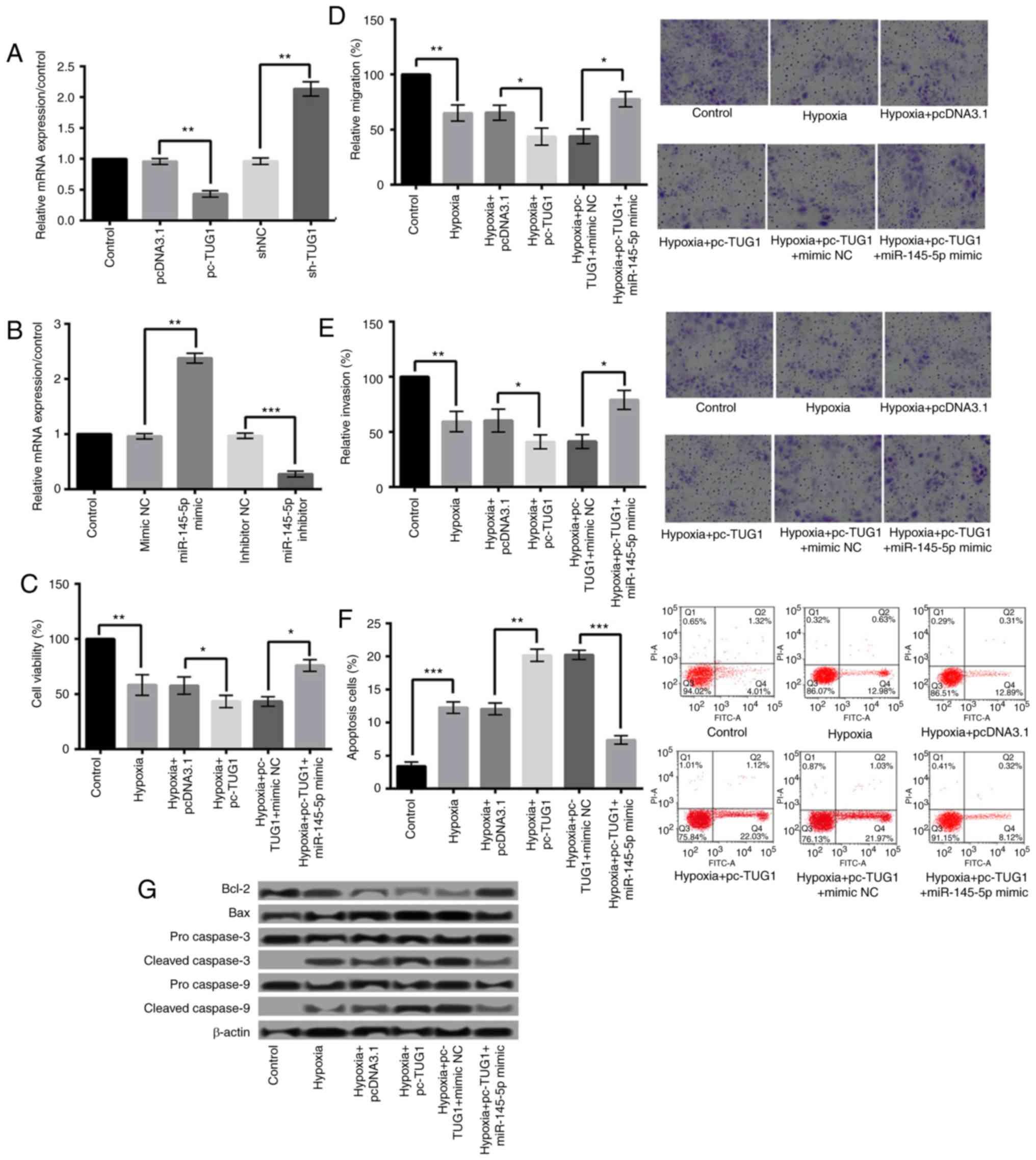 | Figure 4.Overexpression of TUG1 aggravated
hypoxic injury via downregulation of miR-145-5p. (A) The relative
expression of TUG1 following cell transfection. (B) The expression
level of miR-145-5p following H9c2 cell transfection with
miR-145-5p mimic or inhibitor. Hypoxia+pc-TUG1+miR-145-5p mimic
significantly increased the (C) viability, (D) migration and (E)
invasion, and decreased (F) the apoptosis of H9c2 cells when
compared with the hypoxia+pc-TUG1+mimic NC group. Magnification,
×400. (G) miR-145-5p overexpression under hypoxic conditions
increased Bcl-2 expression, and decreased the expression levels of
Bax, and cleaved caspase-3 and caspase-9. *P<0.05, **P<0.01
and ***P<0.001, as indicated. TUG1, taurine upregulated 1
(non-protein coding); miR, microRNA; sh-, small hairpin RNA;
pcDNA3.1, plasmid cytomegalovirus promoter DNA3.1; pc-TUG1, TUG1
overexpression group; NC, negative control; Bcl-2, B-cell lymphoma
2; Bax, Bcl-2-associated X protein. |
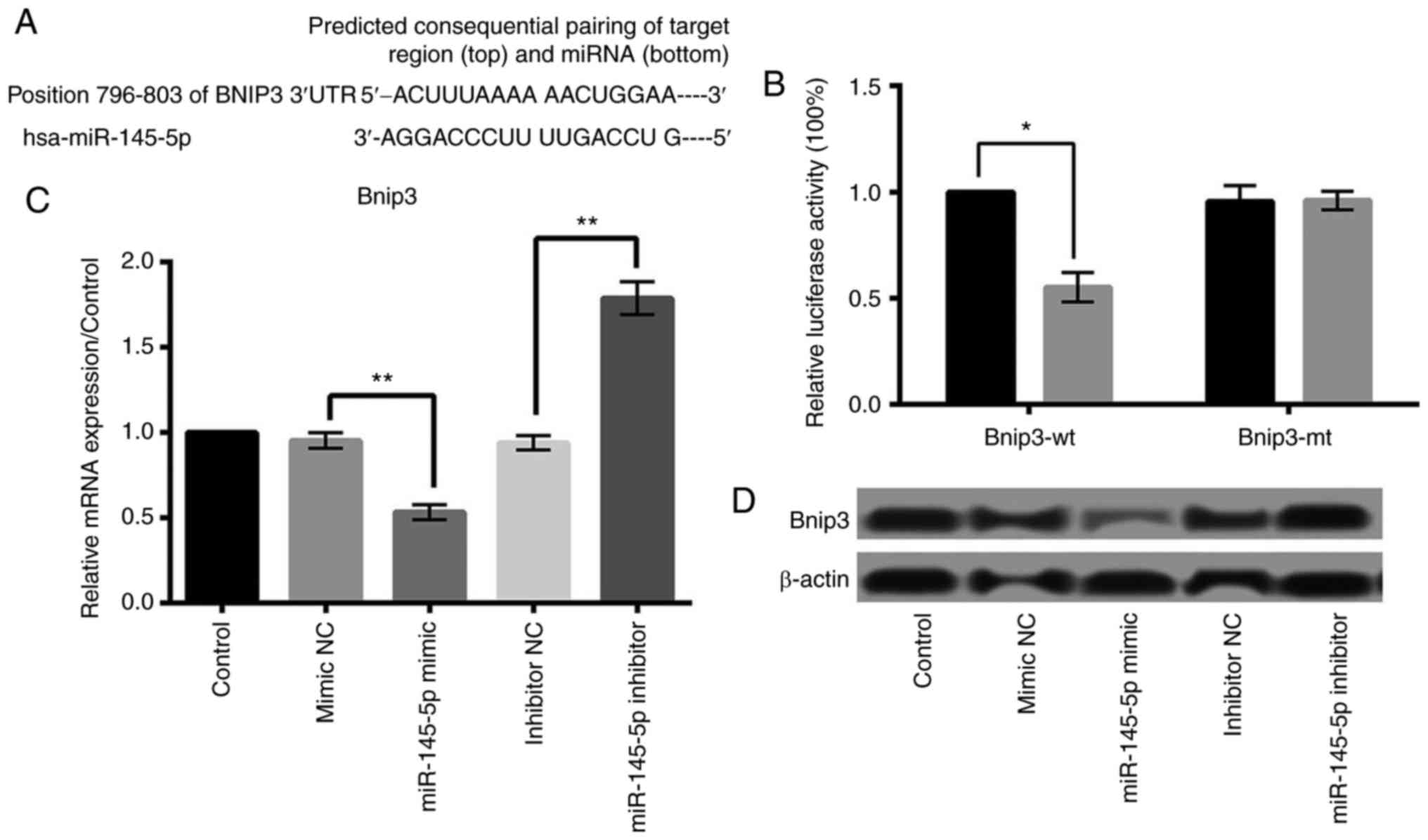
miR-145-5p negatively regulates Bnip3
expression and Bnip3 is a target of miR-145-5p
Study has reported that Bnip3 plays a key role in
apoptosis, necrosis and autophagy of cardiomyocytes (15–18).
Based on the public miRNA database, we found that the 3′-UTR of
Bnip3 was a potential binding site of miR-145-5p, suggesting that
Bnip3 may be a direct target of miR-145-5p in cardiomyocytes
(Fig. 5A). Then we performed
luciferase reporter assay to verify whether Bnip3 was a direct
target of miR-145-5p. As shown in Fig.
5B, miR-145 overexpression significantly reduced the activity
of luciferease gene fused with the Bnip3 wt-3′-UTR (P<0.05).
However, overexpression of miR-145 barely influenced the activity
of luciferase gene fused with the Bnip3 3′-UTR mutant. Furthermore,
qRT-PCR and western blot analyses found that the relative
expression level of Bnip3 was remarkably inhibited by overexpressed
miR-145-5p and was raised by suppressed miR-145-5p (P<0.01)
(Fig. 5C and D). These results
suggested that Bnip3 was a direct target of miR-145-5p in
cardiomyocytes, and was negatively regulated by miR-145-5p.
Overexpression of miR-145-5p protects
against hypoxia-induced injury by downregulation of Bnip3
To further demonstrate that the protective effects
of miR-145-5p on cardiomyocytes were achieved by negatively
regulating Bnip3, we investigated the effects of Bnip3
overexpression on hypoxia-induced injury. qRT-PCR and western blot
were performed to detect the Bnip3 levels after cell transfection.
As shown in Fig. 6A and B,
transfection of pEX-Bnip3 significantly increased the Bnip3
expression, while transfection of si-Bnip3 significantly suppressed
the Bnip3 expression (P<0.001). Subsequent experiments revealed
that the protective effects of miR-145-5p overxpression on
cardiomyocytes were abrogated by overexpressed Bnip3, showing that
overexpressed Bnip3 significantly reduced the viability, migration,
and invasion, and increased the apoptosis of H9c2 cells (Fig. 6C-F) (P<0.01). Additionally, the
expression of Bcl-2 was decreased, while of Bax, cleaved caspase-3
and caspase-9 was increased (Fig.
6G).
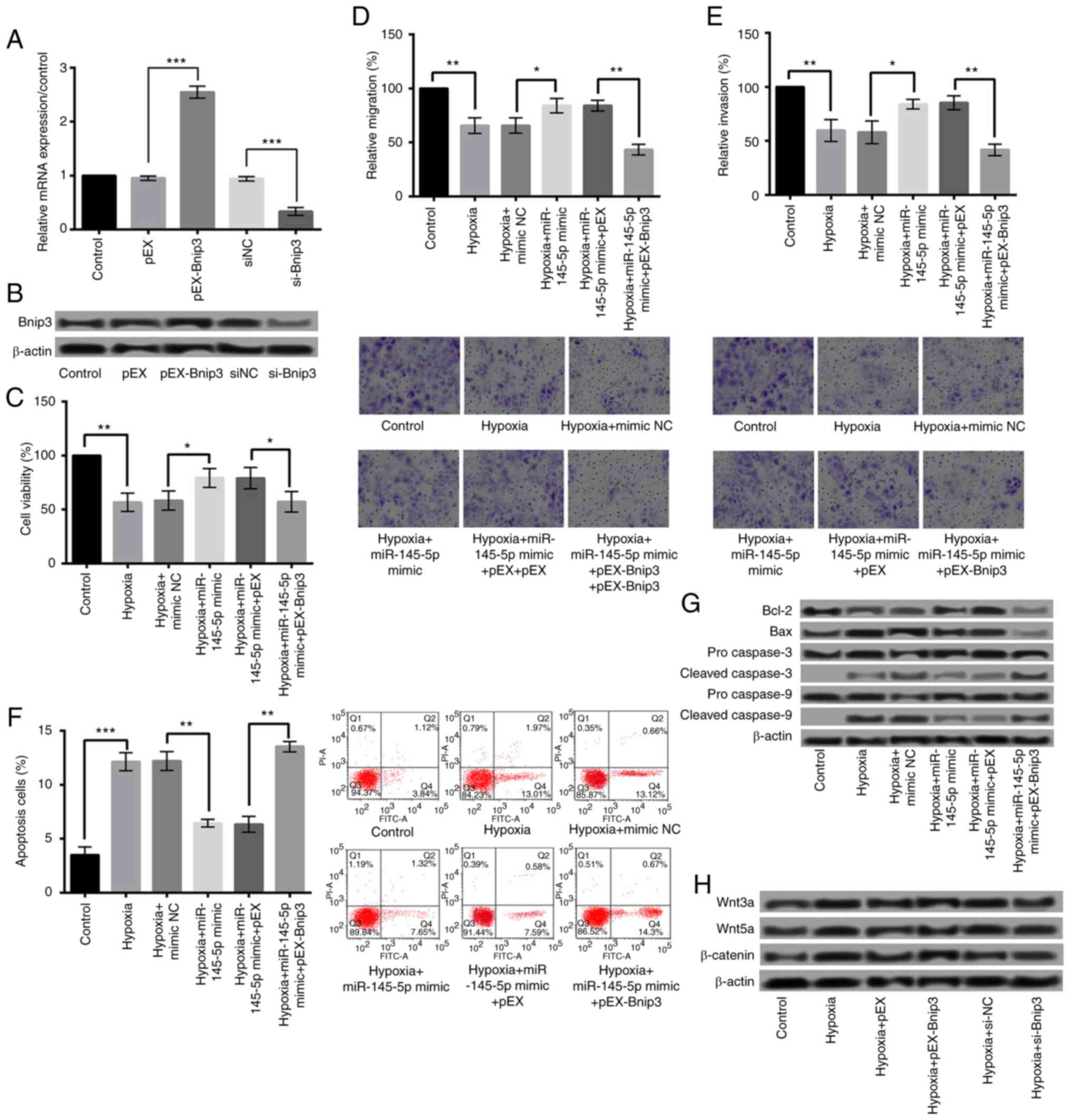 | Figure 6.Overexpression of miR-145-5p was
protective against hypoxia-induced injury via the downregulation of
Bnip3, and the overexpression of Bnip3 aggravated hypoxia-induced
injury via the Wnt/β-catenin signaling pathway. The relative (A)
mRNA and (B) protein expression of Bnip3 following cell
transfection. Overexpressed Bnip3 significantly reduced the (C)
viability, (D) migration and (E) invasion, and increased the (F)
apoptosis of H9c2 cells. Magnification, ×400. (G) The expression of
Bcl-2 was decreased, while that of Bax, and cleaved caspase-3 and
caspase-9 was increased following Bnip3 overexpression. (H) Bnip3
overexpression increased the protein expressions of proteins
associated with the Wnt/β-catenin signaling pathways, including
Wnt3a/5a and β-catenin. *P<0.05, **P<0.01 and ***P<0.001,
as indicated. Bnip3, B-cell lymphoma 2 interacting protein 3; miR,
microRNA; si-, small interfering RNA. |
Overexpression of Bnip3 aggravates
hypoxia-induced injury via Wnt/β-catenin signaling pathways
To explore the underlying mechanisms of Bnip3
overexpression aggravating hypoxia-induced injury, we investigated
the effect of Bnip3 overexpression on Wnt/β-catenin signaling
pathways. As presented in Fig. 6H,
Bnip3 overexpression significantly increased the protein
expressions of Wnt/β-catenin signaling pathways-related proteins,
including Wnt3a/5a and β-catenin. These results suggested that
overexpressed Bnip3 aggravated hypoxia-induced cell injury by
activating Wnt/β-catenin pathways in H9c2 cells.
Discussion
In this study, the effects of TUG1 on the cell
hypoxia injury in H9c2 cells were studied. The results showed that
hypoxia induced injury in H9c2 cells, including inhibiting cell
viability, migration and invasion and promoting cell apoptosis.
Overexpression of TUG1 aggravated hypoxia injury in H9c2 cells.
Further studies showed that miR-145-5p was negatively regulated by
TUG1, and TUG1 overexpression aggravated hypoxia injury by
downregulation of miR-145-5p. Moreover, we found that miR-145-5p
negatively regulated Bnip3 expression and Bnip3 was suggested to be
a target gene of miR-145-5p. Overexpression of Bnip3 aggravated
hypoxia-induced cell injury by activating Wnt/β-catenin pathways in
H9c2 cells. Our study may provide a new strategy for the treatment
of myocardial damage induced by hypoxia.
TUG1 was originally found in taurine-treated mouse
retinal cells. It has been revealed that TUG1 knockdown leads to
malformed outer segments of transfected photoreceptors via
increased apoptosis in the newborn retina (19). Additionally, down-regulation of
TUG1 has also been suggested to promote apoptosis in many cancer
cells (14,20,21).
These studies may suggest the critical role of TUG1 in apoptosis.
Interestingly, extensive investigation associated with
cardiomyocyte ischemic injury found that apoptosis was associated
with lots of forms of cardiac pathology, including myocardial
ischemia (22,23). Notably, TUG1 is found to function
as a miRNA sponge to promote neurons apoptosis under ischemia,
which possibly severed as a new therapeutic target in stroke
(24). In this study,
overexpression of TUG1 was found to significantly decreased the
viability, migration, and invasion, and increased the apoptosis of
hypoxia-induced H9c2 cells, suggesting the important role of TUG1
in myocardial ischemia.
MiR-145 is a tumor suppressor miRNA which suppresses
proliferation and induce apoptosis in various tumor cell lines
(25,26). It is reported that TUG1 can
influence epithelial-to-mesenchymal transition in several cancers
through targeting miR-145 (20,27).
Serum miR-145 is found positively correlated with plasma
high-sensitivity C-reactive protein (hs-CRP) and the combination of
hs-CRP and serum miR-145 may be an effective approach for
predicting acute ischemia stroke (28). Recently, the functions of miR-145
in heart were explored. In the study of Li et al (29), miR-145 was suggested to exert a
protective effect against the oxidative stress-induced apoptosis in
cardiomyocytes. Importantly, they have demonstrated that under
oxidative stress, miR-145 protects against the mitochondria
apoptotic pathway activation in cardiomyocytes via targeting Bnip3
directly. In accordance with their study, our results showed that
TUG1 overexpression aggravated hypoxia injury by downregulation of
miR-145-5p. Moreover, Bnip3 was a target of miR-145-5p and was
negatively regulated by miR-145-5p.
Bnip3, primarily in the mitochondrial outer
membrane, belongs to BH3-only subfamily of Bcl-2 family proteins,
which antagonizes the activity of pro-survival proteins and
promotes apoptosis (30,31). Normally, the Bnip3 expression is
undetectable in most organs, including the heart. However, its
expression level can be increased by hypoxia (32). During myocardial ischemia and
reperfusion, Bnip3 is found to act as a mitochondrial sensor of
oxidative stress (16). Graham
et al (33) also reported
that Bnip3 is overexpressed in heart following acute ischemia, and
in chronic heart failure after myocardial infarction.
Interestingly, in this study, overexpression of Bnip3 was found to
aggravate hypoxia-induced cell injury, which was in consistence
with the studies above. Furthermore, the overexpressed Bnip3
aggravating hypoxia-induced cell injury was found to be achieved by
activating Wnt/β-catenin pathways.
It is reported that interacting cells can form a
‘cellular interactome’ under a defined condition. After cardiac
injury, different populations of cells in the heart can construct a
complex cardiac cellular interactome, which is regulated by many
signaling systems (34). The
Wnt/β-catenin signaling pathway has been demonstrated to play a
critical role in cardiac development, and in orchestrating a
cardiac injury response (35,36).
Therefore, our study further suggested the role of Wnt/β-catenin
signaling pathway in hypoxia-induced injury of cardiomyocytes.
In conclusion, our data suggest that TUG1
overexpression aggravates hypoxia injury of cardiomyocytes by
regulating miR-145-5p-Bnip3 axis to activate Wnt/β-catenin
pathways. Therefore, TUG1 may be used as a diagnostic marker and
therapeutic target in myocardial ischemia. However, there is lack
of in vivo research in myocardial ischemia to better
investigate the role of LncRNA TUG1 in the whole organism. Further
clinical and in vivo studies are still needed to confirm the
results.
References
|
1
|
Reimer KA and Ideker RE: Myocardial
ischemia and infarction: Anatomic and biochemical substrates for
ischemic cell death and ventricular arrhythmias. Human Pathol.
18:462–475. 1987. View Article : Google Scholar
|
|
2
|
Buja LM, Hagler HK and Willerson JT:
Altered calcium homeostasis in the pathogenesis of myocardial
ischemic and hypoxic injury. Cell Calcium. 9:205–217. 1988.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Farber JL, Chien KR and Mittnacht S Jr:
Myocardial ischemia: The pathogenesis of irreversible cell injury
in ischemia. Am J Pathol. 102:271–281. 1981.PubMed/NCBI
|
|
4
|
Buja LM: Myocardial ischemia and
reperfusion injury. Cardiovasc Pathol. 14:1–175. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
American College of Emergency Physicians,
; Society for Cardiovascular Angiography and Interventions, ;
O'Gara PT, Kushner FG, Ascheim DD, Casey DE Jr, Chung MK, de Lemos
JA, Ettinger SM, Fang JC, et al: 2013 ACCF/AHA guideline for the
management of ST-elevation myocardial infarction: Executive
summary: A report of the american college of cardiology
foundation/american heart association task force on practice
guidelines. J Am Coll Cardiol. 61:485–510. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gartler SM and Riggs AD: Mammalian
X-chromosome inactivation. Annu Rev Genet. 17:155–190. 1983.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shi X, Sun M, Liu H, Yao Y and Song Y:
Long non-coding RNAs: A new frontier in the study of human
diseases. Cancer Lett. 339:159–166. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Wapinski O and Chang HY: Long noncoding
RNAs and human disease. Trends Cell Biol. 21:354–361. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Li J, Xuan Z and Liu C: Long non-coding
RNAs and complex human diseases. Int J Mol Sci. 14:18790–18808.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Grote P, Wittler L, Hendrix D, Koch F,
Währisch S, Beisaw A, Macura K, Bläss G, Kellis M, Werber M and
Herrmann BG: The tissue-specific lncRNA Fendrr is an essential
regulator of heart and body wall development in the mouse. Dev
Cell. 24:206–214. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Ishii N, Ozaki K, Sato H, Mizuno H, Saito
S, Takahashi A, Miyamoto Y, Ikegawa S, Kamatani N, Hori M, et al:
Identification of a novel non-coding RNA, MIAT, that confers risk
of myocardial infarction. J Hum Genet. 51:1087–1099. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Yin DD, Zhang EB, You LH, Wang N, Wang LT,
Jin FY, Zhu YN, Cao LH, Yuan QX, De W and Tang W: Downregulation of
lncRNA TUG1 affects apoptosis and insulin secretion in mouse
pancreatic β cells. Cell Physiol Biochem. 35:1892–1904. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Zhang Q, Geng PL, Yin P, Wang XL, Jia JP
and Yao J: Down-regulation of long non-coding RNA TUG1 inhibits
osteosarcoma cell proliferation and promotes apoptosis. Asian Pac J
Cancer Prev. 14:2311–2315. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Han Y, Liu Y, Gui Y and Cai Z: Long
intergenic non-coding RNA TUG1 is overexpressed in urothelial
carcinoma of the bladder. J Surg Oncol. 107:555–559. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Lomonosova E and Chinnadurai G: BH3-only
proteins in apoptosis and beyond: An overview. Oncogene. 27 Suppl
1:S2–S19. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kubli DA, Quinsay MN, Huang C, Lee Y and
Gustafsson AB: Bnip3 functions as a mitochondrial sensor of
oxidative stress during myocardial ischemia and reperfusion. Am J
Physiol Heart Circ Physiol. 295:H2025–H2031. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Kubasiak LA, Hernandez OM, Bishopric NH
and Webster KA: Hypoxia and acidosis activate cardiac myocyte death
through the Bcl-2 family protein BNIP3. Proc Natl Acad Sci USA.
99:pp. 1–12830. 2002; PubMed/NCBI
|
|
18
|
Guo K, Searfoss G, Krolikowski D, Pagnoni
M, Franks C, Clark K, Yu KT, Jaye M and Ivashchenko Y: Hypoxia
induces the expression of the pro-apoptotic gene BNIP3. Cell Death
Differ. 8:367–376. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Young TL, Matsuda T and Cepko CL: The
noncoding RNA taurine upregulated gene 1 is required for
differentiation of the murine retina. Curr Biol. 15:501–512. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Tan J, Qiu K, Li M and Liang Y:
Double-negative feedback loop between long non-coding RNA TUG1 and
miR-145 promotes epithelial to mesenchymal transition and
radioresistance in human bladder cancer cells. FEBS Lett.
589:3175–3181. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Liu Y, Yang S and Zhang X: WITHDRAWN:
Down-regulation of long non-coding RNA TUG1 suppresses melanoma
cell proliferation and induces apoptosis via up-regulating
microRNA-9. Biochem Biophys Res Commun. 2013.
|
|
22
|
Buja LM: Modulation of the myocardial
response to ischemia. Lab Invest. 78:1345–1373. 1998.PubMed/NCBI
|
|
23
|
Buja LM and Entman ML: Modes of myocardial
cell injury and cell death in ischemic heart disease. Circulation.
98:1355–1357. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Chen S, Wang M, Yang H, Mao L, He Q, Jin
H, Ye ZM, Luo XY, Xia YP and Hu B: LncRNA TUG1 sponges microRNA-9
to promote neurons apoptosis by up-regulated Bcl2l11 under
ischemia. Biochem Biophys Res Commun. 485:167–173. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Spizzo R, Nicoloso MS, Lupini L, Lu Y,
Fogarty J, Rossi S, Zagatti B, Fabbri M, Veronese A, Liu X, et al:
miR-145 participates with TP53 in a death-promoting regulatory loop
and targets estrogen receptor-alpha in human breast cancer cells.
Cell Death Differ. 17:246–254. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu Q, Liu LZ, Qian X, Chen Q, Jiang Y, Li
D, Lai L and Jiang BH: MiR-145 directly targets p70S6K1 in cancer
cells to inhibit tumor growth and angiogenesis. Nucleic Acids Res.
40:761–774. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Lei H, Gao Y and Xu X: LncRNA TUG1
influences papillary thyroid cancer cell proliferation, migration
and EMT formation through targeting miR-145: Acta. Biochim Biophys
Sin (Shsnghai). 49:1–597. 2017.
|
|
28
|
Jia L, Hao F, Wang W and Qu Y: Circulating
miR-145 is associated with plasma high-sensitivity C-reactive
protein in acute ischemic stroke patients. Cell Biochem Funct.
33:314–319. 2015. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Li R, Yan G, Li Q, Sun H, Hu Y, Sun J and
Xu B: MicroRNA-145 protects cardiomyocytes against hydrogen
peroxide (H2O2)-induced apoptosis through targeting the
mitochondria apoptotic pathway. PLoS One. 7:e449072012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ray R, Chen G, Vande Velde C, Cizeau J,
Park JH, Reed JC, Gietz RD and Greenberg AH: BNIP3 heterodimerizes
with Bcl-2/Bcl-X(L) and induces cell death independent of a Bcl-2
homology 3 (BH3) domain at both mitochondrial and nonmitochondrial
sites. J Biol Chem. 275:1439–1448. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Yasuda M, Theodorakis P, Subramanian T and
Chinnadurai G: Adenovirus E1B-19K/BCL-2 interacting protein BNIP3
contains a BH3 domain and a mitochondrial targeting sequence. J
Biol Chem. 273:12415–12421. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Bruick RK: Expression of the gene encoding
the proapoptotic Nip3 protein is induced by hypoxia. Proc Natl Acad
Sci USA. 97:pp. 9082–9087. 2000; View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Graham RM, Frazier DP, Thompson JW, Haliko
S, Li H, Wasserlauf BJ, Spiga MG, Bishopric NH and Webster KA: A
unique pathway of cardiac myocyte death caused by hypoxia-acidosis.
J Exp Biol. 207:3189–3200. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Deb A: Cell-cell interaction in the heart
via Wnt/β-catenin pathway after cardiac injury. Cardiovasc Res.
102:214–223. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bergmann MW: WNT signaling in adult
cardiac hypertrophy and remodeling: Lessons learned from cardiac
development. Circ Res. 107:1198–1208. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Gessert S and Kühl M: The multiple phases
and faces of wnt signaling during cardiac differentiation and
development. Circ Res. 107:186–199. 2010. View Article : Google Scholar : PubMed/NCBI
|