Introduction
Increasing data have revealed that atrial fibrosis
is the fundamental characteristic of the structural pathology
associated with atrial fibrillation (AF) (1–3).
Fibrosis is a consequence of an imbalance between the degradation
and deposition of cardiac extracellular matrix (ECM). Fibroblast
proliferation and differentiation into collagen-secreting
myofibroblasts contribute to atrial fibrosis via excess ECM
deposition, which promotes re-entry and enhances automaticity
(4,5). Although it is clear that fibrosis is
associated with AF, the precise role of fibrosis in the
pathogenesis of AF remains to be fully elucidated.
Following decades of investigations, increasing
evidence has revealed that inflammation is important the
pathogenesis of AF. Markers of pro-inflammatory cytokines,
including C-reactive protein, tumor necrosis factor-α (TNF-α),
interleukin-6 (IL-6) and macrophage migration inhibitory factor
(MIF), are increased in patients with AF (6–9).
This increases the possibility that inflammation may contribute to
atrial fibrosis and atrial structural remodeling, which contribute
to AF. Previous studies have demonstrated that inflammation is
associated with atrial fibrosis. The number of immune cells
resident in tissues and higher levels of serum inflammatory markers
are associated with extensive left atrium fibrosis and enlargement
in patients with AF (10–12).
In the authors' previous study, it was demonstrated
that MIF, an important pro-inflammatory cytokine, is involved in
the electrical remodeling of atrium myocytes through regulating the
L- and T-type calcium channel current (8,13).
However, the role of MIF in atrial fibrosis remains to be fully
elucidated. The present study aimed to investigate whether MIF
affects the proliferation and secretory functions of cardiac
fibroblasts (CFs).
Materials and methods
Patients
The present study was performed in accordance with
the principles outlined in the Declaration of Helsinki and was
approved by the research Ethics Committee of Guangdong General
Hospital, Guangdong Academy of Medical Sciences (no. GDREC2013047H;
Guangzhou, China). All patients provided written informed consent
(version no. 20130307). Patients with pneumonia or other infectious
diseases were excluded from the study. Left atrial appendages were
obtained from patients undergoing open-heart surgery (from March
2013 to March 2016) in Guangdong General Hospital in Guangzhou
(Guangdong, China). The specimens were collected prior to
initiation of cardiopulmonary bypass, immediately snap-frozen in
liquid nitrogen, and stored at −80°C until the day of the
experiments. Tissues from 11 patients with chronic AF (from 32–67
years) and a control group of 10 patients (from 30–61 years) with
sinus rhythm (SR) were also used in the present study. Patient data
are presented in Table I.
 | Table I.Baseline characteristics of
patients. |
Table I.
Baseline characteristics of
patients.
| Characteristic | SR | AF |
|---|
| Patients (n) | 10 | 11 |
| Men (n) | 4 | 4 |
| Age (years) | 42.8±11.1 | 46.9±14.4 |
| LAD (mm) | 48.2±8.4 | 50.1±4.4 |
| EF (%) | 65.0±7.3 | 62.1±6.3 |
| AVR (n) | 5 | 5 |
| MVR (n) | 5 | 6 |
| β-blocker (n) | 1 | 2 |
| Digitalis (n) | 1 | 6 |
Culture of primary CFs
The experiments using CFs were approved by the
research Ethics Committee of Guangdong General Hospital, Guangdong
Academy of Medical Sciences (no. GDREC2013047A). The CFs were
isolated from 1–3-day-old Wistar rats (n=24, 14:10 male:female)
which were provided by Sun Yat-sen University and fed at 20–25°C
(12-h light/dark cycle), as previously described (14). Briefly, the heart tissues were
finely minced and mechanically digested with 0.25% Trypsin + EDTA
(Gibco; Thermo Fisher Scientific, Inc., Waltham, MA, USA) using a
rotor in a flask. The cell suspensions were plated in a cell
culture flask for 90 min to separate the fibroblasts and
cardiomyocytes. The majority of CFs adhered to the flask. The cells
were cultured in Dulbecco's modified Eagle's medium (Gibco; Thermo
Fisher Scientific, Inc.) containing 4.5 g/l D-glucose, 10% fetal
bovine serum (Gibco; Thermo Fisher Scientific, Inc.), penicillin
(100 U/ml) and streptomycin (100 µg/ml) in a humidified atmosphere
(95% humidity, 37°C) composed of 95% O2, 5%
CO2. The CFs were used at passages 3–6.
Cell treatments
The cells were treated according to three
experimental designs. In the first, the cells (50–60% confluence)
were treated with different doses of mouse recombinant MIF at 37°C
(rMIF; 20 and 40 nM; R&D Systems Inc., Minneapolis, MN, USA)
for 24 or 48 h. In the second, the cells were pretreated with PP1
(5 µM; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for 1 h. The
MIF treatment groups were pretreated with an equal quantity of
dimethyl sulfoxide (DMSO) and were stimulated with 40 nM MIF for 48
h. In the third, the cells were treated with 40 nM MIF for 15, 30,
45 and 60 min, or with 20 and 40 nM MIF for 24 h.
Cell proliferation assay
The CFs were plated into a 96-well plate at a
density of 4,500/well and proliferation was measured using the cell
proliferation reagent (WST-1) kit (cat. no. 11644807001; Roche
Diagnostics GmbH, Mannheim, Germany) according to the
manufacturer's instructions. The signal in optical density was read
at 450 nm on a plate reader (Mutiskan GO; Thermo Fisher Scientific,
Inc.). The experiment was repeated three times.
Western blot analysis
The cells were lysed in 0.05 M Tris-HCl buffer (pH
8.0), containing 0.15 M sodium chloride, 0.02% sodium azide, 0.1%
sodium dodecyl sulfate (SDS), 1% Nonidet P-40 (NP-40), and a
protease inhibitor cocktail set (Calbiochem; EMD Millipore,
Billerica, MA, USA). The cell lysates were centrifuged at 12,000 ×
g for 15 min at 4°C and protein concentrations were determined by
bicinchoninic acid Protein Assay kit (Pierce; Thermo Fisher
Scientific, Inc.). The samples were diluted with 4X loading buffer
(Bio-Rad Laboratories, Inc., Hercules, CA, USA) and heated at 95°C
for 5 min. The proteins (30 µg) were fractionated on 8%
SDS-polyacrylamide gels for Src, phosphorylated (p-)Src, collagen
type 1, α1 (Col-1A1), collagen type 3, α1 (Col-3A1), matrix
metalloproteinase (MMP)-2/-9 and transforming growth factor (TGF)-β
or 10% SDS polyacrylamide gels for MIF. The proteins were then
transferred onto nitrocellulose membranes (GE Healthcare Life
Sciences, Chalfont, UK) according to standard protocols. The
membranes were blocked with dried skimmed milk powder in
TBS-Tween-20 (TBST) for 1 h at room temperature, prior to overnight
incubation at 4°C with primary rabbit polyclonal antibodies against
Src (1:1,000, cat. no. ab47405; Abcam, Cambridge, UK), p-Src
Tyr416 (1:1,000, cat. no. 2101; Cell Signaling
Technology, Inc., Danvers, MA, USA), Col-1A1 (1:1,000, cat. no.
ab34710), Col-3A1 (1:1,000, cat. no. ab7778) (both from Abcam),
MMP-9 (1:1,000, cat. no. AB19016; EMD Millipore, Billerica, MA,
USA) and TGF-β (1:1,000, cat. no. 3711; Cell Signaling Technology,
Inc.) and MMP-2 (1:2,000, cat. no. AB19015; EMD Millipore;). The
signals were normalized to the protein levels of GAPDH (1:1,000,
cat. no. 2118) or β-actin (1:3,000, cat. no. 3700) (both from Cell
Signaling Technology, Inc.). Following washing in TBST, the
membranes were incubated for 1 h with horseradish
peroxidase-conjugated anti-rabbit or anti-mouse IgG
(1:5,000-1:10,000; Abcam) in blocking solution. The protein bands
were visualized using electrochemiluminescence reagents (Pierce;
Thermo Fisher Scientific, Inc.), and images were evaluated
densitometrically using ImageJ 1.39 U software (National Institutes
of Health, Bethesda, MD, USA).
Drugs
The rMIF was dissolved in sterile PBS containing
0.1% bovine serum albumin (Sigma-Aldrich; Merck KGaA). PP1 was
purchased from Sigma-Aldrich; Merck KGaA. The kinase inhibitor was
dissolved in DMSO (Sigma-Aldrich; Merck KGaA). The concentration of
DMSO in the working solutions did not exceed 1.0%.
Statistical analysis
All data are expressed as the mean ± standard error
of the mean. Student's t-test was used as evaluate pairwise
statistical significances of differences between two group means,
and one-way analysis of variance was used for multiple groups with
SPSS 21 statistical software (IBM Corp, Armonk, NY, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
Expression of Col-1A1, Col-3A1,
MMP-2/-9, TGF-β and MIF in left atrial homogenates of patients with
SR and AF
The protein levels of Col-1A1, Col-3A1, MMP-2/-9,
TGF-β and MIF were detected in left atrial homogenates from
patients with AF and SR controls (for patient characteristics;
Table I). The protein expression
levels of Col-1A1, Col-3A1, MMP-2/-9, TGF-β and MIF were normalized
to those of GAPDH in each sample. With the exception of Col-1A1,
the protein levels of Col-3A1, MMP-2/9, TGF-β1 and MIF were
significantly higher in the left atrial tissues from patients with
AF (n=11), compared with the SR (n=10) group (Col-1A1, 0.81±0.12,
vs. 0.51±0.09, P=0.062; Col-3A1, 0.88±0.12, vs. 0.32±0.06, P=0.001;
MMP-2, 1.06±0.09, vs. 0.73±0.05, P=0.036; MMP-9, 1.06±0.15, vs.
0.57±0.09, P=0.013; TGF-β, 0.55±0.11, vs. 0.30±0.04, P=0.049; MIF,
1.20±0.15, vs. 0.76±0.14, P=0.044), as shown in Fig. 1A-G. These results indicated that,
as an important proinflammatory cytokines, MIF may be involved in
the regulation of the atrial fibrosis, which contributes to the
onset of AF.
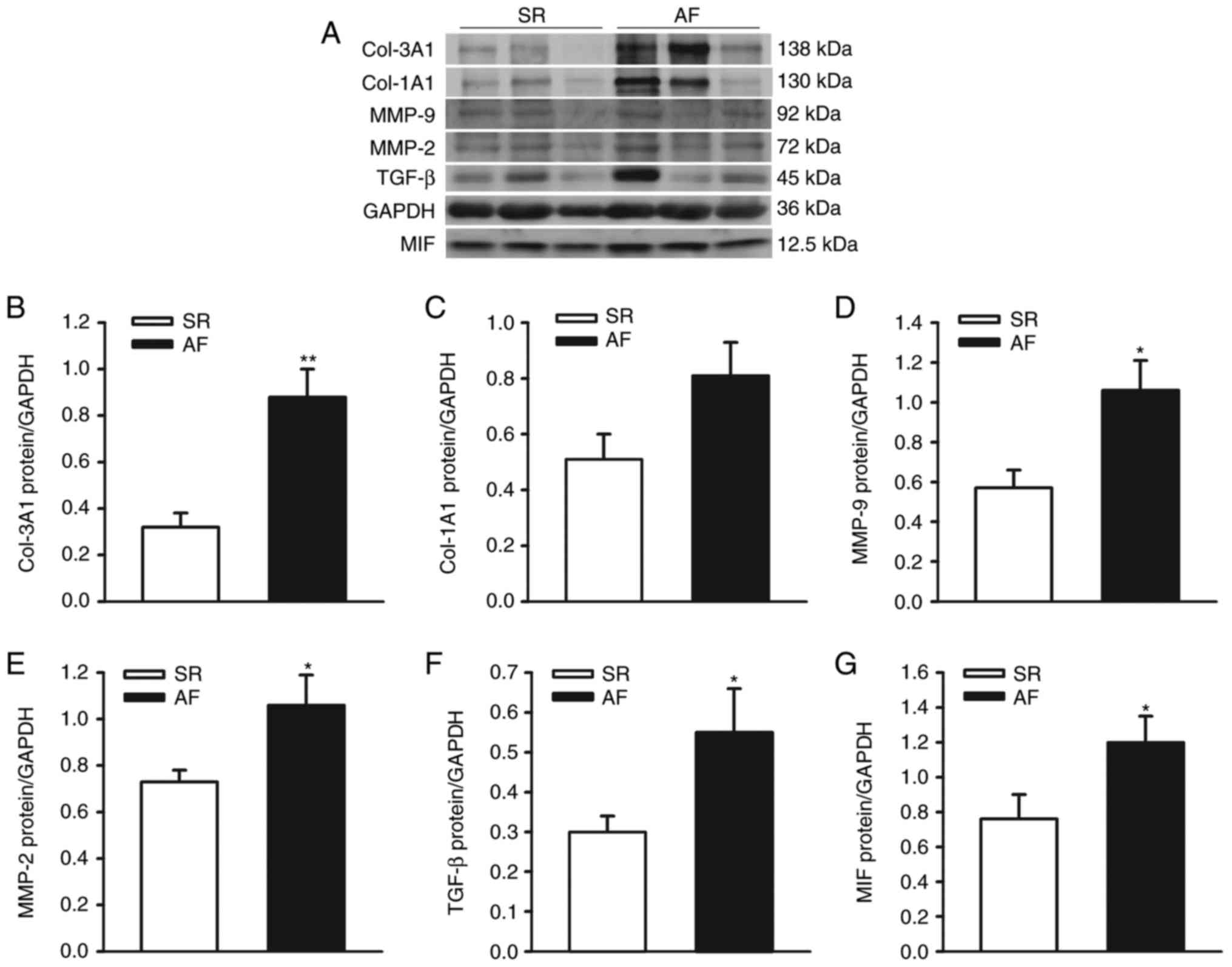 | Figure 1.Expression of ECM and MIF in left
atrial appendages from patients with chronic AF and SR. (A)
Representative immunoblots and protein levels of ECM (Col-1A1,
Col-3A1, MMP-2/-9 and TGF-β) and MIF in left atrial appendages from
patients with AF and SR. GAPDH was used as an internal control.
Densitometric analysis of protein levels of (B) Col-3A1, (C)
Col-1A1, (D) MMP-9, (E) MMP-2, (F) TGF-β and (G) MIF in patients
with AF and SR controls (n=10 in SR group, n=11 in AF group).
**P<0.01 and *P<0.05 vs. SR. AF, atrial fibrillation; SR,
sinus rhythm; ECM, extracellular matrix; Col-1A1, collagen type 1,
α1; Col-3A1, collagen type 3, α1; MMP-2/-9, matrix
metalloproteinase-2/-9; TGF-β, transforming growth factor-β; MIF,
migration inhibitory factor. |
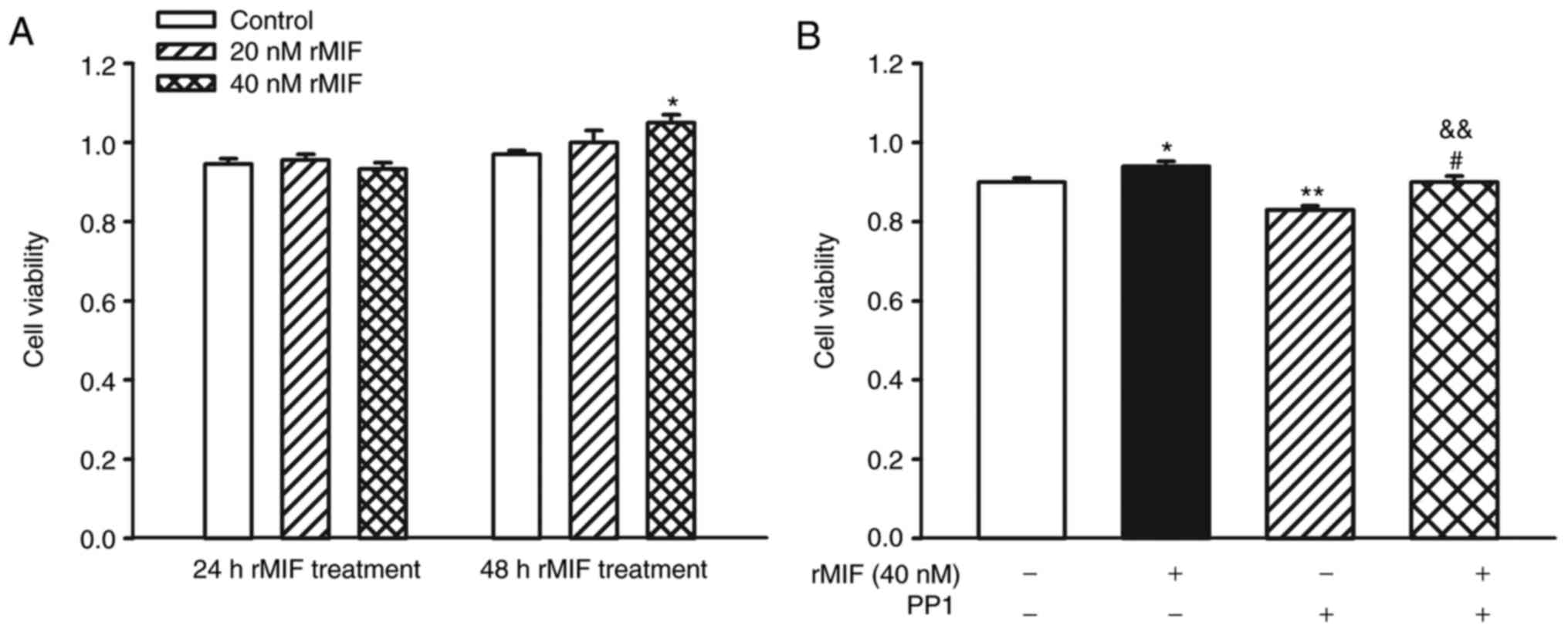
MIF promotes the proliferation of
CFs
The proliferation of CFs is important in myocardial
fibrosis. The present study investigated the proliferation of CFs
using CCK-8 assays, and it was found that 24-h MIF treatment had no
effect on the proliferation of CFs, whereas cells exhibited rapid
growth following stimulation for 48 h with 40 nM MIF (Fig. 2A).
Src kinase inhibitor alleviates the
promoting effect of MIF on CF proliferation
Src, a member of the protein tyrosine kinase (PTK)
family, has been suggested to be involved in cell proliferation.
The present study examined whether Src kinases were involved in
MIF-induced CF proliferation using a specific Src antagonist (PP1).
The CFs were pretreated with PP1 (5 µM) for 1 h prior to rMIF
stimulation. PP1 completely inhibited the MIF-induced CF
proliferation (Fig. 2B). These
results suggested that MIF induced cell proliferation through Src
kinase-dependent mechanisms in CFs.
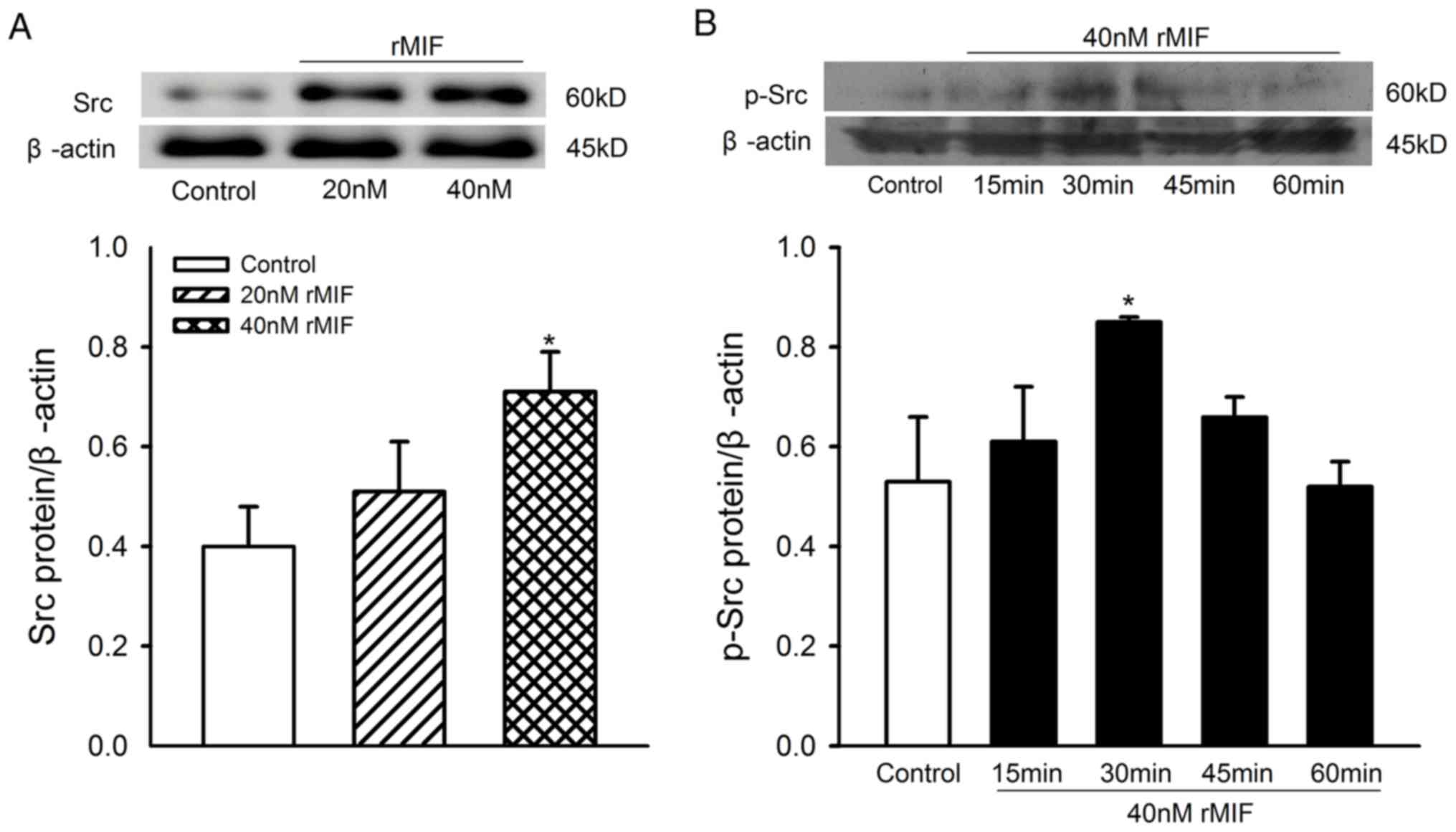
Effect of rMIF on the expression of
Src in CFs
The results reported above suggested that the Src
kinase signaling pathway may be responsible for the proliferation
of CFs induced by rMIF. To confirm the specificity of the signaling
pathways involved in the proliferation of CFs induced by rMIF,
western blot analysis was performed to analyze the levels of Src
and p-Src in CFs following treatment with MIF. The CFs were treated
with 20 or 40 nM of rMIF for 24 h, or 40 nM rMIF for 0, 15, 30, 45
and 60 min, and the levels of Src and p-Src were measured. The
higher concentration of rMIF (40 nM) was found to produce a marked
increase in the protein levels of Src, compared with those in the
unstimulated cells and the 20 nM rMIF-treated group of cells, and
Src was activated by 40 nM rMIF at 30 min (Fig. 3A and B).
MIF increases the levels of Col-1A1,
Col-3A1, MMP-2/-9 and TGF-β in CFs
To investigate the role of MIF on the secretion
function of CFs, the present study also examined the effects of MIF
on collagen deposition, and the expression of MMP and profibrotic
TGF-β in CFs. The MIF dose-response was assessed using
concentrations of 20 and 40 nM for 24 h, and it was found that the
expression levels of Col-1A1, Col-3A1, MMP-2/-9 and TGF-β increased
significantly in the 40 nM rMIF treatment group, but not in 20 nM
rMIF treatment group, with the exception of MMP-9 which did
increase significantly at 20 nM (Fig.
4A-F).
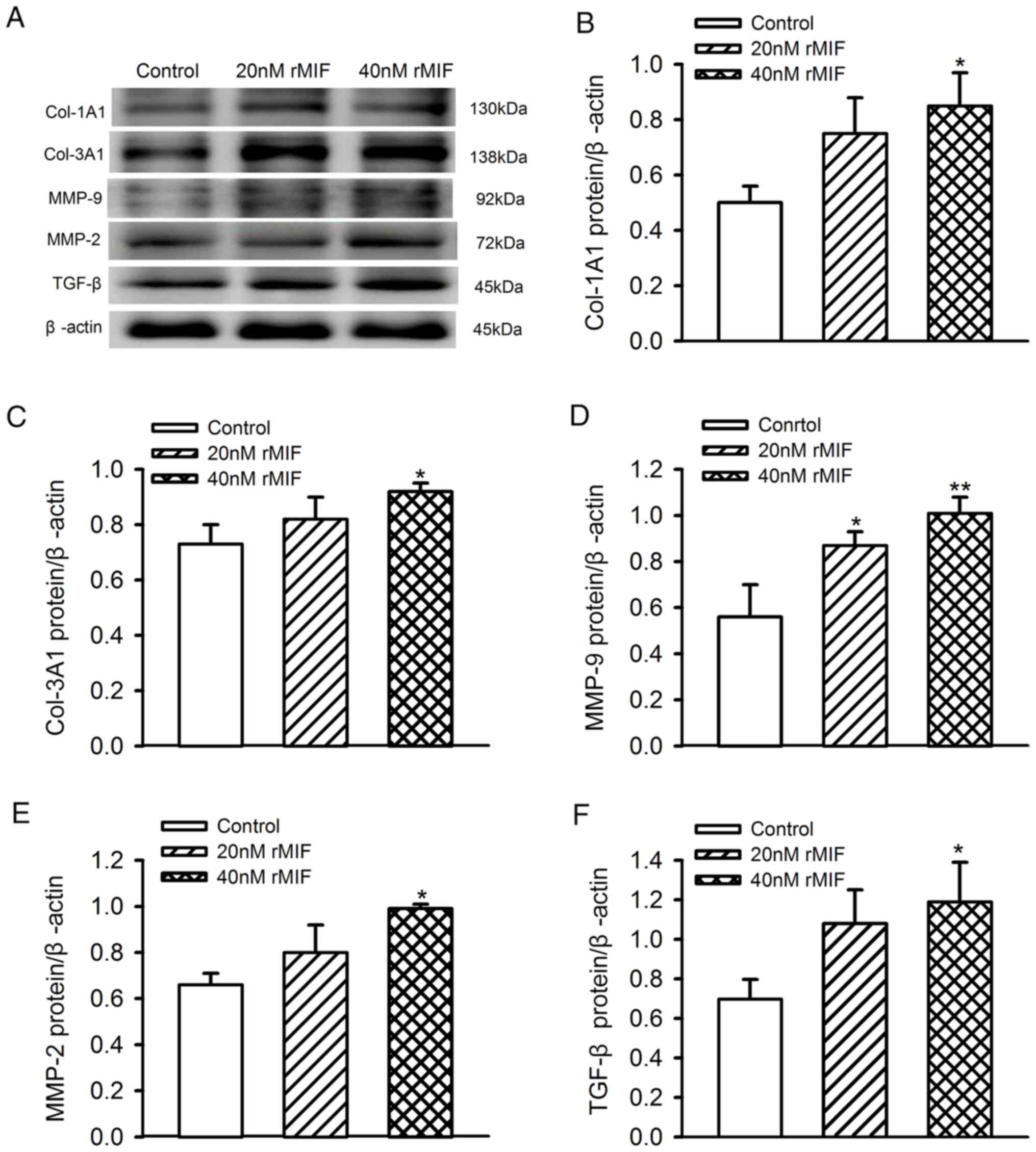 | Figure 4.Expression levels of Col-1A1, Col-3A1,
MMP-2/-9 and TGF-β in CFs following rMIF treatment (20 and 40 nM)
for 24 h. (A) Representative western blot analysis of ECM protein
in treated cells; β-actin was used as the internal control.
Densitometric analysis of protein expression of (B) Col-1A1, (C)
Col-3A1, (D) MMP-9, (E) MMP-2 and (F) TGF-β in treated cells and
controls. **P<0.01 and *P<0.05 vs. control. Each blot
represents one of four experiments. rMIF, recombinant migration
inhibitory factor; Col-1A1, collagen type 1, α1; Col-3A1, collagen
type 3, α1; MMP-2/-9, matrix metalloproteinase-2/-9; TGF-β,
transforming growth factor-β. |
Discussion
The results of the present study demonstrated the
following: i) Expression levels of collagen, MMP-2/-9 and TGF-β
were significantly increased in the left atrial tissues of patients
with AF; ii) long-term treatment of rMIF promoted the proliferation
of CFs; iii) PTK inhibitor inhibited the proliferation of CFs and
reversed the increase in cell proliferation induced by rMIF; and
iv) rMIF promoted the secretory function of CFs.
Atrial fibrosis is an important factor in initiating
and maintaining AF, which is characterized by marked alterations in
Col 1 and Col 3 synthesis/degradation, associated with disturbed
MMP/tissue inhibitors of MMP systems and increased levels of TGF-β.
Previous studies have revealed that the ECM changes are more severe
in patients with permanent AF, compared with those in patients with
SR (3,15). In the present study, it was also
found that, compared with the SR controls, the protein levels of
Col-1A1/3A1, MMP-2/-9 and TGF-β were significantly increased in the
left atrial tissues of patients with AF.
It has been established in previous years that
inflammation contributes to the atrial fibrosis and structural
remodeling of AF. In the left atrium of patients with AF, the
regions infiltrated by immune cells are positively correlated with
left atrial fibrosis and left atrial dimension (10). In vitro, the direct
cell-cell interaction between human monocytes and CFs can stimulate
TGF-β-mediated myofibroblast activity and increase the local ECM
remodeling (16). Certain
inflammatory markers are indicators for the degree of left atrial
fibrosis. As a novel marker of inflammation, high serum levels of
YKL-40 are associated with the presence and more extensive left
atrial fibrosis in patients with lone AF (12). In post-operative AF in rats with
sterile pericarditis, anti-interleukin-17A monoclonal antibody has
been shown to alleviate the inflammation and fibrosis, prolong
refraction and markedly suppress the development of AF (17).
MIF, an important pro-inflammatory cytokine, is
known to be involved in the inflammatory cardiovascular diseases.
MIF can cause progress and instability of atherosclerotic plaques,
increases the expression of intercellular adhesion molecule-1, and
markedly induces the expression of MMP-9 (18,19).
MIF is also involved in the pathological process of myocarditis
(20), and in cardiac dysfunction
following burn injury and sepsis (21). It has also been demonstrated that
the increased expression of MIF contributes to the development of
electrical remodeling of atrial myocytes through downregulating the
protein expression of L- and T-type calcium channels, and impairing
the channel function (8,13). However, the role of MIF in atrial
fibrosis, and the proliferation and function of CFs remains to be
fully elucidated. To investigate whether MIF is involved in the
pathogenesis of atrial fibrosis, the present study detected the
protein expression levels of MIF in left atrial homogenates from
patients with AF or SR controls. It was demonstrated that the
protein expression of MIF was significantly elevated in the atrial
tissues from patients with AF, which was positively correlated with
the increased expression of ECM. These data implicate MIF in the
upregulation of ECM in AF.
In addition to excess ECM deposition, fibrosis is
associated with fibroblast proliferation and differentiation into
collagen-secreting myofibroblasts. To confirm whether MIF regulates
the proliferation and function of CFs, and to address the possible
mechanisms, the effects of rMIF on the proliferation and function
of CFs were examined in the present study. These experiments
indicated that treatment with human rMIF (40 nM) for 48 h
significantly promoted the proliferation of CFs.
Src, one of the proto-oncogenes encoding a
membrane-associated, non-receptor PTK, has been implicated in the
regulation of a wide range of cellular functions, including cell
proliferation, survival and migration (22). The activation of Src can result in
the promotion of survival and proliferation pathways, and even
induce malignant tumors (v-src) (23). Src can be activated by several
factors, including high glucose and inflammatory factors TNF-α,
IFN-γ and MIF (8,24–26).
In the present study, it was found that the specific Src
antagonist, PP1, inhibited MIF-induced CF proliferation. In
addition, 40 nM rMIF was found to produce a marked increase in the
levels of Src and the activation of Src kinase, compared with that
in cells of the unstimulated group and 20 nM rMIF treatment group.
These results indicated that MIF promoted the proliferation of CFs
through the Src kinase signaling pathway.
The present study also investigated the effect of
MIF on the function of CFs, and found that rMIF treatment (40 nM)
for 24 h stimulated the production of fibrosis-related factors,
including Col-1A1/3A1, MMP-2/-9 and TGF-β. Therefore, it was
hypothesized that MIF, an important regulator of inflammation,
stimulates the expression of Src kinase, which in turn promotes the
proliferation of CFs and the production of factors associated with
fibrosis, which contributes to the atrial fibrosis observed in
AF.
There were a number of potential limitations in the
present study. Firstly, atrial fibroblasts behave differently to
ventricular fibroblasts in vitro and in vivo, with
atrial fibroblasts showing more marked fibrotic and oxidative
responses than ventricular fibroblasts (27,28).
In the present study, the cellular experiments were performed with
mouse CFs, which included atrial and ventricular fibroblasts,
whereas the use of atrial fibroblasts is likely to be superior in
investigations of AF. Secondly, with the increase of cell
generation, the proportion of myofibroblasts increases,
particularly for later passages (>7). Therefore, cells with a
similar proportion of myofibroblasts (passages 3–6) were preferable
for use in the present study.
Acknowledgements
This study was supported by grants from the National
Natural Science Foundation of China (grant nos. 81370295, 81470440
and 81670314), the Guangdong Natural Science Foundation (grant no.
S2013010016256) and the Medical Science Foundation of Guangdong
(grant no. A2013049).
References
|
1
|
Li D, Fareh S, Leung TK and Nattel S:
Promotion of atrial fibrillation by heart failure in dogs: Atrial
remodeling of a different sort. Circulation. 100:87–95. 1999.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Nattel S: How does fibrosis promote atrial
fibrillation persistence: In silico findings, clinical observations
and experimental data. Cardiovasc Res. 110:295–297. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Polyakova V, Miyagawa S, Szalay Z, Risteli
J and Kostin S: Atrial extracellular matrix remodelling in patients
with atrial fibrillation. J Cell Mol Med. 12:189–208. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Yue L, Xie J and Nattel S: Molecular
determinants of cardiac fibroblast electrical function and
therapeutic implications for atrial fibrillation. Cardiovasc Res.
89:744–753. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Miragoli M, Salvarani N and Rohr S:
Myofibroblasts induce ectopic activity in cardiac tissue. Circ Res.
101:755–758. 2007.PubMed/NCBI
|
|
6
|
Conway DS, Buggins P, Hughes E and Lip GY:
Prognostic significance of raised plasma levels of interleukin-6
and C-reactive protein in atrial fibrillation. Am Heart J.
148:462–466. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Guo Y, Lip GY and Apostolakis S:
Inflammation in atrial fibrillation. J Am Coll Cardiol.
60:2263–2270. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Rao F, Deng CY, Wu SL, Xiao DZ, Yu XY,
Kuang SJ, Lin QX and Shan ZX: Involvement of Src in L-type
Ca2+ channel depression induced by macrophage migration
inhibitory factor in atrial myocytes. J Mol Cell Cardiol.
47:586–594. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Deng H, Xue YM, Zhan XZ, Liao HT, Guo HM
and Wu SL: Role of tumor necrosis factor-alpha in the pathogenesis
of atrial fibrillation. Chin Med J (Engl). 124:1976–1982.
2011.PubMed/NCBI
|
|
10
|
Yamashita T, Sekiguchi A, Suzuki S,
Ohtsuka T, Sagara K, Tanabe H, Kunihara T, Sawada H and Aizawa T:
Enlargement of the left atrium is associated with increased
infiltration of immune cells in patients with atrial fibrillation
who had undergone surgery. J Arrhythm. 31:78–82. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sonmez O, Ertem FU, Vatankulu MA, Erdogan
E, Tasal A, Kucukbuzcu S and Goktekin O: Novel fibro-inflammation
markers in assessing left atrial remodeling in non-valvular atrial
fibrillation. Med Sci Monit. 20:463–470. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Canpolat U, Aytemir K, Hazirolan T, Özer N
and Oto A: Serum YKL-40 as a marker of left atrial fibrosis
assessed by delayed enhancement MRI in lone atrial fibrillation.
Pacing Clin Electrophysiol. 38:1386–1395. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rao F, Deng CY, Wu SL, Xiao DZ, Huang W,
Deng H, Kuang SJ, Lin QX, Shan ZX, Liu XY, et al: Mechanism of
macrophage migration inhibitory factor-induced decrease of T-type
Ca(2+) channel current in atrium-derived cells. Exp Physiol.
98:172–182. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang WK, Wang B, Lu QH, Zhang W, Qin WD,
Liu XJ, Liu XQ, An FS, Zhang Y and Zhang MX: Inhibition of
high-mobility group box 1 improves myocardial fibrosis and
dysfunction in diabetic cardiomyopathy. Int J Cardiol. 172:202–212.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Barth AS, Merk S, Arnoldi E, Zwermann L,
Kloos P, Gebauer M, Steinmeyer K, Bleich M, Kääb S, Hinterseer M,
et al: Reprogramming of the human atrial transcriptome in permanent
atrial fibrillation: Expression of a ventricular-like genomic
signature. Circ Res. 96:1022–1029. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Mewhort HE, Lipon BD, Svystonyuk DA, Teng
G, Guzzardi DG, Silva C, Yong VW and Fedak PW: Monocytes increase
human cardiac myofibroblast-mediated extracellular matrix
remodeling through TGF-β1. Am J Physiol Heart Circ Physiol.
310:H716–H724. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Fu XX, Zhao N, Dong Q, Du LL, Chen XJ, Wu
QF, Cheng X, Du YM and Liao YH: Interleukin-17A contributes to the
development of post-operative atrial fibrillation by regulating
inflammation and fibrosis in rats with sterile pericarditis. Int J
Mol Med. 36:83–92. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kong YZ, Yu X, Tang JJ, Ouyang X, Huang
XR, Fingerle-Rowson G, Bacher M, Scher LA, Bucala R and Lan HY:
Macrophage migration inhibitory factor induces MMP-9 expression:
Implications for destabilization of human atherosclerotic plaques.
Atherosclerosis. 178:207–215. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Schmeisser A, Marquetant R, Illmer T,
Graffy C, Garlichs CD, Bockler D, Menschikowski D, Braun-Dullaeus
R, Daniel WG and Strasser RH: The expression of macrophage
migration inhibitory factor 1alpha (MIF 1alpha) in human
atherosclerotic plaques is induced by different proatherogenic
stimuli and associated with plaque instability. Atherosclerosis.
178:83–94. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Matsui Y, Okamoto H, Jia N, Akino M, Uede
T, Kitabatake A and Nishihira J: Blockade of macrophage migration
inhibitory factor ameliorates experimental autoimmune myocarditis.
J Mol Cell Cardiol. 37:557–566. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Willis MS, Carlson DL, Dimaio JM, White
MD, White DJ, Adams GA IV, Horton JW and Giroir BP: Macrophage
migration inhibitory factor mediates late cardiac dysfunction after
burn injury. Am J Physiol Heart Circ Physiol. 288:H795–H804. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Cary LA, Klinghoffer RA, Sachsenmaier C
and Cooper JA: SRC catalytic but not scaffolding function is needed
for integrin-regulated tyrosine phosphorylation, cell migration and
cell spreading. Mol Cell Biol. 22:2427–2440. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Chen Q, Zhou Z, Shan L, Zeng H, Hua Y and
Cai Z: The importance of Src signaling in sarcoma. Oncol Lett.
10:17–22. 2015.PubMed/NCBI
|
|
24
|
Cai T, Kuang Y, Zhang C, Zhang Z, Chen L,
Li B, Li Y, Wang Y, Yang H, Han Q and Zhu Y: Glucose-6-phosphate
dehydrogenase and NADPH oxidase 4 control STAT3 activity in
melanoma cells through a pathway involving reactive oxygen species,
c-SRC and SHP2. Am J Cancer Res. 5:1610–1620. 2015.PubMed/NCBI
|
|
25
|
Lin CC, Pan CS, Wang CY, Liu SW, Hsiao LD
and Yang CM: Tumor necrosis factor-alpha induces VCAM-1-mediated
inflammation via c-Src-dependent transactivation of EGF receptors
in human cardiac fibroblasts. J Biomed Sci. 22:532015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Boekhoudt GH, McGrath AG, Swisher JF and
Feldman GM: Immune complexes suppress IFN-γ-induced responses in
monocytes by activating discrete members of the SRC kinase family.
J Immunol. 194:983–989. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Burstein B, Libby E, Calderone A and
Nattel S: Differential behaviors of atrial versus ventricular
fibroblasts: A potential role for platelet-derived growth factor in
atrial-ventricular remodeling differences. Circulation.
117:1630–1641. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yeh YH, Kuo CT, Chang GJ, Qi XY, Nattel S
and Chen WJ: Nicotinamide adenine dinucleotide phosphate oxidase 4
mediates the differential responsiveness of atrial versus
ventricular fibroblasts to transforming growth factor-β. Circ
Arrhythm Electrophysiol. 6:790–798. 2013. View Article : Google Scholar : PubMed/NCBI
|