Introduction
Colon cancer is one of the most common digestive
tract tumors, and is the second most common cancer in women and
third most common cancer in men, worldwide (1). Rapid progression of colon cancer, and
the way in which it metastasizes to local tissues and distant
organs has been systematically reviewed and analyzed in a number of
reports (2,3). In addition, previous reports have
proposed available treatment modalities for colon cancer and have
evaluated the accuracy of mini-probe endoscopic ultrasound in
assigning clinical stages to colon cancer (4–6).
However, appropriate management of diagnosis, treatments and
prognosis of colorectal cancer has not been improved and is not
clearly understood (5,6). Metastasis and invasion of colon
cancer cells causes problems for clinicians attempting to treat the
disease and worsens the disease in patients (7,8).
Inhibition of colon cancer cell growth and metastasis prolongs
survival of patients with colon cancer (9). Therefore, the underlying potential
mechanisms of colon cancer cell growth, metastasis and invasion
have been explored in target therapy for human cancer (10,11).
Tunicamycin is a nucleoside antibiotic (12,13).
It is regarded as an inhibitor of glycosylation that disrupts the
protein folding machinery in eukaryotic cells, which causes
accumulation of unfolded proteins in the endoplasmic reticulum
(14). Tunicamycin has been
reported to exhibit anti-cancer potential in the published
literature. A report demonstrated that tunicamycin inhibits growth
and promotes apoptosis of human prostate cancer cells via glucose
regulated protein 78-mediated apoptosis (15). Shiraishi et al (16) suggested that tunicamycin may
enhance tumor necrosis factor-related apoptosis-inducing ligand
(TRAIL)-induced apoptosis in human prostate cancer cells. Ling
et al (17) revealed that
tunicamycin may activate endoplasmic reticulum stress and inhibit
epidermal growth factor receptor N-glycosylation in human non-small
cell lung cancer cells. Additionally, research suggested that
inhibition of N-linked glycosylation by tunicamycin may suppress
E-cadherin-mediated proliferation, migration and invasion in human
colon cancer cells (18).
In the present study, the inhibitory effects of
tunicamycin in colon cancer cells, and the potential underlying
mechanism, was investigated. As controlling tumor cell growth and
metastasis is imperative for the survival of patients with colon
cancer, molecular bioinformatics has enabled scientists to screen
for targeted molecules that offer individual tailored therapies for
patients with cancer or other human diseases (19,20).
Therefore, the present study analyzed the anti-cancer efficacy of
tunicamycin in colon cancer cells in vitro, and in xenograft
mice. Results suggested that tunicamycin significantly inhibits
growth and aggressiveness of colon cancer cells via downregulation
of the extracellular signal-regulated kinase (ERK)/c-JUN N-terminal
kinase (JNK)-mediated AKT/mammalian target of rapamycin (mTOR)
signaling pathway. This research may provide molecular targets for
colon cancer treatment.
Materials and methods
Ethics statement
The present study was performed in strict accordance
with the recommendations in the Guide for the Care and Use of
Laboratory Animals (21). All
experimental protocols and animal procedures were approved by the
Committee on the Ethics of Animal Experiments Defense Research of
Jingmen No. 2 People's Hospital (Jingmen, China) Surgery and
euthanasia were performed to minimize suffering.
Cells and reagents
Colon tumor cell lines SW620 and SW480 were
purchased from American Type Culture Collection (Manassas, VA,
USA). All tumor cells were cultured in Eagle's Minimum Essential
medium (MEM; Sigma-Aldrich; Merck KGaA, Darmstadt, Germany)
supplemented with 10% fetal calf serum (Invitrogen; Thermo Fisher
Scientific, Inc., Waltham, MA, USA). All cells were cultured at
37°C and 5% CO2.
MTT assay
SW620 cells (1×105 cells/well) were
incubated at room temperature with tunicamycin (0–2.5 mg/ml) in
96-well plates for 24, 48, 72 and 96 h in triplicate for each
condition or with PBS as a control. After the indicated time
incubation, 20 µl MTT (5 mg/ml) in PBS was added to each well for 4
h. Most of the medium was removed and 100 µl dimethyl sulfoxide was
added into the wells to solubilize the crystals. The optical
density was measured using a plate reader at a wavelength of 450
nm.
Tumor cell colony formation assay
SW620 cells (1×104) were plated in 6-well
plates. Tunicamycin (2 mg/ml) was added to plates, or PBS was added
as a control, and then cultured at 37°C. After 72 h culture, SW620
cells colonies were stained with crystal violet (0.005%) for 30 min
and images were captured with a CX31 light microscope (Olympus
Corporation, Tokyo, Japan). Clonal numbers of SW620 cells were
analyzed by Alpha Innotech Corporation imaging software (version
3.30; San Leandro, CA, USA).
Cell invasion and migration
assays
SW620 cells (1×105 cells/well) were
incubated with tunicamycin (2 mg/ml) for 48 h. For invasion assay,
SW620 cells were suspended at a density of 5×105 in 500
µl serum-free MEM. SW620 cells were transferred to the tops of BD
BioCoat Matrigel invasion chambers (BD Biosciences, Franklin Lakes,
NJ, USA) according to the manufacturer's protocol. For migration
assay, cells were subjected to a control insert (BD Biosciences)
instead of a Matrigel invasion chamber, 6 h at 37°C. Chambers were
then incubated for 24 h at room temperature. Subsequently, cells
were fixed with ice-cold methanol for 5 min then washed twice with
PBS, the membrane was stained with crystal violet (1%) for 5 min at
room temperature. Top chambers were cleaned with a cotton swab and
the lower chamber was photographed under a light microscope (CX31
Olympus Corporation; magnification, ×200). Images of cells adhered
to the lower surface of the chamber were captured (five random
fields). Each experiment was performed in triplicate.
Endogenous overexpression of ERK
SW620 cells were cultured until 85% confluent and
the culture media was then removed. Human ERK cDNA plasmids
(Invitrogen; Thermo Fisher Scientific, Inc.) were transfected into
293T cells for 48 h to generate a lentivirus using
Lipofectamine® 2000 (Sigma-Aldrich; Merck KGaA),
according to the manufacturer's protocol. The viral supernatant was
subsequently collected and used to infect the SW620 cells. Further
analysis was performed 72 h post-transfection. Control groups
included the mock treatment group and vector-transfected group.
Reverse transcription-quantitative polymerase chain reaction
(RT-qPCR) and western blotting were performed in order to determine
the efficiency of transfection (21). Total RNA was extracted from SW620
cells (1×105 cells/well) with TRIzol reagent (Abcam,
Cambridge, UK) according to the manufacturer's protocol. Total RNA
(5 µg) was reverse transcribed into cDNA using an oligo-(dT) primer
and M-MLV reverse transcriptase (Invitrogen; Thermo Fisher
Scientific, Inc.) for qPCR analysis. qPCR was performed in a final
volume of 10 µl, which consisted of 5 µl SsoFast™ EvaGreen Supermix
(Bio-Rad Laboratories, Inc., Hercules, CA, USA), 1 µl cDNA (1:50
dilution) and 2 µl each of the forward and reverse primers (1 mM).
The specific primers (Invitrogen; Thermo Fisher Scientific, Inc.)
were as follows: ERK sense, 5′-CAAAGGTGGATCAGATTCAAG-3′ and
antisense, 5′-GGTGAGCATTATCACCCAGAA-3′; GAPDH sense,
5′-CAAAGGTGGATCAGATTCAAG-3′ and antisense,
5′-GGTGAGCATTATCACCCAGAA-3′. The thermal cycling procedure was as
follows: 95°C for 4 min, followed by 40 cycles of 95°C for 25 sec,
55°C for 30 sec and 72°C for 20 sec with 2 sec for plate reading,
and melting curve analysis from 65 to 95°C. GAPDH was used as the
control for normalizing gene expression. The relative
quantification of the gene of interest was determined using the
comparative ΔCq method (22).
Xenograft tumor model
A total of 80 specific pathogen-free male Balb/c
(6–8 weeks old) nude mice were purchased from Slack Life Co., Ltd.
(Shanghai, China). All mice were housed at preference temperature
(22–24°C) under a 12-h light-dark cycle and fed ad libitum at
40-70% humidity. SW620 cells (1×106) were mixed with 100
µl PBS and injected subcutaneously in the flanks of Balb/c mice (n
= 60). Xenograft mice were divided into two groups of 40, the
treatment group received treatment with tunicamycin (10 mg/kg) by
one intravenous injection on day 6 after tumor implantation
(diameter, 5–8 mm) and the control group received 100 µl PBS by
intravenous injection. The tumor volumes were calculated according
to a previous study (23). The
tumor volumes were calculated according to the following formula:
length × width2 × 0.52. On day 25, mice (n=20 in each
group) were sacrificed under 1.5% pentobarbital sodium (1 ml/kg;
Lianshuoinc, Shanghai, China) for further analysis. Residual mice
(n=20 in each group) were continued to be housed in order to
analyze the long-term survival rate (considered to be 120 days). In
the survival rate experiments, the largest tumor size was ~2,000
mm3. Mice were sacrificed with 1.5% pentobarbital sodium
(100 mg/kg) by intravenous injection when tumor diameter reached 12
mm. On day 25, mice (n=10 in each group) were sacrificed to make
further analysis. The remaining mice (n=10 in each group) were
continued to house to analyze the long-term survival rate (120
days). The remaining mice were sacrificed with 1.5% pentobarbital
sodium (100 mg/kg) by intravenous injection at the end of the
experiment.
Western blotting
Protein samples from colorectal tumors and SW620
cells were homogenized using radioimmunoprecipitation assay lysis
buffer (Invitrogen; Thermo Fisher Scientific, Inc.) and were
centrifuged at 7,103 × g at 4°C for 10 min. The protein
concentrations of the cell extracts were then measured using
Bradford protein dye reagent (Bio-Rad Laboratories, Inc.). A total
of 30 µg/lane protein was loaded and separated by SDS-PAGE (12%
gel) and transferred to nitrocellulose membranes. The membranes
were blocked with 5% skimmed milk for 1 h at room temperature,
washed in Tris-buffered saline containing 0.1% Tween-20 (TBST) and
incubated with the following primary antibodies: Anti-FIB (1:2,000;
ab4566; Abcam), anti-VIM (1:1,000; ab8978, Abcam), anti-Eca
(1:1,000; ab11512; Abcam), anti-Bad (1:2,000; ab32445; Abcam),
anti-Caspase9 (1:1,000; ab32539; Abcam), anti-Apaf-1 (1:2,000;
ab2001; Abcam), anti-Cyto c (1:2,000; ab13575; Abcam), anti-Cyclin
D1 (1:2,000; ab134175; Abcam), anti-CDK1 (1:2,000; ab133327;
Abcam), anti-CDK2 (1:2,000; ab32147; Abcam), anti-Ki67 (1:1,000;
ab16667; Abcam), anti-PCNA (1:1,000; ab18197; Abcam), anti-t-ERK
(1:2,000; ab196883; Abcam), anti-p-ERK (1:2,000; ab214362; Abcam),
anti-t-JNK (1:1,000; ab179461; Abcam), anti-p-JNK (1:1,000;
ab76572; Abcam), anti-β-catenin (1:1,000; ab16051; Abcam),
anti-Dkk1 (1:1,000; ab109416; Abcam), anti-t-AKT (1:1,000; ab8978;
Abcam), anti-p-AKT (1:1,000; ab8933; Abcam), anti-mTOR (1:1,000;
ab87540; Abcam), and anti-β-actin (1:5,000; ab8226; Abcam)
overnight at 4°C. The labeled membranes were then washed three
times with TBST, incubated for 2 h at room temperature with
secondary anti-mouse primary IgG (1:1,500; ab6785; Abcam) and
anti-rabbit primary IgG (1:1,500; ab6721; Abcam) conjugated with
horseradish peroxidase. The protein bands labeled with the
antibodies were visualized using the SuperSignal West Pico
Chemiluminescent Substrate Trial Kit (Pierce; Thermo Fisher
Scientific, Inc.). Images were obtained using the ChemiDoc XRS
system with Quantity One software (Bio-Rad Laboratories, Inc.).
Protein expression was analyzed using BandScan software (version
5.0; Glyko, Inc., Novato, CA, USA). All experiments were repeated
≥3 times.
Immunohistochemistry
Colon tumors from xenograft mice were fixed using
formaldehyde (10%) at room temperature for 24 h and were then
embedded in paraffin. The tissues were cut into 4-µm sections and
mounted on glass slides. The sections were rinsed with PBS and
placed in a solution containing primary antibodies directed against
Ki67 (1:1,000; ab16667; Abcam), PCNA (1:1,000; ab18197; Abcam),
Caspase3 (1:2,000; ab90437; Abcam), Caspase9 (1:1,000; ab32539;
Abcam), Bcl-2 (1:1,000; ab692; Abcam), ERK (1:2,000; ab196883;
Abcam), JNK (1:1,000; ab179461; Abcam), mTOR (1:1,000; ab87540;
Abcam) and AKT (1:1,000; ab8978; Abcam) incubated overnight at 4°C.
Subsequently, sections were incubated with the appropriate
secondary antibodies, mouse primary IgG (1:1,500; ab6785; Abcam)
and rabbit primary IgG (1:1,500; ab6721; Abcam), for 2 h at room
temperature were added to specimens prior to visualization. The
sections were then washed with PBS and observed by fluorescent
video microscopy (BZ-9000; Keyence Corporation, Osaka, Japan) and a
Ventana BenchMark automated staining system (Roche Applied Science,
Penzberg, Germany) was used for observation of protein expression
levels. A negative control was employed wherein mice were injected
with 100 µl PBS.
Apoptosis assay
SW620 cells were cultured at 37°C and 5%
CO2 until 90% confluent. Apoptosis was assessed by
incubating the cells with tunicamycin (2.0 mg/ml) for 48 h. After
incubation, SW620 cells were trypsinized and collected. The cells
were then washed in cold PBS, resuspended to 1×106
cells/ml in PBS, labeled with Annexin V-fluorescein isothiocyanate
(FITC) and propidium iodide using an Annexin V-FITC kit from BD
Biosciences, and analyzed on a FACScan flow cytometer (BD
Biosciences) using Cell Quest acquisition software (version 2.9; BD
Biosciences). The treatments were performed in triplicate, and the
percentage of labeled cells undergoing apoptosis in each group was
determined and calculated using Cell Quest acquisition
software.
Cell cycle analysis
Prior to testing, SW620 cells were treated with
tunicamycin and PBS for 48 h, as aforementioned. Following this,
1×105 cells were collected and fixed with ice-cold
ethanol (70%; 10 min on ice) prior to washing, re-suspension in
cold PBS and incubation at 37°C for 30 min with 10 mg/ml RNase and
1 mg/ml propidium iodide (Sigma-Aldrich; Merck KGaA). DNA content
was analyzed by a FACScan Flow cytometer (BD Biosciences, San Jose,
CA, USA). The percentage of cells in each phase of the cell cycle
was determined using Cell Quest acquisition software (version 2.9;
BD Biosciences).
Statistical analysis
Data are expressed as the mean ± standard deviation
of triplicate experiments and analyzed by using a Student t test or
one-way analysis of variance followed by the Tukey's honest
significant difference post hoc test. All data were analyzed using
SPSS software version 19.0 (IBM Corp., Armonk, NY, USA), GraphPad
Prism version 5.0 software (GraphPad Software, Inc., La Jolla, CA,
USA) and Microsoft Excel (Microsoft Corporation, Redmond, WA, USA).
P<0.05 was considered to indicate a statistically significant
difference.
Results
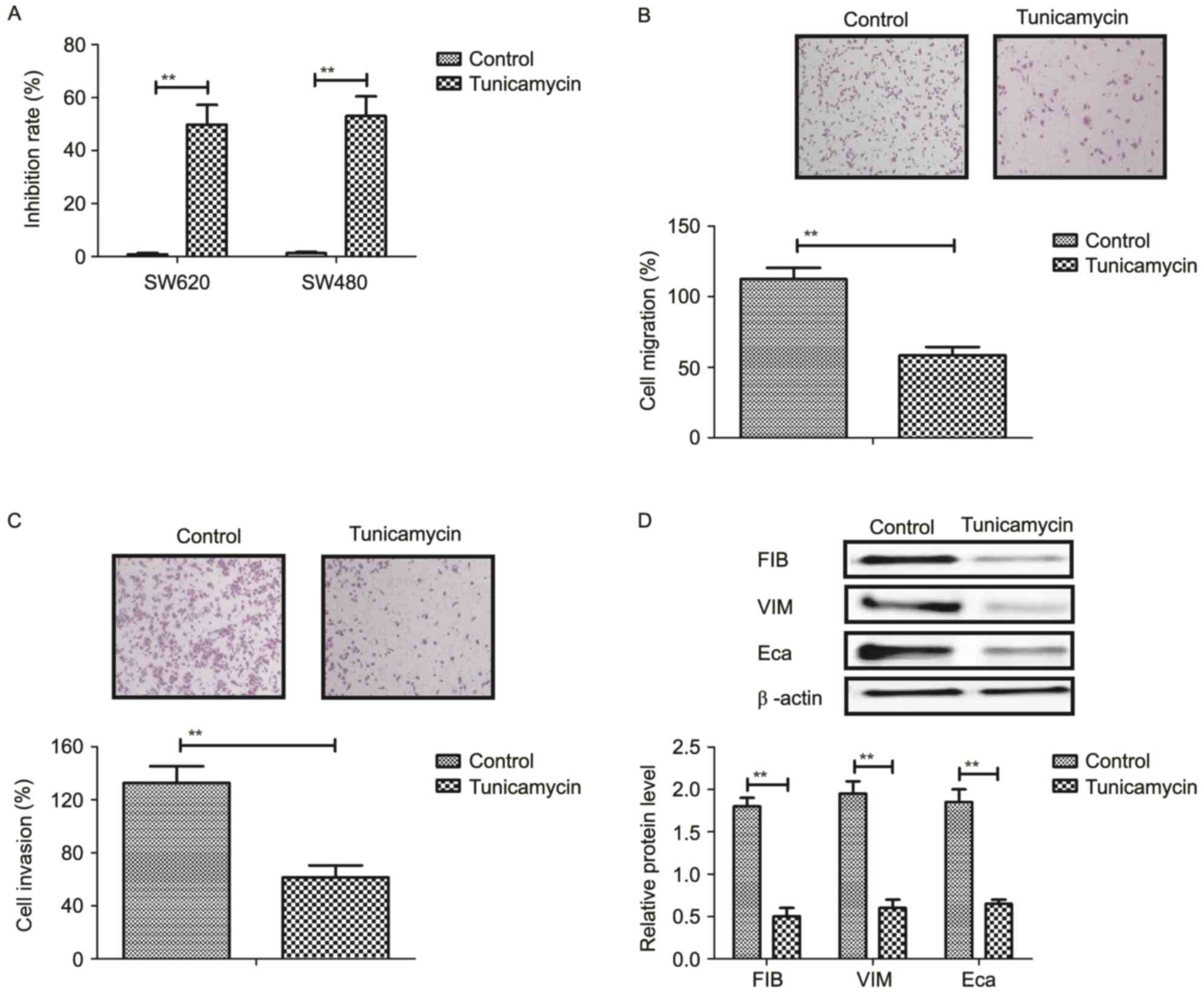
Tunicamycin treatment inhibits growth
and aggressiveness of colon cancer cells
The inhibitory effects of tunicamycin on growth and
aggressiveness of colon cancer cells were investigated. As
demonstrated in Fig. 1A,
tunicamycin significantly inhibited SW620 and SW480 cell growth
(P<0.01). Migration and invasion assays demonstrated that
tunicamycin treatment suppressed aggressiveness of SW620 cells
(P<0.01; Fig. 1B and C).
Western blotting demonstrated that fibronectin, vimentin and
E-cadherin expression levels were decreased by tunicamycin
treatment (P<0.01; Fig. 1D).
These results suggested that tunicamycin treatment may inhibit
growth and aggressiveness of colon cancer cells.
Tunicamycin treatment promotes
apoptosis of colon cancer cells via the mitochondrial apoptotic
signaling pathway
Results revealed that tunicamycin treatment promoted
apoptosis of colon cancer cells (Fig.
2A). Western blotting demonstrated that expression levels of
apoptotic peptidase activating factor-1 and cytochrome c were
increased by tunicamycin treatment compared with control cells in
SW620 cells (Fig. 2B). Bcl-2
associated agonist of cell death and caspase-9 expression levels
were increased in SW620 cells treated with tunicamycin compared
with control cells, as determined by western blotting (Fig. 2C). Immunofluorescence revealed that
tumor protein p53 (P53) and B-cell lymphoma-2 (Bcl-2) expression
levels were downregulated by tunicamycin treatment in SW620 cells,
compared with the control (Fig.
2D). These results suggested that tunicamycin treatment may
promote apoptosis of colon cancer cells via the mitochondrial
apoptosis signaling pathway.
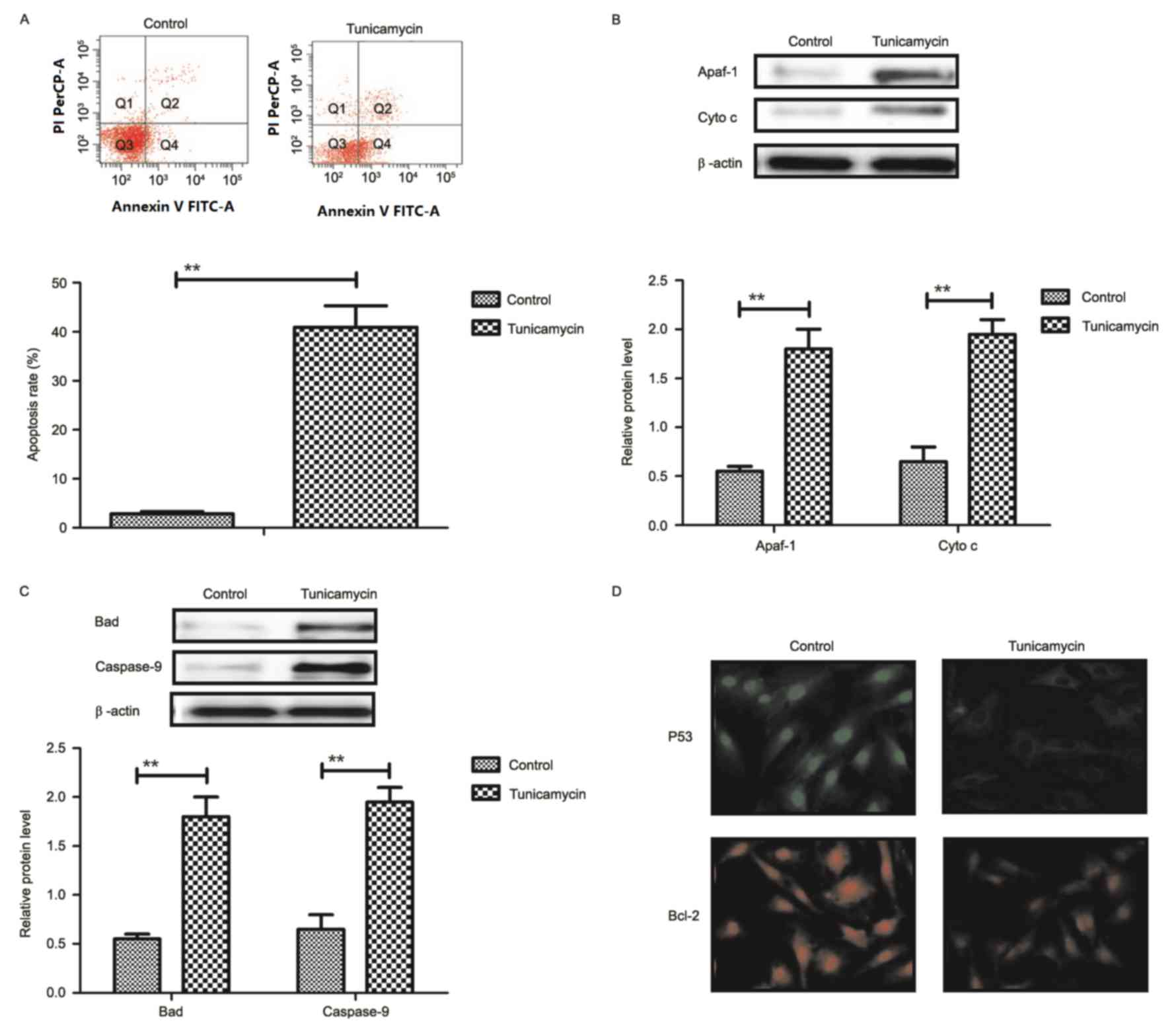 | Figure 2.Tunicamycin treatment induces
apoptosis of colon cancer cells through the mitochondrial apoptotic
signaling pathway. (A) Tunicamycin treatment promoted apoptosis of
SW620 cells. Cell apoptosis was detected by flow cytometry. The
cells were assessed by flow cytometry following Annexin V staining.
Q4-2, early apoptotic cells positively stained for Annexin V-FITC,
and negative for PI; Q3-2, normal cells not stained by Annexin
V-FITC or PI; Q2-2, necrotic cells and late apoptotic cells stained
by both Annexin V-FITC and PI; (B) Effects of tunicamycin treatment
on expression levels of Apaf-1 and cytochrome c in SW620 cells. (C)
Tunicamycin treatment increased Bad and caspase-9 expression levels
in SW620 cells. (D) Tunicamycin treatment promoted P53 and Bcl-2
expression levels in SW620 cells determined by immunofluorescence.
Data are expressed as mean ± standard deviation of triplicate
experiments. **P<0.01. Cyto c, cytochrome c; Apaf-1, apoptotic
peptidase activating factor-1; Bad, Bcl-2 associated agonist of
cell death; P53, tumor protein p53; Bcl-2, B-cell lymphoma-2; PI,
propidium iodide; FITC, fluorescein isothiocyanate. |
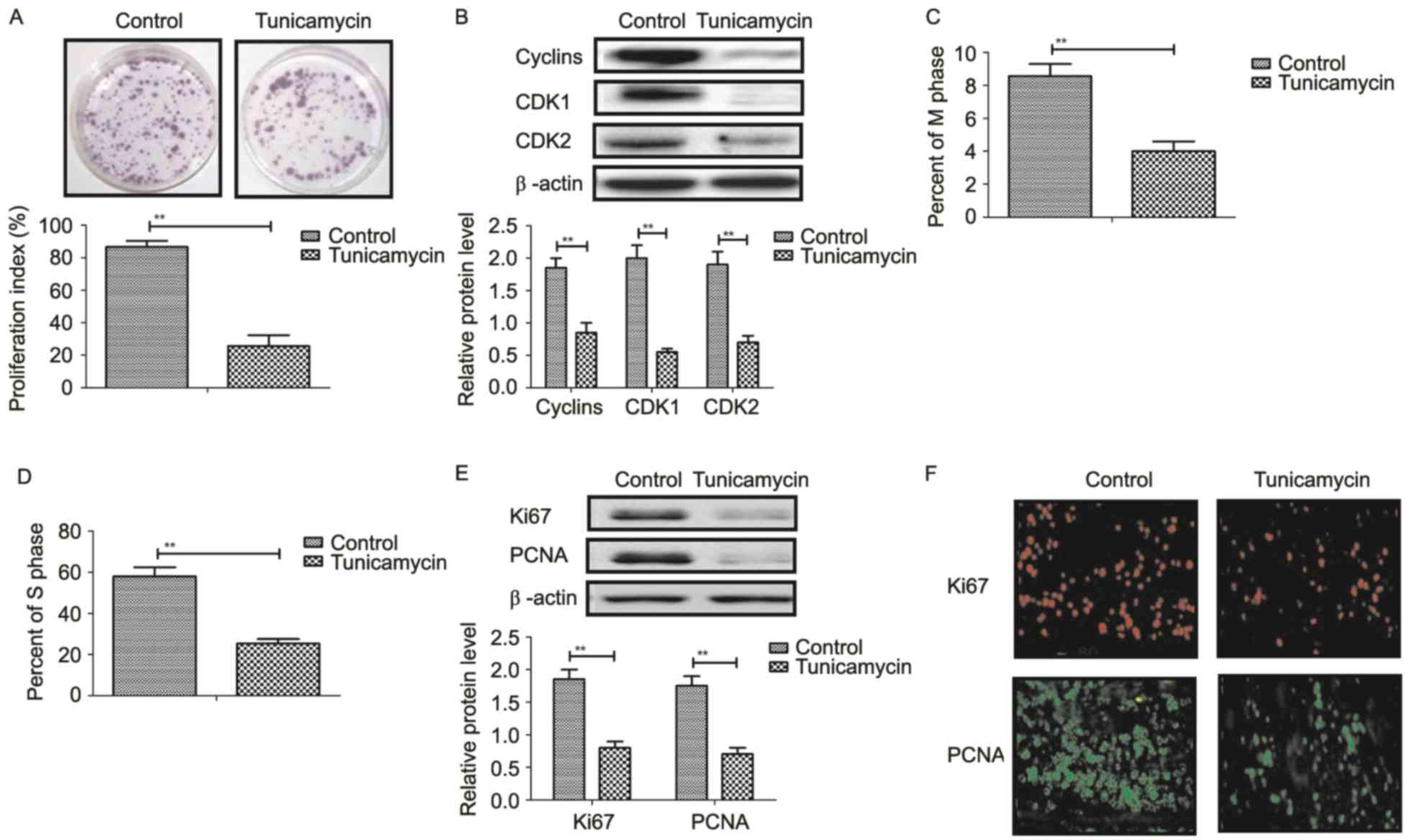
Tunicamycin treatment suppresses
proliferation and arrests the cell cycle of colon cancer cells
Tunicamycin treatment suppressed proliferation of
SW620 cells compared to control cells (Fig. 3A). Western blot revealed that
tunicamycin treatment significantly inhibited the expression levels
of cyclin D1, cyclin dependent kinase (CDK)1 and CDK2 in SW620
cells compared with the control (Fig.
3B). Cells cycle showed that tunicamycin treatment suppressed
entry to M phase and S phase in SW620 cells (Fig. 3C and D). Western blotting and
immunofluorescence revealed that tunicamycin administration
decreased the expression levels of Ki67 and proliferating cell
nuclear antigen (PCNA) in SW620 cells (Fig. 3E and F). These results suggested
that tunicamycin treatment suppressed proliferation and arrested
the cell cycle in colon cancer cells.
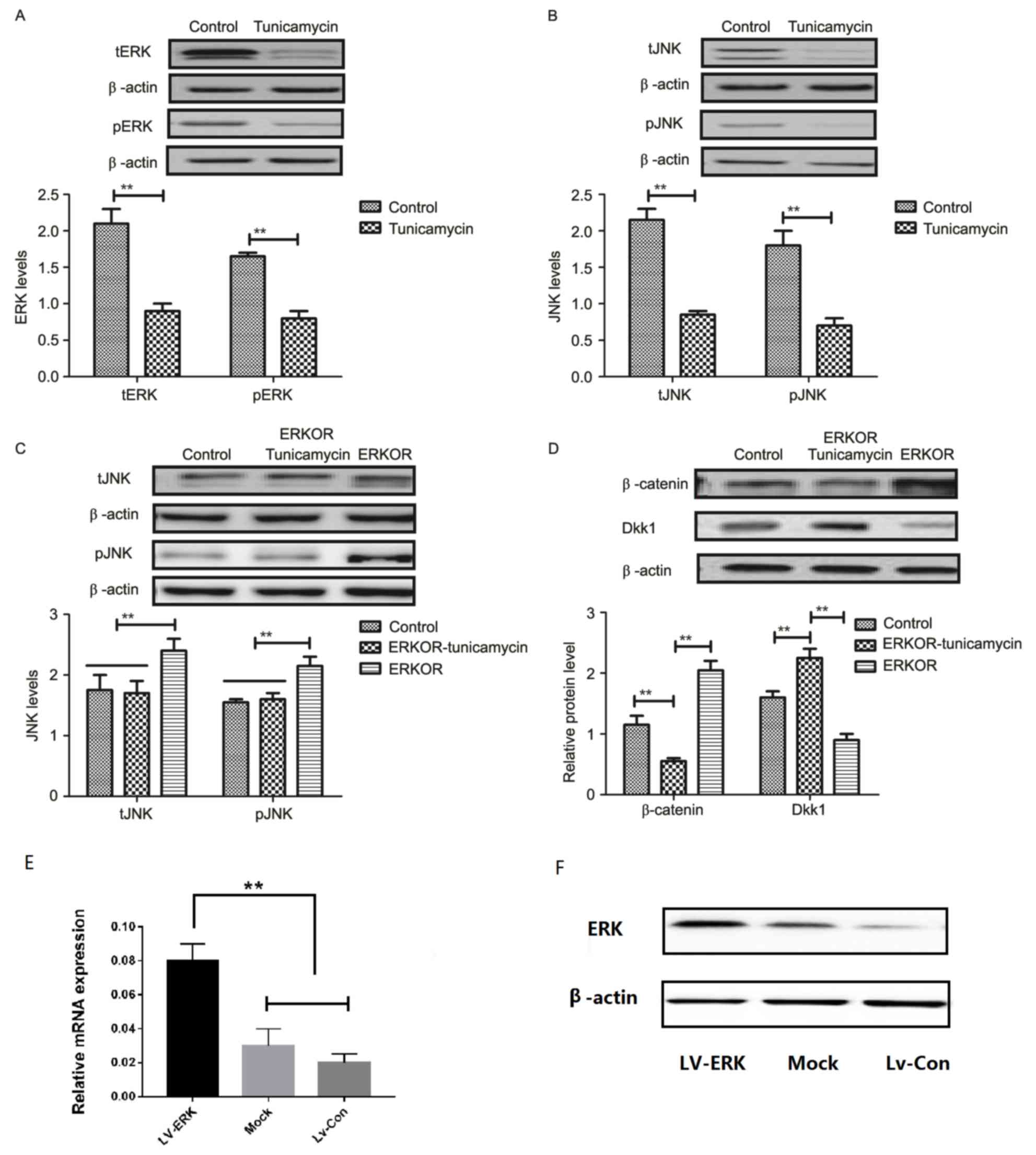
Tunicamycin treatment suppresses the
ERK-JNK signaling pathway in colon cancer cells
To analyze the underlying molecular mechanism of
growth and apoptosis, components of the ERK-JNK signaling pathway
in colon cancer cells were investigated. Expression and
phosphorylation of ERK and JNK were downregulated in
tunicamycin-treated SW620 cells, compared with control cells
(Fig. 4A and B). ERK
overexpression (ERKOR) significantly promoted JNK expression and
phosphorylation in SW620 cells, compared with control cells and the
ERKOR Tunicamycin group. In addition, the ERKOR Tunicamycin group
appeared to not promote JNK expression and phosphorylation in SW620
cells when compared with control cells (Fig. 4C). Results demonstrated that ERK
overexpression promoted expression of beta-catenin, and
downregulated Dickkopf-1 in SW620 cells, compared with the control
and ERKOR Tunicamycin group. Also, ERKOR Tunicamycin group
inhabited expression of β-catenin and upregulated Dickkopf-1 in
SW620 cells, compared with the control and ERKOR group (Fig. 4D). As determined by RT-qPCR and
western blotting, the cells in the LV-ERK group stably
overexpressed ERK at both the mRNA and protein level, as compared
with the mock treatment group or vector-transfected group (Fig. 4E and F). These results suggested
that tunicamycin treatment may suppress ERK-JNK signaling pathway
in colon cancer cells.
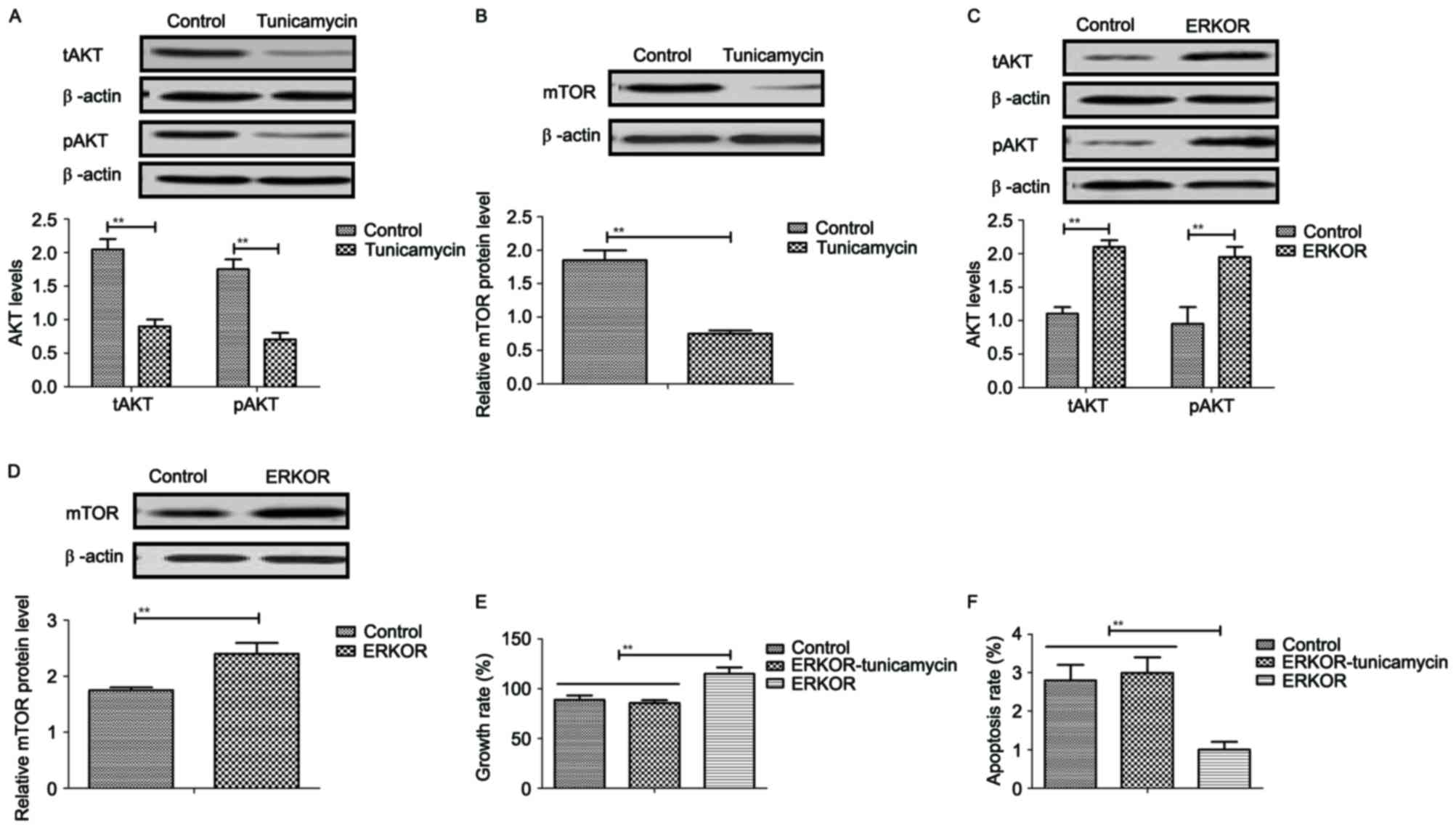
Tunicamycin treatment inhibits growth
and promotes apoptosis of colon cancer cells via the
ERK-JNK-mediated AKT/mTOR signaling pathway
The potential underlying mechanism of
tunicamycin-mediated growth inhibition and apoptosis in colon
cancer cells was investigated. Results revealed that tunicamycin
treatment inhibited AKT expression and phosphorylation in SW620
cells, compared with the control (P<0.01; Fig. 5A). mTOR expression levels were also
inhibited by tunicamycin in SW620 cells compared with the control
(P<0.01; Fig. 5B). ERKOR
increased AKT expression and phosphorylation in SW620 cells
compared with control cells (Fig.
5C). Results demonstrated that ERK overexpression increased
mTOR expression levels in SW620 cells compared with the control
(Fig. 5D). Tunicamycin abolished
ERK overexpression-induced growth and ERK overexpression-induced
inhibition of apoptosis in SW620 cells (P<0.01; Fig. 5E and F). These results suggested
that Tunicamycin treatment may inhibit growth and promote apoptosis
of colon cancer cells via the ERK-JNK-mediated AKT/mTOR signaling
pathway.
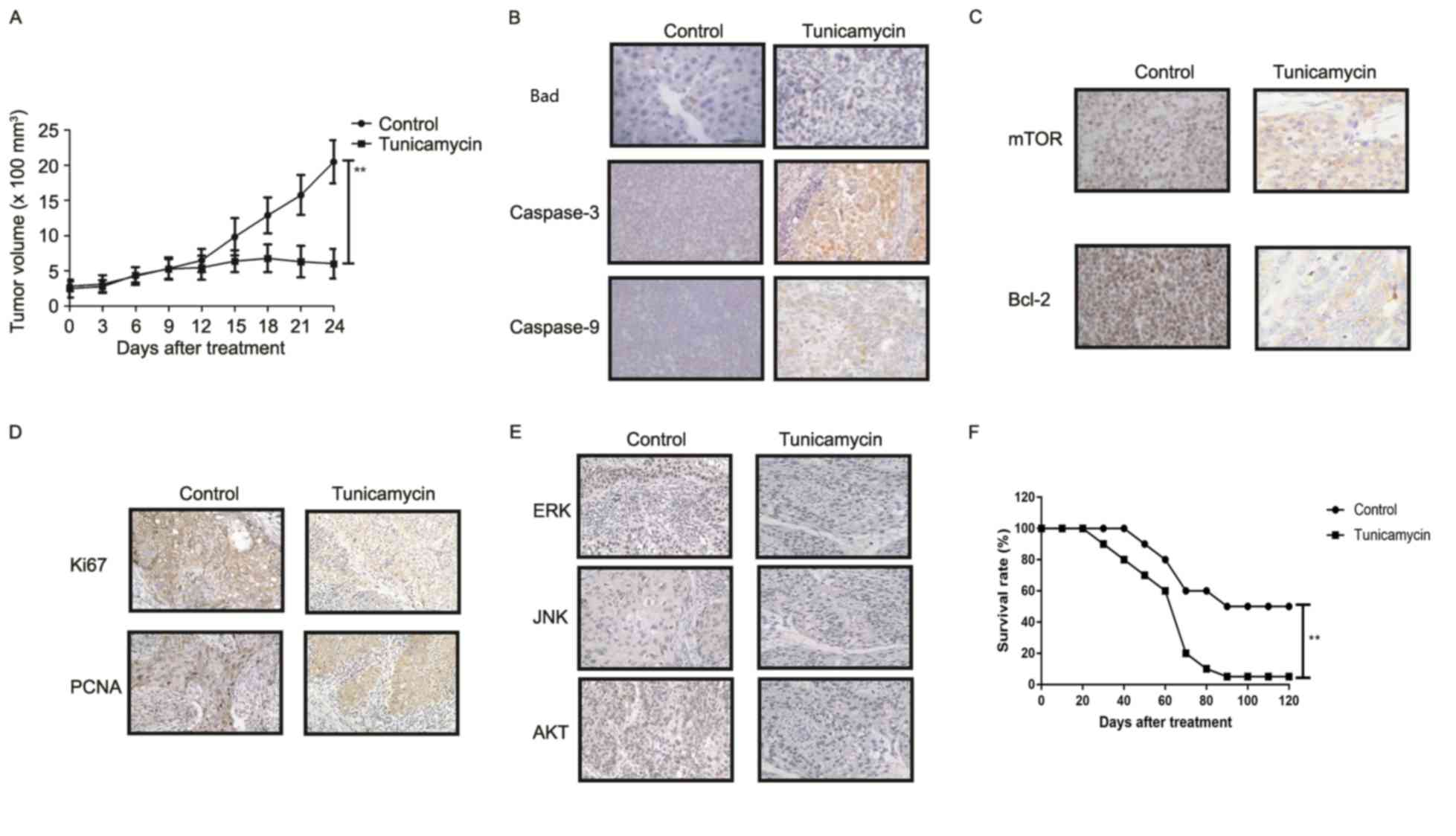
Tunicamycin treatment inhibits tumor
growth and prolongs survival of colon-bearing mice
The anti-tumor effect of tunicamycin treatment was
investigated in SW620-bearing nude mice. Tunicamycin treatment (10
mg/kg) significantly inhibited tumor growth in a 25-day observation
compared with the control group (Fig.
6A). Immunohistochemistry revealed that tunicamycin treatment
increased apoptosis (bad), caspase-3 and caspase-9 expression
levels in tumor tissues compared with the PBS group (Fig. 6B). Tunicamycin treatment decreased
the expression levels of mTOR and Bcl-2 in tumor tissues (Fig. 6C). Tunicamycin treatment decreased
Ki67 and PCNA expression levels in tumor tissues (Fig. 6D). Immunohistochemistry
demonstrated that tunicamycin treatment decreased ERK, JNK and AKT
expression in tumor tissues (Fig.
6E). Tunicamycin treatment prolonged the survival of
SW620-bearing mice in a 120-day experiment (P<0.01; Fig. 6F). These results suggested that
tunicamycin exhibits inhibitory effects on tumor growth and
prolongs survival of SW620-bearing mice.
Discussion
Colon cancer is a common malignant digestive tract
tumor, and usually occurs at the junction between the rectum and
sigmoid colon (24). Systematic
review has demonstrated that peritoneal carcinomatosis from colon
cancer blocks cytoreduction and intraperitoneal chemotherapy, which
requires further investigation (25). Advanced stage colon carcinoma
possesses an aggressive ability to attack adjacent and distant
cells and/or organs (26,27). Diagnosis, treatments and prognosis
of colon cancer has been improved, chemotherapies, radiation
therapies and surgery for colon cancer have been developed
clinically (28). However,
appropriate management of treatments of colorectal cancer has not
been improved and is not clearly understood due to local migration
and long-distance metastasis (26). Tunicamycin may inhibit tumor growth
and enhance apoptosis in human cancer cells (16,18).
In this study, the inhibitory effects of tunicamycin treatment in
colon cancer cells were investigated in vitro and in
vivo. Tunicamycin-mediated apoptosis and growth inhibition and
the potential underlying mechanism, were investigated. Results
revealed that tunicamycin treatment inhibited growth, proliferation
and aggressiveness, and promoted apoptosis of colon cancer cells.
In vivo assays demonstrated that tunicamycin treatment
significantly suppresses tumor growth and prolongs survival of
tumor bearing mice, suggesting tunicamycin may be a potential
anti-cancer agent for colon carcinoma therapy.
Induction of apoptosis and death of tumor cells to
inhibit growth and aggressiveness in patients is the ultimate goal
of neoplastic therapy. A study demonstrated that downregulation of
nuclear factor-κB may induce apoptosis and cell cycle arrest in
HCT116 human colon cancer cells (29). Wan et al (30) suggested that upregulation of the
caspase-3-dependent pathway inhibits growth of human colon cancer
cells and induced apoptosis. In addition, a report revealed that
downregulation of the Bcl-2 associated X apoptosis regulator/Bcl-2
ratio and upregulation of caspase-9-dependent pathway may promote
apoptosis of HT-29 human colon cancer cells (31). Furthermore, Travica et al
(32) demonstrated that colon
cancer-specific cytochrome P450 2W1 may convert duocarmycin
analogues into potent tumor cytotoxins, which further induces
apoptosis of HT29, DLD-1 and LoVo colon cancer cells (33). In the present study, the results
revealed that tunicamycin induced apoptosis of colon cancer cells
by downregulation of Bcl-2 and P53 expression levels. Results also
suggested that caspase-3 and caspase-9 expression levels were
upregulated by tunicamycin treatment, which markedly induced
apoptosis of colon cancer cells in tumor-bearing mice.
Clinical treatments are required for the inhibition
of migration and invasion, to prolong survival of patients with
colon carcinoma (34,35). Reports have suggested that
tunicamycin serves an important role in tumorigenesis, growth,
proliferation, aggressiveness and apoptosis via regulation of the
P38 mitogen-activated protein kinase signaling pathway (36,37).
Research has suggested that upregulation of the ERK-JNK signaling
pathway promotes metastasis of colon cancer cells via crosstalk
between C-C motif chemokine ligand 7 and C-C motif chemokine
receptor 3 (38). The findings of
the present study suggested that tunicamycin treatment inhibits
growth and metastasis of colon carcinoma cells by downregulation of
ERK-JNK signaling pathways. Additionally, Tandutinib may inhibit
the AKT/mTOR signaling pathway to suppress colon cancer growth
in vitro and in vivo, which is consistent with
previously published reports (39–41).
It was also observed that tunicamycin treatment decreased Ki67 and
PCNA expression levels in tumor tissues, which was associated with
apoptosis and increased survival of tumor bearing mice (42).
In conclusion, the results of the current study
suggested that tunicamycin treatment efficiently inhibits colon
carcinoma cell growth and aggressiveness via downregulation of
expression levels of vimentin, FIB and Ecacollagen type I and Slug.
Tunicamycin treatment may promote apoptosis of colon carcinoma
cells via the ERK-JNK-mediated AKT/mTOR signaling pathway, which
further contributes to inhibition of tumor growth both in
vitro and in vivo, and prolong survival of SW620-bearing
mice. These findings suggested that tunicamycin may be a potential
anti-cancer agent for treatment of colon carcinoma.
References
|
1
|
Siegel R, Desantis C and Jemal A:
Colorectal cancer statistics, 2014. CA Cancer J Clin. 64:104–117.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Hirai HW, Tsoi KK, Chan JY, Wong SH, Ching
JY, Wong MC, Wu JC, Chan FK, Sung JJ and Ng SC: Systematic review
with meta-analysis: Faecal occult blood tests show lower colorectal
cancer detection rates in the proximal colon in
colonoscopy-verified diagnostic studies. Aliment Pharmacol Ther.
43:755–764. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Fahrner R, Theis B, Ardelt M, Rauchfuss F,
Schüle S and Settmacher U: Rapid progressive colon cancer
metastasized to the right epididymis and liver: Report of a case
and review of the literature. Int J Colorectal Dis. 31:721–722.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gall TM, Markar SR, Jackson D, Haji A and
Faiz O: Mini-probe ultrasonography for the staging of colon cancer:
A systematic review and meta-analysis. Colorectal Dis. 16:O1–O8.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Kim HD, Ha KS, Woo IS, Jung YH, Han CW and
Kim TJ: Tumor lysis syndrome in a patient with metastatic colon
cancer after treatment with 5-fluorouracil/leucovorin and
oxaliplatin: Case report and literature review. Cancer Res Treat.
46:204–207. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Petrelli F, Tomasello G, Borgonovo K,
Ghidini M, Turati L, Dallera P, Passalacqua R, Sgroi G and Barni S:
Prognostic survival associated with left-sided vs right-sided colon
cancer: A systematic review and meta-analysis. JAMA Oncol. Oct
27–2016.(Epub ahead of print).
|
|
7
|
Moilanen JM, Kokkonen N, Löffek S,
Väyrynen JP, Syväniemi E, Hurskainen T, Mäkinen M, Klintrup K,
Mäkelä J, Sormunen R, et al: Collagen XVII expression correlates
with the invasion and metastasis of colorectal cancer. Hum Pathol.
46:434–442. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Fan Z, Cui H, Xu X, Lin Z, Zhang X, Kang
L, Han B, Meng J, Yan Z, Yan X and Jiao S: MiR-125a suppresses
tumor growth, invasion and metastasis in cervical cancer by
targeting STAT3. Oncotarget. 6:25266–25280. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Siani LM and Garulli G: Laparoscopic
complete mesocolic excision with central vascular ligation in right
colon cancer: A comprehensive review. World J Gastrointest Surg.
8:106–114. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Ma J, Ma P, Zhao C, Xue X, Han H, Liu C,
Tao H, Xiu W, Cai J and Zhang M: B7-H3 as a promising target for
cytotoxicity T cell in human cancer therapy. Oncotarget.
7:29480–29491. 2016.PubMed/NCBI
|
|
11
|
Hamamoto R and Nakamura Y: Dysregulation
of protein methyltransferases in human cancer: An emerging target
class for anticancer therapy. Cancer Sci. 107:377–384. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Matsui H, Ito H, Taniguchi Y, Takeda S and
Takahashi R: Ammonium chloride and tunicamycin are novel toxins for
dopaminergic neurons and induce Parkinson's disease-like phenotypes
in medaka fish. J Neurochem. 115:1150–1160. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Reszka N, Krol E, Patel AH and Szewczyk B:
Effect of tunicamycin on the biogenesis of hepatitis C virus
glycoproteins. Acta Biochim Pol. 57:541–546. 2010.PubMed/NCBI
|
|
14
|
Nami B, Donmez H and Kocak N:
Tunicamycin-induced endoplasmic reticulum stress reduces in vitro
subpopulation and invasion of CD44+/CD24- phenotype breast cancer
stem cells. Exp Toxicol Pathol. 68:419–426. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Miyake H, Hara I, Arakawa S and Kamidono
S: Stress protein GRP78 prevents apoptosis induced by calcium
ionophore, ionomycin, but not by glycosylation inhibitor,
tunicamycin, in human prostate cancer cells. J Cell Biochem.
77:396–408. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Shiraishi T, Yoshida T, Nakata S, Horinaka
M, Wakada M, Mizutani Y, Miki T and Sakai T: Tunicamycin enhances
tumor necrosis factor-related apoptosis-inducing ligand-induced
apoptosis in human prostate cancer cells. Cancer Res. 65:6364–6370.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Ling YH, Li T, Perez-Soler R and Haigentz
M Jr: Activation of ER stress and inhibition of EGFR
N-glycosylation by tunicamycin enhances susceptibility of human
non-small cell lung cancer cells to erlotinib. Cancer Chemother
Pharmacol. 64:539–548. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
de Freitas Junior JC, Silva Bdu R, de
Souza WF, de Araújo WM, Abdelhay ES and Morgado-Diaz JA: Inhibition
of N-linked glycosylation by tunicamycin induces
E-cadherin-mediated cell-cell adhesion and inhibits cell
proliferation in undifferentiated human colon cancer cells. Cancer
Chemother Pharmacol. 68:227–238. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
BukurovaIu A, Khankin SL, Krasnov GS,
Grigor'eva ES, Mashkova TD, Lisitsin NA, Karpov VL and Beresten'
SF: Comparison of 2D analysis and bioinformatics search efficiency
for colon cancer marker identification. Mol Biol (Mosk).
44:375–381. 2010.(In Russian). PubMed/NCBI
|
|
20
|
Thompson BA, Goldgar DE, Paterson C,
Clendenning M, Walters R, Arnold S, Parsons MT, Michael DW,
Gallinger S, Haile RW, et al: A multifactorial likelihood model for
MMR gene variant classification incorporating probabilities based
on sequence bioinformatics and tumor characteristics: A report from
the colon cancer family registry. Hum Mutat. 34:200–209. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Renshaw A and Elsheikh TM: A validation
study of the Focalpoint GS imaging system for gynecologic cytology
screening. Cancer Cytopathol. 121:737–738. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Bai FL, Yu YH, Tian H, Ren GP, Wang H,
Zhou B, Han XH, Yu QZ and Li DS: Genetically engineered Newcastle
disease virus expressing interleukin-2 and TNF-related
apoptosis-inducing ligand for cancer therapy. Cancer Biol Ther.
15:1226–1238. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Stipa F, Burza A, Curinga R, Santini E,
Delle Site P, Avantifiori R and Picchio M: Laparoscopic colon and
rectal resections with intracorporeal anastomosis and trans-vaginal
specimen extraction for colorectal cancer. A case series and
systematic literature review. Int J Colorectal Dis. 30:955–962.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Nadler A, McCart JA and Govindarajan A:
Peritoneal carcinomatosis from colon cancer: A systematic review of
the data for cytoreduction and intraperitoneal chemotherapy. Clin
Colon Rectal Surg. 28:234–246. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Pandor A, Eggington S, Paisley S,
Tappenden P and Sutcliffe P: The clinical and cost-effectiveness of
oxaliplatin and capecitabine for the adjuvant treatment of colon
cancer: Systematic review and economic evaluation. Health Technol
Assess. 10:iii–iv, xi-xiv, 1–185. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Millat B, Rougier P, Aparicio T, Guimbaud
R and Chaussade S: Conference review. Colon cancer: What treatment
in 2004? The point in five questions. Ann Chir. 130:277–283.
2005.(In French).
|
|
28
|
Chibaudel B, Bonnetain F, Tournigand C and
de Gramont A: Maintenance treatment in metastatic colorectal
cancer. Lancet Oncol. 16:e583–e584. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim MK, Kang YJ, Kim DH, Hossain MA, Jang
JY, Lee SH, Yoon JH, Chun P, Moon HR, Kim HS, et al: A novel
hydroxamic acid derivative, MHY218, induces apoptosis and cell
cycle arrest through downregulation of NF-κB in HCT116 human colon
cancer cells. Int J Oncol. 44:256–264. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Wan Y, Xin Y, Zhang C, Wu D, Ding D, Tang
L, Owusu L, Bai J and Li W: Fermentation supernatants of
Lactobacillus delbrueckii inhibit growth of human colon cancer
cells and induce apoptosis through a caspase 3-dependent pathway.
Oncol Lett. 7:1738–1742. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Lee JS, Jung WK, Jeong MH, Yoon TR and Kim
HK: Sanguinarine induces apoptosis of HT-29 human colon cancer
cells via the regulation of Bax/Bcl-2 ratio and caspase-9-dependent
pathway. Int J Toxicol. 31:70–77. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Travica S, Pors K, Loadman PM, Shnyder SD,
Johansson I, Alandas MN, Sheldrake HM, Mkrtchian S, Patterson LH
and Ingelman-Sundberg M: Colon cancer-specific cytochrome P450 2W1
converts duocarmycin analogues into potent tumor cytotoxins. Clin
Cancer Res. 19:2952–2961. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Rawłuszko AA, Sławek S, Gollogly A,
Szkudelska K and Jagodziński PP: Effect of butyrate on aromatase
cytochrome P450 levels in HT29, DLD-1 and LoVo colon cancer cells.
Biomed Pharmacother. 66:77–82. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Dowling CM, Herranz Ors C and Kiely PA:
Using real-time impedance-based assays to monitor the effects of
fibroblast-derived media on the adhesion, proliferation, migration
and invasion of colon cancer cells. Biosci Rep. 34:pii: e001262014.
View Article : Google Scholar
|
|
35
|
Kim J, Kang HS, Lee YJ, Lee HJ, Yun J,
Shin JH, Lee CW, Kwon BM and Hong SH: EGR1-dependent PTEN
upregulation by 2-benzoyloxycinnamaldehyde attenuates cell invasion
and EMT in colon cancer. Cancer Lett. 349:35–44. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Heckmann D, Maier P, Laufs S, Li L,
Sleeman JP, Trunk MJ, Leupold JH, Wenz F, Zeller WJ, Fruehauf S and
Allgayer H: The disparate twins: A comparative study of CXCR4 and
CXCR7 in SDF-1α-induced gene expression, invasion and
chemosensitivity of colon cancer. Clin Cancer Res. 20:604–616.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Radziwon-Balicka A, Santos-Martinez MJ,
Corbalan JJ, Corbalan JJ, O'Sullivan S, Treumann A, Gilmer JF,
Radomski MW and Medina C: Mechanisms of platelet-stimulated colon
cancer invasion: Role of clusterin and thrombospondin 1 in
regulation of the P38MAPK-MMP-9 pathway. Carcinogenesis.
35:324–332. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Lee YS, Kim SY, Song SJ, Hong HK, Lee Y,
Oh BY, Lee WY and Cho YB: Crosstalk between CCL7 and CCR3 promotes
metastasis of colon cancer cells via ERK-JNK signaling pathways.
Oncotarget. 7:36842–36853. 2016.PubMed/NCBI
|
|
39
|
Chen J, Shao R, Li F, Monteiro M, Liu JP,
Xu ZP and Gu W: PI3K/Akt/mTOR pathway dual inhibitor BEZ235
suppresses the stemness of colon cancer stem cells. Clin Exp
Pharmacol Physiol. 42:1317–1326. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Zhang X, Shi H, Tang H, Fang Z, Wang J and
Cui S: miR-218 inhibits the invasion and migration of colon cancer
cells by targeting the PI3K/Akt/mTOR signaling pathway. Int J Mol
Med. 35:1301–1308. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Ponnurangam S, Standing D, Rangarajan P
and Subramaniam D: Tandutinib inhibits the Akt/mTOR signaling
pathway to inhibit colon cancer growth. Mol Cancer Ther.
12:598–609. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Emenaker NJ, Calaf GM, Cox D, Basson MD
and Qureshi N: Short-chain fatty acids inhibit invasive human colon
cancer by modulating uPA, TIMP-1, TIMP-2, mutant p53, Bcl-2, Bax,
p21 and PCNA protein expression in an in vitro cell culture model.
J Nutr. 131 11 Suppl:3041S–3046S. 2001. View Article : Google Scholar : PubMed/NCBI
|