Introduction
Excessive alcohol consumption is an important health
issue globally, with ~3.3 million mortalities occurring annually
worldwide as a result of alcohol consumption (1). The National Institute on Alcohol
Abuse and Alcoholism defines binge drinking as a pattern of
drinking that elevates blood alcohol concentration levels to ≥0.08
g/dl (2). Harmful effects of binge
drinking, including enhanced coronary calcification, plaque
formation and circulatory dysfunction, have previously been
reported (3). Furthermore, binge
drinking has been revealed to increase mortality in patients with
acute myocardial infarction, or cancers of the oropharynx and
esophagus. In addition, binge drinking is reported to substantially
aggravate liver injury in individuals that chronically abuse
alcohol (4). Therefore, a
comprehensive understanding of the underlying mechanisms of binge
drinking-induced injury and its association with patient health is
of great importance.
The liver has an important role in the regulation of
host immune responses to various pathogens and toxins, and contains
various types of immune cells (5,6).
Natural killer T (NKT) cells account for 20–30% of liver
lymphocytes in mice and are involved in the pathogenesis of various
immune-mediated liver diseases (7–10).
Furthermore, NKT cells are reported to contribute to liver damage
following chronic alcohol consumption via the promotion of hepatic
neutrophil infiltration, which is one of the most important immune
cells associated with the induction of alcohol-induced liver injury
(11–13).
Small heterodimer partner (SHP), which is encoded by
the NR0B2 gene, is an orphan nuclear receptor, the ligand of which
remains undetermined, and lacks a DNA-binding domain, unlike the
majority of nuclear receptors. SHP acts as a corepressor by
directly interacting with other transcription factors, and it is
also involved in the metabolism of cholesterol, bile acid, and
fatty acids, in addition to roles in glucose and homocysteine
homeostasis (14,15). SHP is expressed in various organs,
including the liver (16,17), and certain studies have
demonstrated an association between alcohol-associated liver injury
and SHP expression in chronic alcohol-administered models (15,18).
These previous studies demonstrated that SHP-knockout (KO) mice
exhibited reduced inflammation via rapid detoxification of ethanol
following administration of alcohol; however, the role of SHP in an
acute binge drinking animal model, to the best of our knowledge,
has not been previously investigated. Therefore, in the present
study, the role of SHP in the severity of acute binge
drinking-induced liver injury, using a SHP-KO animal model, was
investigated.
Materials and methods
Animal experiments
Male SHP-KO mice (total n=50) and wild-type (WT)
C57BL/6 mice (total n=50; 8–10 weeks old; body weight, 23–27 g)
were obtained from the Korea Research Institute of Bioscience and
Biotechnology (KRIBB; Daejeon, Korea). All mice were housed and
maintained in a specific pathogen-free facility of Laboratory
Animal Resource Center of KRIBB (temperature of 22±2°C, relative
humidity of 50–60%, ammonia concentration less than 1 ppm, and 12-h
light/dark cycle) and were given ad libitum access to food
and water. All mice received a single dose of ethanol [6 g/kg body
weight, 25% (v/v) solution, n=30 for each SHP-KO mice or WT mice]
or PBS (control, n=20 for each SHP-KO mice or WT mice)
intragastrically to induce acute alcohol-associated liver injury,
according to a previously reported method (19,20),
and were euthanized 12 h post-treatment. A total of 9–11 mice were
used for both control groups and 15 mice used for both alcohol
groups. All experimental procedures were approved by the
Institutional Animal Care and Use Committee of the KRIBB (Daejeon,
Korea) and were performed in accordance with the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals (Guide for the care and use of laboratory animals, 8th
edition) (21).
Blood analysis
At 12 h post-alcohol or vehicle administration,
blood samples were collected and centrifuged at 9,800 × g for 10
min at 4°C. The isolated plasma was subsequently used for alanine
aminotransferase (ALT) and aspartate aminotransferase (AST)
analysis using an automated blood chemistry analyzer (Hitachi 7150;
Hitachi, Ltd., Tokyo, Japan).
Oil red O staining and lipid
quantification
Liver tissues, which were obtained from animals at
12 h post-alcohol or vehicle administration, were embedded in a
Tissue-Tek optimal cutting temperature compound (Sakura Finetek,
Tokyo, Japan) and sectioned at a thickness of 8-µm using a cryotome
(Sakura Finetek). Cryostat sections of liver tissue were fixed in
10% neutral buffered formalin at room temperature. After fixation,
liver tissue sections were stained with 0.3% oil red O solution for
1 h and counterstained with Mayer's hematoxylin for 30 sec at room
temperature. Images were captured using a light microscope (BX51;
Olympus Corporation, Tokyo, Japan). Total hepatic lipid was
isolated by performing previously reported methods (22). Briefly, frozen livers were
homogenized in 0.9% saline and a 100% chloroform: 100% methanol
(1:2, v/v) solution was subsequently added to the samples. The
solution was vortexed and centrifuged at 890 × g for 20 min. The
chloroform layer was transferred to a glass tube and air-dried
until only the pellet remained. Hepatic triglyceride (TG)
concentrations were analyzed using a commercially available kit
(cat. no. AM 157S-K; Asan Pharmaceuticals Co., Ltd., Seoul, Korea),
in accordance with the manufacturer's protocol.
Lipid peroxidation assay
Hepatic lipid peroxidation levels were determined by
previously described methods (23)
with slight modifications. Briefly, the livers that were obtained
from mice at 12 h after alcohol or vehicle treatment were
homogenized in PBS containing 1 mmol/l butylated hydroxytoluene and
5% trichloroacetic acid solution. Following a cooling period on
ice, the liver extracts were centrifuged at 890 × g for 15 min.
Supernatants were collected, 0.8% thiobarbituric acid (TBA)
solution was added and the suspensions were subsequently boiled at
100°C for 60 min in order to generate the malondialdehyde (MDA)-TBA
complex (red pigment). Samples were subsequently chilled on ice and
centrifuged at 1,600 × g for 10 min. Supernatants were collected
and the absorbance was measured at 535 nm. MDA solution was freshly
prepared via hydrolysis of 1 mmol/l 1,1,3,3-tetramethoxypropane
solution (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) and the
addition of 1 mol/l hydrogen chloride, and was used as the
reference standard.
Determination of total reactive oxygen
species (ROS)
Mouse livers were obtained from mice at 12 h after
alcohol or vehicle treatment and lysed in ice-cold lysis buffer
containing 1% nonyl phenoxypolyethoxylethanol, 0.25% sodium
deoxycholate, 50 mmol/l Tris-HCl, 1 mmol/l
ethylenediaminetetraacetic acid, 120 mmol/l sodium chloride,
protease inhibitor cocktail (Roche Diagnostics, Indianapolis, IN,
USA) and phosphatase inhibitor cocktails (Sigma-Aldrich; Merck
KGaA). Following two cycles of centrifugation at 16,600 × g for 15
min at 4°C, the supernatants were used for total ROS measurement.
The protein concentration was measured using the Bradford assay.
Following this, liver extracts were incubated with 20 µmol/l
2′,7′-dichlorodihydrofluorescein diacetate (Invitrogen; Thermo
Fisher Scientific, Inc., Waltham, MA, USA) at 37°C for 60 min.
Fluorescence intensity was analyzed using a Victor3 1420 Multilabel
Counter (PerkinElmer, Inc., Waltham, MA, USA) at 485 nm excitation
and 530 nm emission wavelengths.
RNA extraction and reverse
transcription-quantitative polymerase chain reaction (RT-qPCR)
Total RNA was isolated from mouse livers, which were
obtained from mice at 12 h after alcohol or vehicle treatment,
using TRIzol reagent (Invitrogen; Thermo Fisher Scientific, Inc.).
Liver tissues were homogenized in TRIzol reagent with stainless
steel beads using TissueLyser (Qiagen GmbH, Hilden, Germany).
Following the addition of 300 µl 100% chloroform, the mixture was
centrifuged at 16,600 × g for 15 min at 4°C and the supernatant was
transferred to a clean tube. The 500 µl 100% isopropanol was
subsequently added into supernatant. Following inversion, the
mixture was centrifuged at 16,600 × g for 5 min at 4°C and the
pellets were washed with 75% (v/v) ethanol. The RNA pellet was
dried at room temperature and dissolved in diethyl
pyrocarbonate-treated water. Following this, cDNA was synthesized
from 1 µg total isolated RNA using a cDNA synthesis kit (iScript™
cDNA synthesis kit; Bio-Rad Laboratories, Inc., Hercules, CA, USA)
according to the manufacturer's protocol. The cycling conditions
were as follows: Priming at 25°C for 5 min and reverse
transcription at 46°C for 20 min and RT inactivation at 95°C for 1
min. Subsequently, qPCR was performed using SYBR Green PCR Master
Mix (AccuPower® 2X Greenstar™ qPCR Master Mix; Bioneer
Co., Daejeon, Korea) and StepOne™ Real time PCR system (Applied
Biosystems; Thermo Fisher Scientific, Inc.). The cycling conditions
were as follows: Pre-denaturation at 95°C for 10 min, followed by
denaturation at 95°C for 10 sec, and annealing and extension at
60°C for 30 sec, for 45 cycles of amplification. PCR was performed
in duplicates. All expression data were normalized to 18S ribosomal
RNA and were calculated using the 2−ΔΔCq method
(24). The results are presented
in terms of fold change compared with the expression in the control
group. The PCR primer pair sequences are detailed in Table I.
 | Table I.Primer sets for reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I.
Primer sets for reverse
transcription-quantitative polymerase chain reaction analysis.
| Accession
number | Gene | Primer
sequence | Product size,
bp |
|---|
| NM_011850.3 | SHP |
5′-TCTGCAGGTCGTCCGACTATT-3 (F) | 168 |
|
|
|
5′-TGTCTTGGCTAGGACATCCA-3′ (R) |
|
| NR_003278.3 | 18s rRNA |
5′-GACACGGACAGGATTGACAGATTGATAG-3 (F) | 129 |
|
|
|
5′-GTTAGCATGCCAGAGTCTCGTTCGTT-3′ (R) |
|
| NM_008361.4 | IL-1β |
5′-CTACAGGCTCCGAGATGAACAAC-3 (F) | 79 |
|
|
|
5′-TCCATTGAGGTGGAGAGCTTTC-3′ (R) |
|
| NM_031168.2 | IL-6 |
5′-GTTGCCTTCTTGGGACTGATG-3′ (F) | 90 |
|
|
|
5′-GGGAGTGGTATCCTCTGTGAAGTCT-3′ (R) |
|
| NM_013693.3 | TNF-α |
5′-TGGCCTCCCTCTCATCAGTT-3′ (F) | 110 |
|
|
|
5′-CCTCCACTTGGTGGTTTGCT-3′ (R) |
|
| NM_011333.3 | CCL2 |
5′-CAGCAAGATGATCCCAATGAGTAG-3′ (F) | 100 |
|
|
|
5′-TCTCTTGAGCTTGGTGACAAAAAC-3′ (R) |
|
| NM_008176.3 | CXCL1 |
5′-TGTCAGTGCCTGCAGACCAT-3′ (F) | 150 |
|
|
|
5′-CAAGGGAGCTTCAGGGTCAA-3′ (R) |
|
| NM_009140.2 | CXCL2 |
5′-GGCTGTTGTGGCCAGTGAA-3′ (F) | 140 |
|
|
|
5′-CGCCCTTGAGAGTGGCTATG-3′ (R) |
|
| NM_021283.2 | IL-4 |
5′-GGAGATGGATGTGCCAAACG-3′ (F) | 80 |
|
|
|
5′-GCACCTTGGAAGCCCTACAG-3′ (R) |
|
| NM_021282.2 | CYP2E1 |
5′-ATCAACCTCGTCCCTTCCAA-3′ (F) | 71 |
|
|
|
5′-GGGATGACATATCCTCGGAACA-3′ (R) |
|
| NM_007409.2 | ADH1 |
5′-GGAGGGGTGGACTTTTCGTT-3′ (F) | 100 |
|
|
|
5′-CTACGACGACGCTTACACCA-3′ (R) |
|
| NM_009656.3 | ALDH2 |
5′-TGTTGTACCGATTGGCGGAT-3′ (F) | 131 |
|
|
|
5′-GCGGAGACATTTCAGGACCA-3′ (R) |
|
Fluorescence-activated cell sorting
(FACS) analysis
Livers were removed from the mice following
euthanasia at 12 h after treatment with alcohol or vehicle,
grounded using a plunger on 70-µm mesh, centrifuged at 39 × g for 5
min at 4°C, and the resultant supernatants were transferred to
clean tubes. Following centrifugation at 431 × g for 5 min at 4°C,
the supernatants were eluted. Pre-warmed 40% Percoll solution was
added and the mixture was centrifuged at 1,265 × g for 30 min.
Following this, red blood cells (RBCs) were lysed using RBC lysis
buffer (BioLegend Inc., San Diego, CA, USA). Following
centrifugation at 712 × g for 15 min at 4°C, liver mononuclear
cells were suspended in FACS buffer (PBS containing 0.5% bovine
serum albumin and 0.05% NaN3). Cells were blocked for 30
min at 4°C with unlabeled CD16/32 antibody (1/50 dilution; clone
93; cat. no. 101302; BioLegend Inc.). The cells were stained for 30
min at 4°C using the following monoclonal antibodies against cell
surface markers: Fluorescein isothiocyanate (FITC)-anti-CD45,
phycoerythrin-anti-lymphocyte antigen 6 complex (Ly6) locus G,
peridinin-chlorophyll protein complex (PerCP) cyanine
(cy)5.5-anti-Ly6 locus C and allophycocyanin (APC)-anti-CD11b for
neutrophils; and FITC-anti-CD3, PerCP cy5.5-anti-CD45 and
APC-anti-killer cell lectin-like receptor subfamily B member 1C for
NKT cells. Stained cells were detected using a Gallios Flow
Cytometer (Beckman Coulter, Inc., Brea, CA, USA) and analyzed using
FlowJo software V10 (Tree Star, Inc., Ashland, OR, USA). The immune
cell percentage data is expressed as stained cell number divided by
the CD45+ cell number. The absolute cell number/g liver
is calculated by multiplying CD45+ cell number/g liver
by stained cell percentage.
Statistical analysis
Data are presented as the mean ± standard error of
the mean. Comparisons among groups were performed using one-way
analysis of variance followed by Tukey's post-hoc test. JMP 5.1
software was used for analysis (SAS Institute, Inc., Cary, NC,
USA). P<0.05 was considered to indicate a statistically
significant difference.
Results
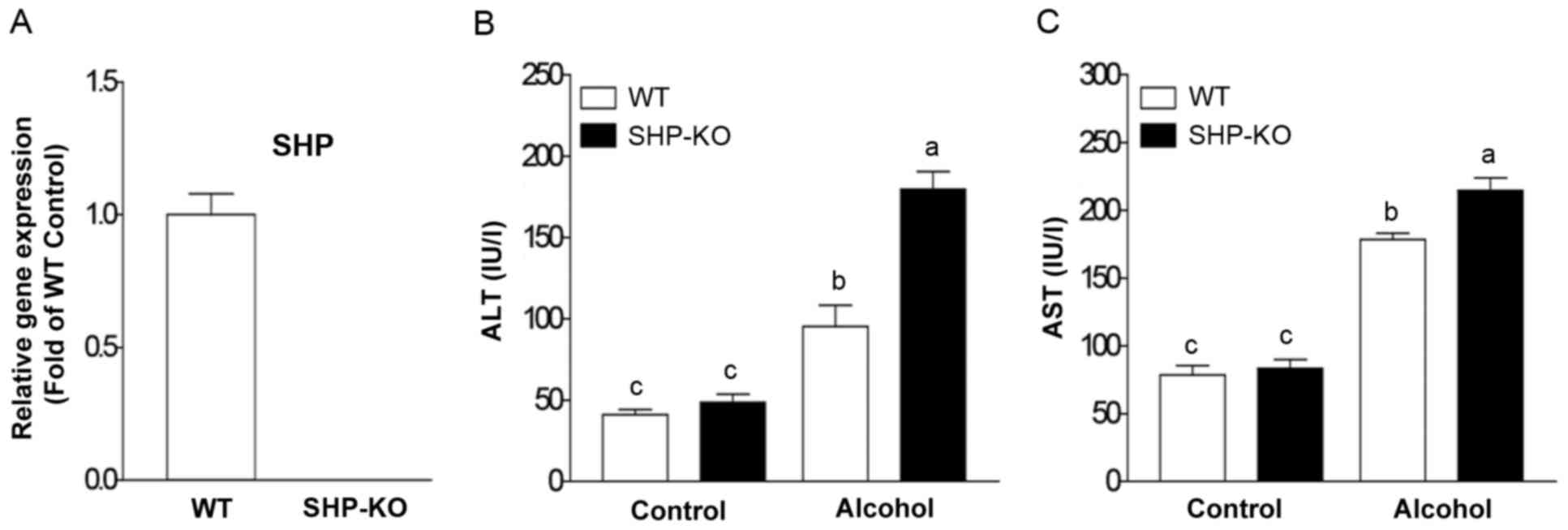
Genetic depletion of SHP aggravates
binge drinking-induced liver injury
In order to investigate the role of SHP in acute
binge drinking-induced liver injury, all mice were administered 6
g/kg body weight of ethanol (alcohol group) or an equal amount of
PBS (control group). SHP depletion in the liver of SHP-KO mice was
confirmed using RT-qPCR (Fig. 1A).
Plasma ALT and AST levels in the WT alcohol group were increased at
12 h post-alcohol administration compared with the control group
(Fig. 1B and C). Notably, the
SHP-KO alcohol group exhibited a significant increase in ALT and
AST compared with the WT alcohol group (Fig. 1B and C). These results indicate
that SHP may serve a protective role in acute binge
drinking-induced liver injury.
Increased binge drinking-induced liver
injury in SHP-KO mice is independent of lipid accumulation and
oxidative stress
Marked lipid accumulation was observed in liver
samples following binge alcohol administration compared with the
control group; however, no marked differences were observed between
the WT and SHP-KO alcohol groups (Fig.
2A). Furthermore, hepatic TG concentrations were enhanced in WT
and SHP-KO alcohol groups compared with the respective control
groups. However, no significant differences in hepatic TG levels
were observed between the WT and SHP-KO alcohol groups (Fig. 2B). In addition, the level of MDA
formation, a marker of lipid peroxidation, was similar between the
WT and SHP-KO alcohol groups (Fig.
2C). Furthermore, alcohol-induced ROS production and the
expression of genes associated with alcohol metabolism, including
cytochrome P450 (CYP)2E1, alcohol dehydrogenase (ADH)1 and aldehyde
dehydrogenase (ALDH)2, were analyzed; however, no significant
differences were demonstrated between WT and SHP alcohol groups
(Figs. 2D and 3). These results indicate that SHP is not
involved in lipid accumulation, lipid peroxidation, ROS production
or alcohol metabolism in response to acute binge drinking exposure
in mice.
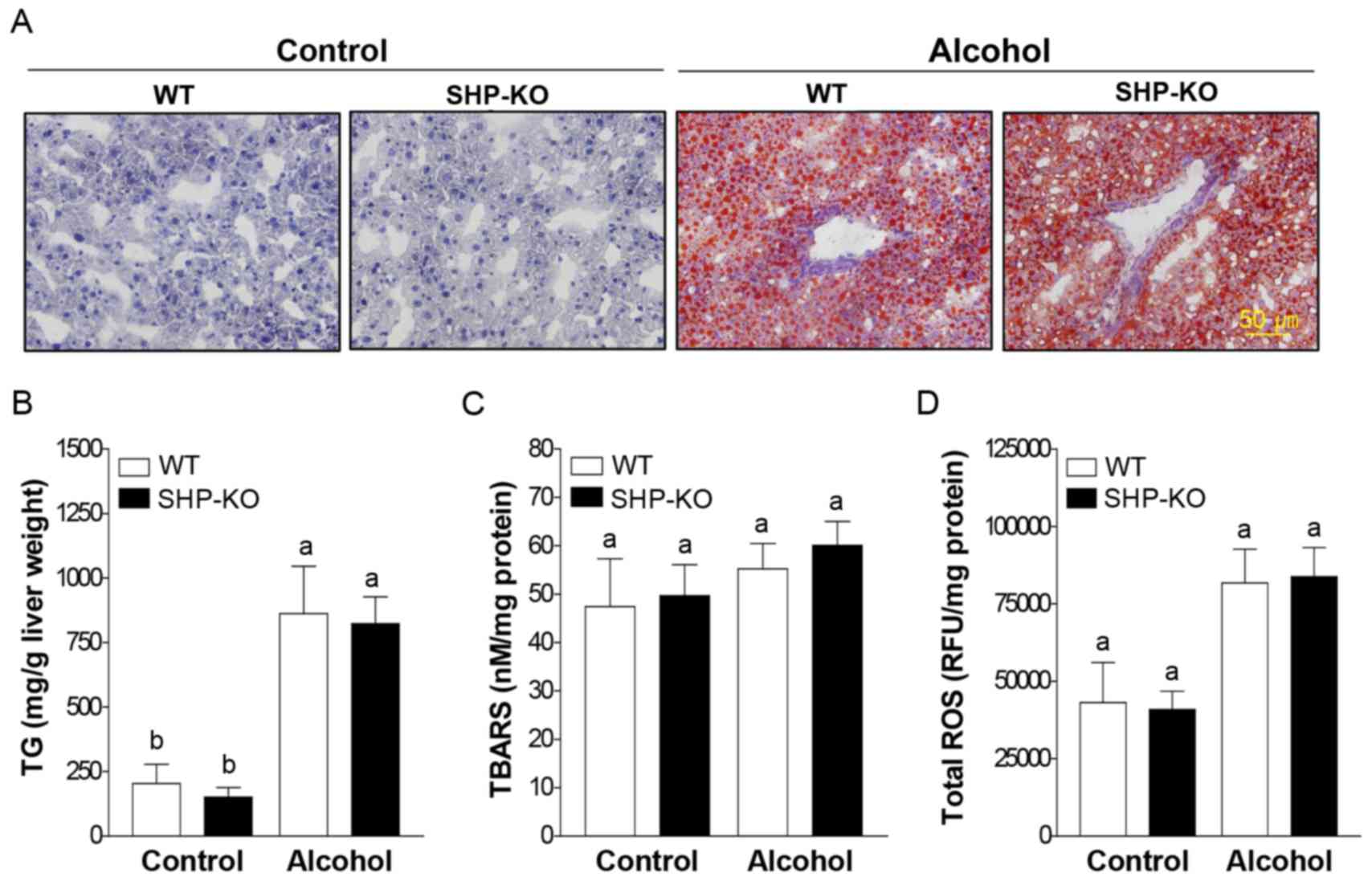 | Figure 2.Lipid accumulation and oxidative
stress in the liver of mice following binge alcohol administration.
Mice were administered a single dose of alcohol (6 g/kg body
weight) or equal amounts of PBS (control group) and were euthanized
12 h post-treatment. (A) Representative images of liver sections
stained with oil red O. Lipid droplets are stained red. For oil red
O staining, n=11 in WT and SHP-KO control groups and n=15 in WT and
SHP-KO alcohol groups. Levels of (B) TG, (C) malondialdehyde
formation and (D) total ROS levels in the liver extracts were
determined. For TG, malondialdehyde lipid peroxidation and total
ROS assays, n=3 in WT and SHP-KO control groups and n=5 in WT and
SHP-KO alcohol groups. Data are presented as the mean ± standard
error of the mean. Comparisons between groups are indicated by
lowercase letters and groups not sharing a common letter are
significantly different to one another at a threshold of P<0.05.
TG, triglyceride; ROS, reactive oxygen species; WT, wild-type; SHP,
small heterodimer partner; KO, knockout; TBARS, thiobarbituric acid
reactive substances; ns, not significant. |
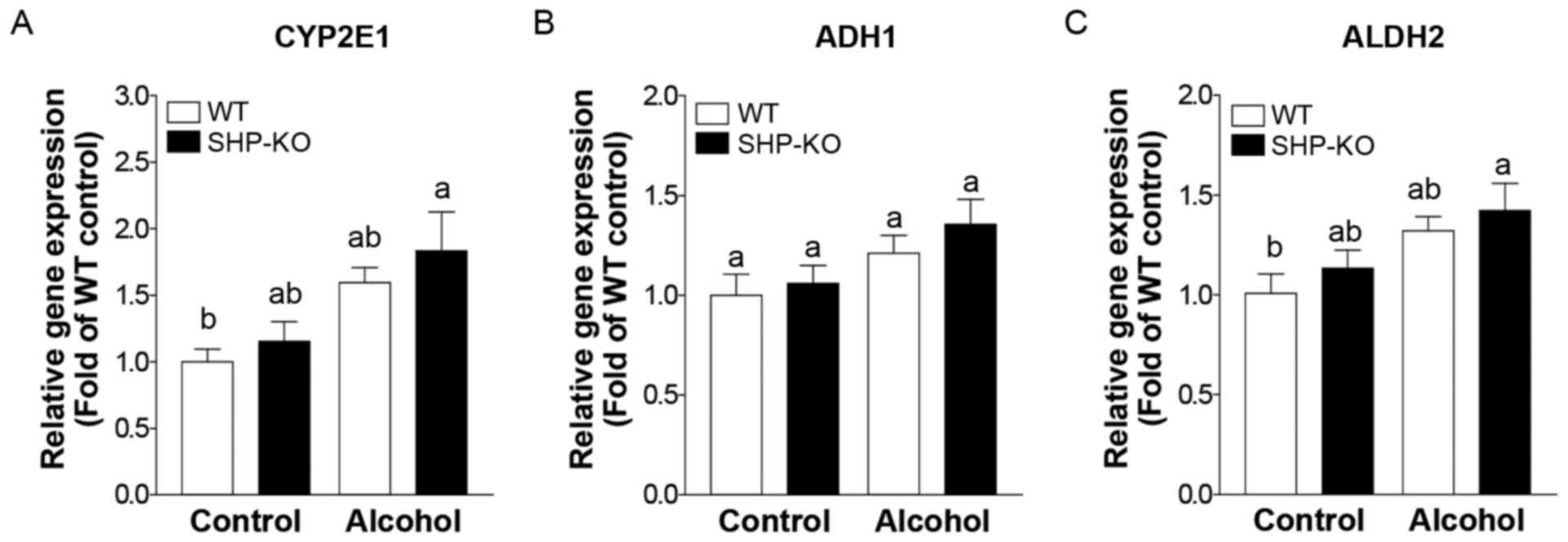 | Figure 3.Expression of genes encoding alcohol
metabolism-associated enzymes following binge alcohol
administration in mice. Mice were administered a single dose of
alcohol (6 g/kg body weight) or equal amounts of PBS (control
group) and were euthanized 12 h post-treatment. mRNA expression
levels of (A) CYP2E1, (B) ADH1 and (C) ALDH2 were measured using
reverse transcription-quantitative polymerase chain reaction. WT
and SHP-KO control groups, n=11; WT and SHP-KO alcohol groups,
n=15. Data are presented as the mean ± standard error of the mean.
Comparisons between groups are indicated by lowercase letters and
groups not sharing a common letter are significantly different to
one another at a threshold of P<0.05. CYP, cytochrome P450; ADH,
alcohol dehydrogenase; ALDH, aldehyde dehydrogenase; WT, wild-type;
SHP, small heterodimer partner; KO, knockout; ns, not
significant. |
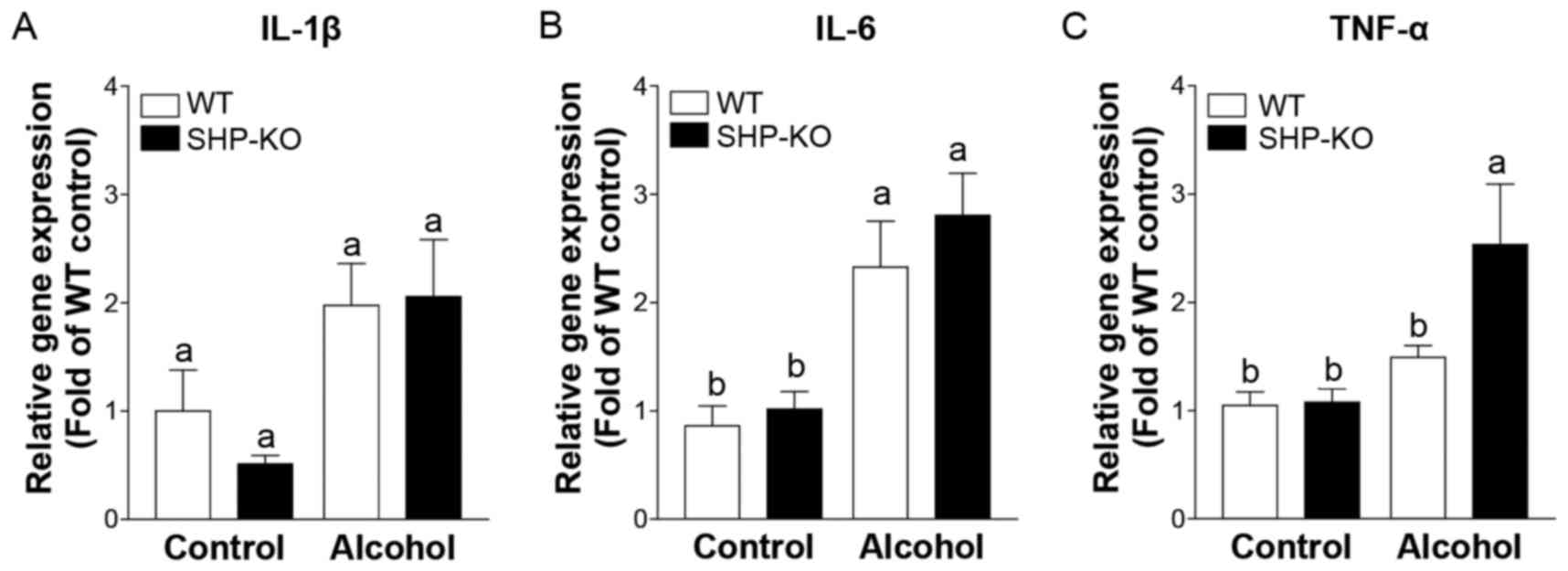
Proinflammatory cytokine expression is
elevated in SHP-KO mice following binge alcohol administration
Inflammation-associated gene expression was
investigated in order to determine the factors that induce severe
liver injury in SHP-KO mice. It was revealed that interleukin
(IL)-1β and IL-6 expression levels were increased following alcohol
administration; however, the differences in the expression levels
between the WT and SHP-KO alcohol groups were not significant
(Fig. 4A and B). Tumor necrosis
factor (TNF)-α expression was also enhanced in WT and SHP-KO
alcohol groups compared with the respective controls, and its
expression was significantly increased in SHP-KO alcohol mice
compared with WT alcohol mice (Fig.
4C). These results indicate that SHP may have a role in acute
binge drinking-induced liver inflammation.
Hepatic NKT cell and neutrophil levels
are increased in SHP-KO mice following binge alcohol
administration
Immune cell populations were analyzed following
acute binge alcohol administration using FACS analysis. The results
demonstrated that the number of neutrophil cells increased in the
livers of both of the alcohol groups compared with the control
groups (Fig. 5A). The percentage
of neutrophils observed was similar between both alcohol groups;
however, the absolute cell numbers were elevated in the SHP-KO
alcohol group compared with the WT alcohol group (Fig. 5A). Acute binge alcohol
administration did not alter the NKT cell population in the livers
of WT mice (Fig. 5B). Notably,
both the percentage and absolute number of NKT cells increased
significantly in the livers of the SHP-KO alcohol group compared
with the WT alcohol group (Fig.
5B). These results indicate that increased recruitment of NKT
cells and neutrophils to the liver may be a causative factor of
exacerbated acute binge drinking-induced liver injury in SHP-KO
mice.
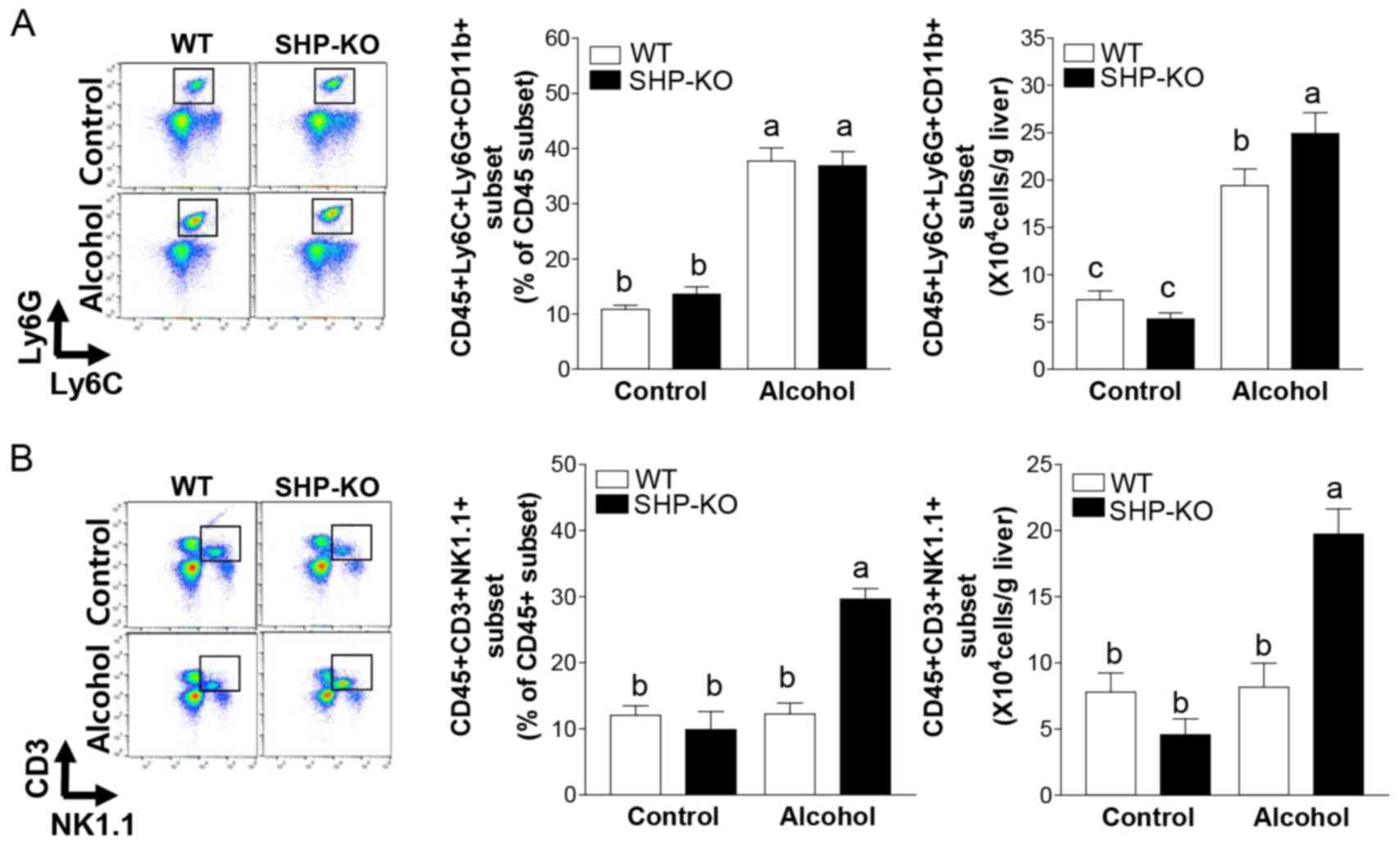 | Figure 5.Immune cell populations in the liver
following binge alcohol administration in mice. Mice were
administered a single dose of alcohol (6 g/kg body weight) or equal
amounts of PBS (control group) and were euthanized 12 h
post-treatment. Liver immune cells were isolated and flow cytometry
analysis was performed. (A) Neutrophils (CD45+,
CD11b+, Ly6C+ and Ly6G+) and (B)
natural killer T cells (CD45+, CD3+ and
NK1.1+) were recruited to the liver in SHP-KO mice
following alcohol administration. WT and SHP-KO control groups,
n=9; WT and SHP-KO alcohol groups, n=15. Data are presented as the
mean ± standard error of the mean. Comparisons between groups are
indicated by lowercase letters and groups not sharing a common
letter are significantly different to one another at a threshold of
P<0.05. Ly6, lymphocyte antigen 6 complex; Ly6C, Ly6 locus C;
Ly6G, Ly6 locus G; NK1.1, killer cell lectin-like receptor
subfamily B member 1C; SHP, small heterodimer partner; KO,
knockout; WT, wild-type. |
Expression levels of cytokines and
chemokines associated with NKT cell and neutrophil migration are
elevated in SHP-KO mice following binge alcohol administration
Chemokine expression levels were investigated in
order to determine the chemotactic factors and cytokines that
elicit increased neutrophil and NKT cell recruitment to the livers
of SHP-KO mice following alcohol administration. Following acute
binge alcohol administration, C-C motif chemokine ligand (CCL)2 and
C-X-C motif chemokine ligand (CXCL)1 expression levels were
upregulated in the livers of both alcohol groups compared with
those exhibited by the control groups, and significantly enhanced
expression levels of CCL2 and CXCL1 were observed in the SHP-KO
alcohol group compared with the WT alcohol group (Fig. 6A and B). Furthermore, although
CXCL2 expression was upregulated in the livers of both of the
alcohol groups compared with the respective controls, the
difference in the expression levels between the SHP-KO group and
the WT group were not significant (Fig. 6C). Additionally, hepatic IL-4
expression was not significantly affected by alcohol administration
in the WT group, and was significantly increased in the SHP-KO
alcohol group compared with the other three groups (Fig. 6D). Taken together, these results
indicate that elevated expression levels of CCL2, CXCL1 and Il-4 in
the liver of SHP-KO binge drinking-exposed mice may promote the
recruitment of NKT cells and neutrophils, and thus exacerbate binge
alcohol-induced liver injury.
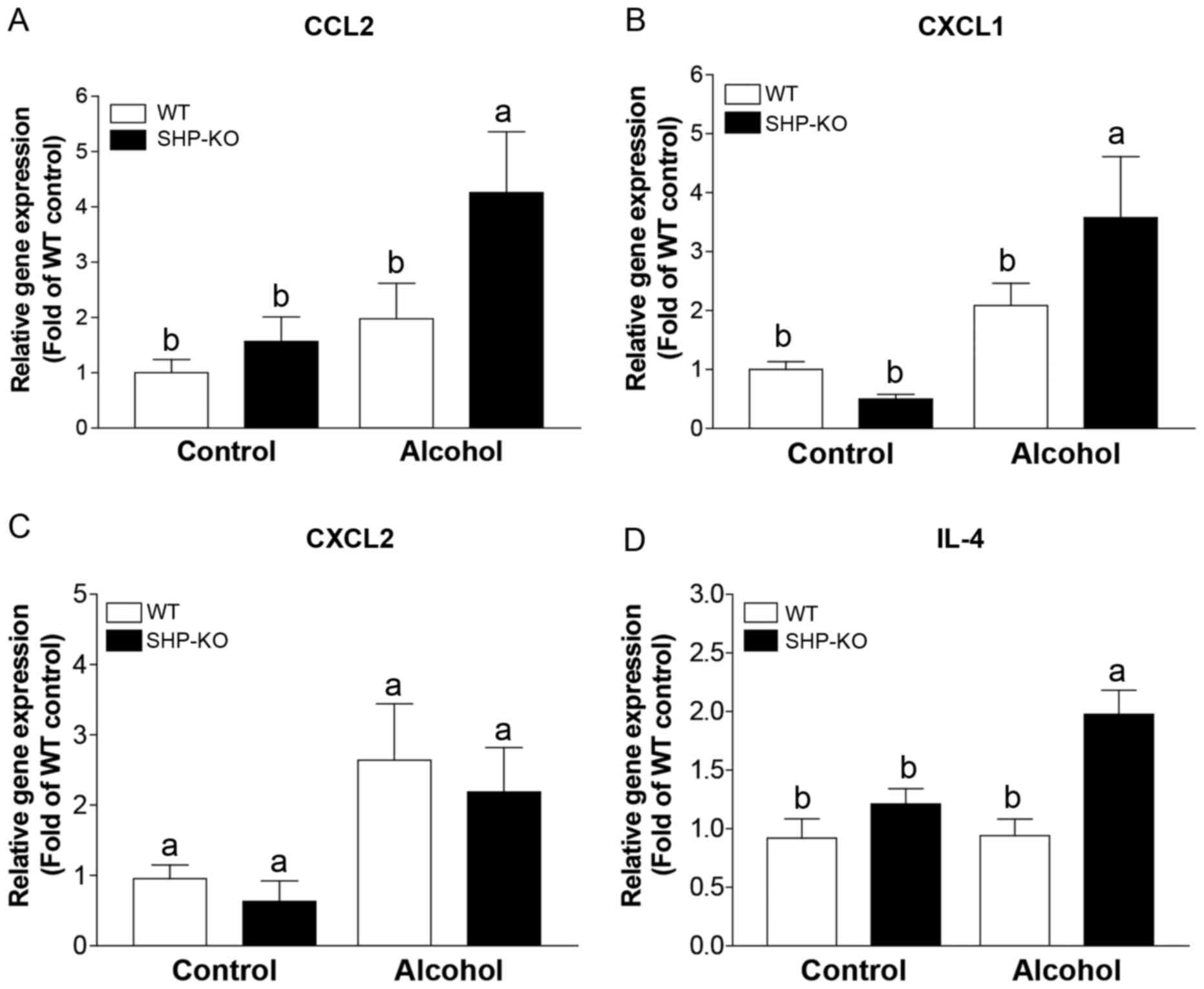 | Figure 6.Liver cytokines and chemokines
associated with immune cell recruitment following binge alcohol
administration. Mice were administered a single dose of alcohol (6
g/kg body weight) or equal amounts of PBS as a control and were
euthanized 12 h later. The expression of immune cell recruitment
associated genes, including (A) CCL2, (B) CXCL1, (C) CXCL2 and (D)
IL-4, was determined in liver tissues using reverse
transcription-quantitative polymerase chain reaction. WT and SHP-KO
control groups, n=9; WT and SHP-KO alcohol groups, n=15. Data are
presented as the mean ± standard error of the mean. Comparisons
between groups are indicated by lowercase letters and groups not
sharing a common letter are significantly different to one another
at a threshold of P<0.05. CCL, C-C motif chemokine ligand; CXCL,
C-X-C motif chemokine ligand; IL, interleukin; WT, wild-type; SHP,
small heterodimer partner; KO, knockout. |
Discussion
In the present study, it has been demonstrated that
acute alcohol binging led to increased infiltration of NKT cells
and neutrophils in the livers of SHP-KO mice, which was associated
with enhanced severity of liver injury in SHP-KO mice compared with
WT mice.
NKT cells, which are a major population of mouse
liver lymphocytes, have been reported to have an important role in
alcohol-induced liver injury (5,6). NKT
cells migrating from peripheral blood to the liver respond to
several chemokines, including CCL2 (25,26).
In the present animal model of acute alcohol binge drinking, CCL2
expression was only significantly upregulated in the liver of the
SHP-KO alcohol group, and not in the WT alcohol group, compared
with the respective control groups. However, it was previously
demonstrated that acute binge ethanol administration did not alter
the NKT cell population in the liver (11). Similarly, in the present study, the
NKT cell population was not significantly altered following acute
binge alcohol administration in the livers of WT mice; however, the
SHP-KO alcohol group demonstrated a significantly increased
infiltration rate of NKT cells to the liver. Furthermore, previous
studies have revealed that NKT cells may participate in the
progression of inflammation by secreting cytokines, including IL-4,
which subsequently induce neutrophil recruitment and survival and
enhance liver injury (27,28). Previous studies also demonstrated
that NKT cells contributed to alcohol-induced liver injury by
promoting neutrophil infiltration, which is a well-established
immune cell responsible for alcoholic steatohepatitis (ASH)
(11–13,29).
In the present study, the percentages of neutrophils were similar
between both alcohol groups, however, the absolute cell numbers
were increased in the SHP-KO alcohol group compared with the WT
alcohol group. The immune cell percentage data is expressed as
stained cell number divided by the CD45+ cell number.
The absolute cell number/g liver is calculated by multiplying
CD45+ cell number/g liver by stained cell percentage. In
the present study, CD45+ cell number/g liver in the
SHP-KO alcohol group was slightly increased compared with the WT
alcohol group (data not shown). Thus, although the percentages of
neutrophils were similar between both alcohol-treated groups, the
absolute cell number was higher in SHP-KO mice. In the current
study, a significant increase in hepatic IL-4 expression was
observed in the SHP-KO alcohol group compared with the WT alcohol
group, in addition to elevated NKT cell infiltration and a higher
migration rate of neutrophils to the liver. Furthermore, hepatic
CXCL1 expression, a well-established chemoattractant of neutrophils
(30,31), was enhanced in the SHP-KO alcohol
group compared with the WT alcohol group. Although the exact
involvement of SHP in immune cell migration and recruitment
mechanisms remain poorly understood, the results of the present
study indicate that the exacerbated acute alcohol-induced liver
injury in SHP-KO mice may be due to increased levels of hepatic
CCL2, CXCL1 and IL-4, and subsequent increases in the infiltration
of NKT cells and neutrophils in the liver. Further studies are
required in order to identify the immune cell-specific roles of
SHP.
SHP is a physiologically important nuclear receptor
that acts as a transcriptional inhibitor of genes associated with
various biological pathways (14,15,32).
In the present study, alcohol-induced liver injury, as measured by
AST and ALT levels in the plasma, was most severe in SHP-KO mice
compared with WT mice. These results indicate that SHP may be
associated with acute alcohol-induced liver injury. ADH1 and CYP2E1
are responsible for metabolizing alcohol to acetaldehyde.
Acetaldehyde is subsequently converted to acetate by ALDH2 in the
liver (33). These alcohol
metabolites were previously reported to induce liver injury via ROS
production and endotoxin influx from the gut due to altered
intestinal permeability (34,35).
Furthermore, alcohol consumption induced lipid accumulation in the
liver by upregulating lipid biosynthesis and lipid oxidation
(36,37). Numerous studies have demonstrated
that lipid accumulation and peroxidation were suppressed in the
livers of chronic alcohol administered SHP-KO mice compared with WT
mice (15,18). Furthermore, these studies indicated
that SHP may be involved in alcohol-associated liver injury via the
regulation of liver-associated lipid metabolism. In the current
acute alcohol binge administration study, the degrees of hepatic
lipid accumulation and lipid peroxidation did not vary between WT
and SHP-KO mice. Previous studies have demonstrated that chronic
and acute alcohol-induced steatosis may share a common mechanism
responsible for the upregulation of lipid biosynthesis and lipid
oxidation, which involves sterol regulatory element-binding protein
1 in fatty acid synthesis and AMP-dependent protein kinase,
peroxisome proliferator-activated receptor (PPAR)α and adiponectin
in lipid oxidation (33,38–40).
To the best of our knowledge, although the underlying mechanisms
responsible for the phenotypic differences resulting from
alcohol-associated hepatic steatosis are largely undetermined,
based on the inconsistent degrees of hepatic lipid accumulation
between acute and chronic alcohol administered SHP-KO mice, it may
be hypothesized that the role of SHP in hepatic lipid metabolism
may vary between acute and chronic alcohol ingestion conditions. In
addition, different alcohol ingestion patterns induced opposite
results under similar conditions; acute and chronic alcohol
consumption led to opposing results in monocytes by regulating the
levels of IL-1 receptor-associated kinase-monocyte (IRAK-M)
(41). Acute alcohol treatment was
demonstrated to inhibit IRAK-M and induce hyporesponsiveness of
monocytes to lipopolysaccharide (LPS) via inhibition of LPS-induced
nuclear factor-κB (NF-κB) and extracellular signal-regulated kinase
(ERK) activation, while chronic alcohol suppressed IRAK-M
expression and activated NF-κB and ERK kinases, which sensitize
monocytes to the effects of LPS (41). Furthermore, alcohol consumption in
patients with alcoholism suffering from acetaminophen poisoning has
been reported to act as protective factor following acute alcohol
abuse, while chronic alcohol abuse represents a risk factor for
acetaminophen-induced hepatotoxicity (42). However, the effects of varied
alcohol ingestion patterns are not clearly defined and thus require
further investigation. Although up to 90% of individuals who
regularly consume large quantities of alcohol may develop
steatosis, only a minority of such individuals develop ASH and
10–20% of regular binge drinkers eventually develop cirrhosis
(43). In the present study, and
according to the results of a previous study (18), SHP-KO mice were susceptible to
acute binge alcohol-induced liver injury and were resistant to
chronic alcohol-induced liver injury. The results of the present
study indicate that SHP may be a candidate genetic regulatory
factor for idiosyncratic reactions of alcohol in humans; however,
further studies are required in order to determine the exact
involvement of SHP in pathogenesis associated with acute and
chronic alcohol consumption conditions.
In the current study, TNF-α expression was
upregulated in both of the alcohol groups, however, the expression
level was significantly higher in SHP-KO alcohol mice compared with
WT alcohol mice. Previous studies have demonstrated that TNF-α is
implicated in acute and chronic alcohol-induced hepatic injury
(19,44) and is considered to be an
apoptosis-mediating cytokine. In the present study, apoptosis was
observed in the WT and SHP-KO alcohol groups; however, the
differences between WT and SHP-KO alcohol groups were not
significant (data not shown). Based on these results, it may be
hypothesized that the enhanced hepatic TNF-α expression in the
present study may have induced an increased severity of liver
damage; however, the damage was insufficient for the induction of
apoptosis following alcohol binge exposure in SHP-KO mice.
Although alcohol consumption is associated with
substantial economic and health problems (1), there is currently no effective
therapy for alcohol-associated liver disease. Cessation of alcohol
consumption, nutrient therapy and antioxidant administration
represent currently available treatments for alcohol-associated
liver disease. Furthermore, nuclear receptors, such as PPARα and
farnesoid X receptor (FXR), have previously been suggested as be
potential therapeutic targets for alcohol-associated liver disease.
Activation of PPARα by an agonist was reported to inhibit hepatic
steatosis, inflammation and fibrosis in alcohol-associated liver
disease via regulation of fatty acid transport and oxidation, the
inflammatory response and hepatic stellate cells activation and
pro-fibrotic genes such as transforming growth factor β 1, while
FXR agonist treatment inhibited lipid accumulation and suppressed
oxidative stress induced by ethanol ingestion (45–47).
In the present study, SHP depletion aggravated acute
alcohol-induced liver injury. Therefore, SHP overexpression may
inhibit liver injury following alcohol ingestion; however, further
studies are required in order to verify this hypothesis. SHP may
serve as a therapeutic target for the treatment of
alcohol-associated liver disease.
In conclusion, to the best of our knowledge, the
present study is the first to demonstrate that SHP depletion
enhanced the migration of NKT cells and neutrophils to the liver,
and aggravated liver injury following acute binge alcohol
administration. The results of the present study indicate that SHP
may be involved in protective mechanisms associated with acute
binge alcohol-induced liver injury, and modulation of SHP may be a
novel therapeutic agent for the treatment of acute binge
drinking-induced liver injury.
Acknowledgements
The authors thank I-B Lee, Y-K Choi, Y J Seo and J-H
Choi for their technical assistance (Laboratory Animal Resource
Center, Korea Research Institute of Bioscience and Biotechnology,
Daejeon, Korea).
Funding
The present study was supported by a grant from the
National Research Foundation of Korea and the Korean government
(grant no. 2016R1A2A1A05004858), and the KRIBB Research Initiative
Program of the Republic of Korea.
Availability of data and materials
The analyzed data sets generated during the study
are available from the corresponding author on reasonable
request.
Authors' contributions
M-J Go, Y-H Kim and C-H Lee designed the experiments
and the study. M-J Go, D-H Choi and Y-H Kim collected data and did
experiments for the study. M-J Go, Y-H Kim and J-R Noh analyzed the
data. J H Hwang, K-S Kim, J-S Lee and C-H Lee contributed to
critical revisions of the text.
Ethics approval and consent to
participate
All experimental procedures were approved by the
Institutional Animal Care and Use Committee of the KRIBB (Daejeon,
Korea) and were performed in accordance with the National
Institutes of Health Guide for the Care and Use of Laboratory
Animals.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
WHO, . Global status report on alcohol and
health 2014. http://www.who.int/substance_abuse/publications/global_alcohol_report/en/2014.
|
|
2
|
National Institute of Alcohol Abuse
Alcoholism, . NIAAA council approves definition of binge drinking.
NIAAA Newsletter. 3:2004.https://pubs.niaaa.nih.gov/publications/Newsletter/winter2004/Newsletter_Number3.pdf
|
|
3
|
Goslawski M, Piano MR, Bian JT, Church EC,
Szczurek M and Phillips SA: Binge drinking impairs vascular
function in young adults. J Am Coll Cardiol. 62:201–207. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Llerena S, Arias-Loste MT, Puente A,
Cabezas J, Crespo J and Fábrega E: Binge drinking: Burden of liver
disease and beyond. World J Hepatol. 7:2703–2715. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Swain MG: Hepatic NKT cells: Friend or
foe? Clin Sci (Lond). 114:457–466. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Gao B, Radaeva S and Park O: Liver natural
killer and natural killer T cells: Immunobiology and emerging roles
in liver diseases. J Leukoc Biol. 86:513–528. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Jiang W, Sun R, Zhou R, Wei H and Tian Z:
TLR-9 activation aggravates concanavalin A-induced hepatitis via
promoting accumulation and activation of liver CD4+ NKT cells. J
Immunol. 182:3768–3774. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Sköld M and Behar SM: Role of
CD1d-restricted NKT cells in microbial immunity. Infect Immun.
71:5447–5455. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Wilson SB and Delovitch TL: Janus-like
role of regulatory iNKT cells in autoimmune disease and tumour
immunity. Nat Rev Immunol. 3:211–222. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Godfrey DI, Stankovic S and Baxter AG:
Raising the NKT cell family. Nat Immunol. 11:197–206. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Mathews S, Feng D, Maricic I, Ju C, Kumar
V and Gao B: Invariant natural killer T cells contribute to
chronic-plus-binge ethanol-mediated liver injury by promoting
hepatic neutrophil infiltration. Cell Mol Immunol. 13:206–216.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Maricic I, Sheng H, Marrero I, Seki E,
Kisseleva T, Chaturvedi S, Molle N, Mathews SA, Gao B and Kumar V:
Inhibition of type I natural killer T cells by retinoids or
following sulfatide-mediated activation of type II natural killer T
cells attenuates alcoholic liver disease in mice. Hepatology.
61:1357–1369. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wang H, Feng D, Park O, Yin S and Gao B:
Invariant NKT cell activation induces neutrophil accumulation and
hepatitis: Opposite regulation by IL-4 and IFN-γ. Hepatology.
58:1474–1485. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lee YS, Chanda D, Sim J, Park YY and Choi
HS: Structure and function of the atypical orphan nuclear receptor
small heterodimer partner. Int Rev Cytol. 261:117–158. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Tsuchiya H, da Costa KA, Lee S, Renga B,
Jaeschke H, Yang Z, Orena SJ, Goedken MJ, Zhang Y, Kong B, et al:
Interactions between nuclear receptor SHP and FOXA1 maintain
oscillatory homocysteine homeostasis in mice. Gastroenterology.
148:1012–1023.e14. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Seol W, Choi HS and Moore DD: An orphan
nuclear hormone receptor that lacks a DNA binding domain and
heterodimerizes with other receptors. Science. 272:1336–1339. 1996.
View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Lee HK, Lee YK, Park SH, Kim YS, Park SH,
Lee JW, Kwon HB, Soh J, Moore DD and Choi HS: Structure and
expression of the orphan nuclear receptor SHP gene. J Biol Chem.
273:14398–14402. 1998. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Park JE, Lee M, Mifflin R and Lee YK:
Enhanced ethanol catabolism in orphan nuclear receptor SHP-null
mice. Am J Physiol Gastrointest Liver Physiol. 310:G799–G807. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Zhou Z, Wang L, Song Z, Lambert JC,
McClain CJ and Kang YJ: A critical involvement of oxidative stress
in acute alcohol-induced hepatic TNF-alpha production. Am J Pathol.
163:1137–1146. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chen P, Wang Z, Zeng L, Yang X, Wang S,
Dong W, Jia A, Cai C and Zhang J: A novel soluble beta-glucan
salecan protects against acute alcohol-induced hepatotoxicity in
mice. Biosci Biotechnol Biochem. 75:1990–1993. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Council NR: Guide for the care and use of
laboratory animals. The National Academies Press; Washington, DC:
2010, PubMed/NCBI
|
|
22
|
Bligh EG and Dyer WJ: A rapid method of
total lipid extraction and purification. Can J Biochem Physiol.
37:911–917. 1959. View
Article : Google Scholar : PubMed/NCBI
|
|
23
|
Ohkawa H, Ohishi N and Yagi K: Assay for
lipid peroxides in animal tissues by thiobarbituric acid reaction.
Anal Biochem. 95:351–358. 1979. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Kawakami K, Kinjo Y, Uezu K, Yara S,
Miyagi K, Koguchi Y, Nakayama T, Taniguchi M and Saito A: Monocyte
chemoattractant protein-1-dependent increase of V alpha 14 NKT
cells in lungs and their roles in Th1 response and host defense in
cryptococcal infection. J Immunol. 167:6525–6532. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Metelitsa LS, Wu HW, Wang H, Yang Y, Warsi
Z, Asgharzadeh S, Groshen S, Wilson SB and Seeger RC: Natural
killer T cells infiltrate neuroblastomas expressing the chemokine
CCL2. J Exp Med. 199:1213–1221. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Biburger M and Tiegs G:
Alpha-galactosylceramide-induced liver injury in mice is mediated
by TNF-alpha but independent of Kupffer cells. J Immunol.
175:1540–1550. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Brennan PJ, Brigl M and Brenner MB:
Invariant natural killer T cells: An innate activation scheme
linked to diverse effector functions. Nat Rev Immunol. 13:101–117.
2013. View
Article : Google Scholar : PubMed/NCBI
|
|
29
|
Cui K, Yan G, Xu C, Chen Y, Wang J, Zhou
R, Bai L, Lian Z, Wei H, Sun R and Tian Z: Invariant NKT cells
promote alcohol-induced steatohepatitis through interleukin-1β in
mice. J Hepatol. 62:1311–1318. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Sawant KV, Xu R, Cox R, Hawkins H, Sbrana
E, Kolli D, Garofalo RP and Rajarathnam K: Chemokine CXCL1-mediated
neutrophil trafficking in the Lung: Role of CXCR2 activation. J
Innate Immun. 7:647–658. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sawant KV, Poluri KM, Dutta AK, Sepuru KM,
Troshkina A, Garofalo RP and Rajarathnam K: Chemokine CXCL1
mediated neutrophil recruitment: Role of glycosaminoglycan
interactions. Sci Rep. 6:331232016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Wu N, Kim KH, Zhou Y, Lee JM, Kettner NM,
Mamrosh JL, Choi S, Fu L and Moore DD: Small Heterodimer Partner
(NR0B2) coordinates nutrient signaling and the circadian clock in
mice. Mol Endocrinol. 30:988–995. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Ceni E, Mello T and Galli A: Pathogenesis
of alcoholic liver disease: Role of oxidative metabolism. World J
Gastroenterol. 20:17756–17772. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Rao R: Endotoxemia and gut barrier
dysfunction in alcoholic liver disease. Hepatology. 50:638–644.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Szabo G: Gut-liver axis in alcoholic liver
disease. Gastroenterology. 148:30–36. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Baraona E and Lieber CS: Effects of
ethanol on lipid metabolism. J Lipid Res. 20:289–315.
1979.PubMed/NCBI
|
|
37
|
Wang HJ, Gao B, Zakhari S and Nagy LE:
Inflammation in alcoholic liver disease. Ann Rev Nutr. 32:343–368.
2012. View Article : Google Scholar
|
|
38
|
Ji C, Chan C and Kaplowitz N: Predominant
role of sterol response element binding proteins (SREBP) lipogenic
pathways in hepatic steatosis in the murine intragastric ethanol
feeding model. J Hepatol. 45:717–724. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yin HQ, Kim M, Kim JH, Kong G, Kang KS,
Kim HL, Yoon BI, Lee MO and Lee BH: Differential gene expression
and lipid metabolism in fatty liver induced by acute ethanol
treatment in mice. Toxicol Appl Pharmacol. 223:225–233. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Qiu P, Li X, Kong DS, Li HZ, Niu CC and
Pan SH: Herbal sgr formula prevents acute ethanol-induced liver
steatosis via inhibition of lipogenesis and enhancement fatty acid
oxidation in mice. Evid Based Complement Alternat Med.
2015:6135842015. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Mandrekar P, Bala S, Catalano D, Kodys K
and Szabo G: The opposite effects of acu and chronic alcohol on
lipopolysaccharide-induced inflammation are linked to IRAK-M in
human monocytes. J Immunol. 183:1320–1327. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Schmidt LE, Dalhoff K and Poulsen HE:
Acute versus chronic alcohol consumption in acetaminophen-induced
hepatotoxicity. Hepatology. 35:876–882. 2002. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mathurin P and Bataller R: Trends in the
management and burden of alcoholic liver disease. J Hepatol. 62 1
Suppl:S38–S46. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Rodríguez-Rodríguez E, González-Reimers E,
Santolaria-Fernández F, Milena-Abril A, Rodríguez-Moreno F,
Oramas-Rodríguez J and Martínez-Riera A: Cytokine levels in acute
alcoholic hepatitis: A sequential study. Drug Alcohol Depend.
39:23–27. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Lívero FA and Acco A: Molecular basis of
alcoholic fatty liver disease: From incidence to treatment. Hepatol
Res. 46:111–123. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Nan YM, Wang RQ and Fu N: Peroxisome
proliferator-activated receptor α, a potential therapeutic target
for alcoholic liver disease. World J Gastroenterol. 20:8055–8060.
2014. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Lívero FA, Stolf AM, Dreifuss AA,
Bastos-Pereira AL, Chicorski R, de Oliveira LG, de Souza CE,
Fabossi IA, Rabitto IS, Gremski LH, et al: The FXR agonist 6ECDCA
reduces hepatic steatosis and oxidative stress induced by ethanol
and low-protein diet in mice. Chem Biol Interact. 217:19–27. 2014.
View Article : Google Scholar : PubMed/NCBI
|