Introduction
Post-resuscitation myocardial dysfunction is an
important morbidity of post-resuscitation syndrome. The mortality
rate following cardiopulmonary resuscitation (CPR) in
out-of-hospital cardiac arrest (CA) is ~75%, one-third of which is
thought to be caused by myocardial dysfunction (1). Post-resuscitation myocardial
dysfunction always occurs following return of spontaneous
circulation (ROSC) and has long-term effects. Left ventricular
ejection fractions prior to and following CA are associated with
the prognosis of CA (2). In
addition, one study reported a potentially harmful effect of
epinephrine (EP) on post-ROSC hemodynamics, with a lower cardiac
index in patients that received higher cumulative EP doses during
CPR (3).
Ischemia-reperfusion injury is one of the
pathological mechanisms associated with myocardial dysfunction
following resuscitation (2). This
injury leads to the activation of the systemic and local
renin-angiotensin (Ang) system (RAS), subsequently leading to
increased serum levels of renin and Ang II, and activation of Ang
II receptors (4). Furthermore, EP
stimulates the β1 receptor of glomerular parietal cells, promotes
renin secretion and induces the upregulation of serum renin and Ang
II levels (5). The binding of Ang
II to Ang II receptor type 1 (AT1R) causes myocardial cell damage
and heart failure (6). However,
Ang II receptor type 2 (AT2R) counteracts the canonical signaling
of RAS that is mediated by AT1R. It has been previously
demonstrated that AT2R inhibits ligand-induced AT1R signaling in a
protein kinase C-dependent manner (7), and the AT2R agonist C21 significantly
improves post-myocardial infarct cardiac function (8).
Erythropoietin (EPO) has been previously reported to
protect cardiac function and lessen post-resuscitation myocardial
dysfunction (5,9–11).
Specifically, 5,000 U/kg EPO reduced the area of myocardial
necrosis and restored cardiac function to an almost normal level
within a week (11). In addition,
1,200 U/kg EPO reduced post-resuscitation myocardial stunning of
ventricular fibrillation in pig models of ROSC (9). Similar results were obtained in rats
models of asphyxia-induced CA (5)
and hemorrhagic shock (10).
Furthermore, additional studies have indicated that EPO treatment
reduces the activity of the RAS (12–14),
and also reduces inflammation and oxidative stress (12).
Based on the established roles of the RAS in
myocardial ischemia-reperfusion injury following CA, it may be
hypothesized that EPO may alleviate post-resuscitation myocardial
dysfunction by regulating the RAS, which, to the best of our
knowledge, has not been previously investigated. Therefore, the
present study aimed to elucidate whether EPO improves
post-resuscitation myocardial dysfunction and how it affects the
RAS. The cardiac function, serum renin and Ang II levels, and the
myocardial expression of renin, AT1R and AT2R, in rats following CA
resuscitation with or without EPO pretreatment (administered when
CPR began) were compared. The effect of EP addition on CA
resuscitation was also observed.
Materials and methods
Experimental design and grouping
The present study was approved by the Animal Ethics
Committee of Guizhou Provincial People's Hospital (approval no.
2012026; Guiyang, China) and performed in accordance with the
International Ethical Guidelines for Animal Research (15). Male (n=75) and female (n=75)
Sprague-Dawley rats (8 weeks old) with a body weight of 350–450 g
were provided by the Experimental Animal Center of Guiyang College
of Traditional Chinese Medicine (License no. SCXK Chongqing
2012-0005; Guiyang, China). Animals were housed at a constant
temperature of 22°C, humidity of 50%, and CO2
concentration of 0.04%. Rats received food and water ad
libidum the night before the experiment, but were fasted and
water-deprived during the experiments. Light was kept constant
during the experiment.
Sprague-Dawley rats were randomly divided into the
following five groups: Sham-operated group (sham group, n=30); CA
resuscitation group (vehicle group, n=30); CA resuscitation + EP
group (EP group, n=30); CA resuscitation + EPO group (EPO group,
n=30); and CA resuscitation + EP + EPO group (EP + EPO group,
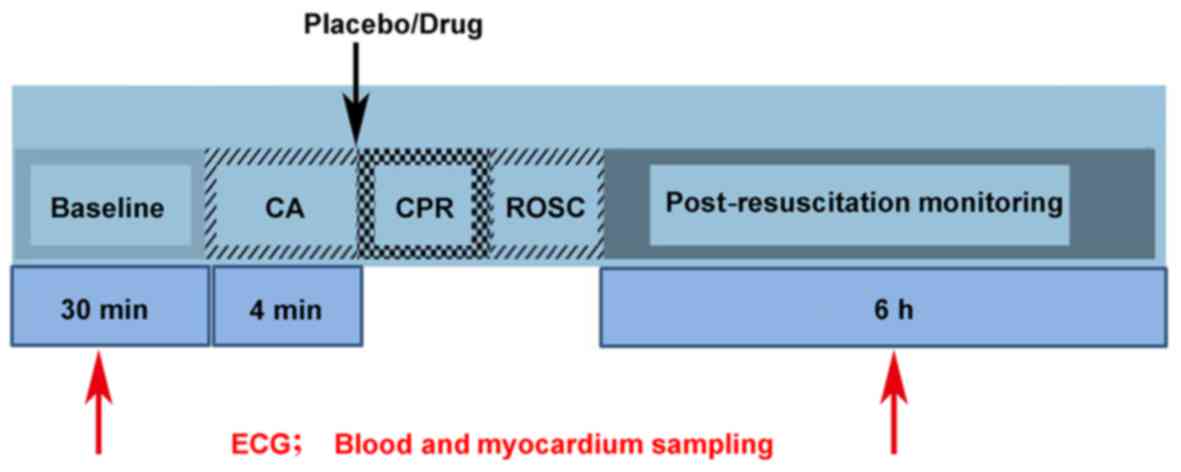
n=30). The process of CA resuscitation included CA, CPR and ROSC. A
diagram indicating the process is presented in Fig. 1. Electrocardiograms were obtained
at baseline (prior to surgery) and at 0, 1, 2, 4 and 6 h after ROSC
(n=6 per group for each time-point; however, the same batch of
animals were used for electrocardiogram measurements at 0 and 1 h
after ROSC). Samples of blood and cardiac tissues were obtained
from each group at baseline and at 2, 4 and 6 h after ROSC (n=6 per
group for each time-point).
CA resuscitation and cardiac function
monitoring
The night before the operation, the rats were
fasted, except for water, and intraperitoneally injected with 45
mg/kg chloral hydrate for anesthesia, 10 mg/kg of which was
administered every hour to maintain its effect.
Initially, low-volume (or lung protective)
mechanical ventilation was performed for 30 min (i.e. baseline,
prior to surgery) to ensure hemodynamic stability in all five
groups and to avoid lung injury (16). Animals with a mean arterial
pressure (MAP) <80 mmHg, those with excessive surgical bleeding
or those with a surgical time >40 min were excluded.
CA was caused by asphyxiation, which was induced by
turning off the ventilator and by clamping the endotracheal tube.
Bradycardia, hypotension and cardiac failure with an MAP <10
mmHg that occurred shortly after asphyxiation were defined as CA
(17). At 4 min after CA,
ventilation was restored when chest compression was performed using
a Modified Brunswick Animal Heart-Lung Resuscitator (Landswick
medical technology, Co. Ltd., Guangzhou, China). The chest
compression rate was 200/min with a depth half the chest
anteroposterior diameter; the pressing and relaxation times were
similar. Chest compression was adjusted to the coronary perfusion
pressure, which is >30 mmHg. ROSC was characterized by a
continuous MAP of 60 mmHg (17).
Resuscitation was terminated if ROSC did not appear after 6 min of
continuous chest compressions. The sham operation group underwent:
anesthesia, endotracheal intubation and mechanical ventilation.
During the whole operation, an incandescent lamp was employed to
maintain the rectal temperature at 36.5–37.5°C. A 14-gauge cannula
was used for percutaneous tracheal intubation under a mechanical
ventilation of 80 breaths/min, a tidal volume of 0.65 ml/100 g and
a fractional inspired oxygen of 100% (ALC-V9 Animal Ventilator;
Alcott Biotech Co., Ltd., Shanghai, China). A PE-50 tube filled
with normal saline was inserted into the right femoral artery to
measure the arterial blood pressure, and another PE-50 tube filled
with normal saline was inserted into the left ventricle through the
right common carotid artery for confirmation of left ventricular
pressure waveform. Another saline-filled PE-50 tube was inserted
into the right internal jugular vein for fluid infusion. The
central venous pressure was recorded. An BL-420S biological data
acquisition and analysis system (Techman Software Co., Ltd.,
Chengdu, China; en.tme.com.cn/products_detail/productId=45.html) was
connected to monitor electrocardiograms, monitoring the heart rate
(HR), MAP, left ventricular systolic pressure (LVSP), left
ventricular end-diastolic pressure (LVEDP), maximal ascending rate
of left ventricular pressure during left ventricular isovolumic
contraction (+LVdP/dt max, which represents the left ventricular
systolic function) and maximal descending rate of left ventricular
pressure during left ventricular isovolumic relaxation (-LVdP/dt
max, which represents the left ventricular diastolic function).
Drug administration
The drugs were administrated when CPR began.
Following CA, the vehicle and sham groups received intravenous
injection of 2 ml/kg normal saline as a control, the EP group was
intravenously injected with 0.02 mg/kg (dosing volume, 2 ml/kg) EP
(Fuzhou Neptunus Fuyao Pharmaceutical Co., Ltd., Fuzhou, China),
the EPO group was intravenously injected with 5,000 U/kg
recombinant human (rh)EPO (5)
(dosing volume, 2 ml/kg; Shandong Ahua Biochemical Co., Ltd.,
Liaocheng, China) and the EP + EPO group was intravenously injected
with 5,000 U/kg rhEPO (dosing volume, 1 ml/kg) + 0.02 mg/kg EP
(dosing volume, 1 ml/kg).
The randomization code for drug information was
delivered in a sealed envelope to the operator. The assessors were
blind to grouping and drug information. Experimental failure was
reported to the investigator, who then broke the code to determine
which group the animal belonged to.
Sampling of blood and cardiac
tissues
Blood samples (6 ml) were extracted from the jugular
vein and centrifuged at 3,000 × g, for 10 min at 4°C to obtain the
supernatant serum. The rats were sacrificed by an intravenous
injection of a lethal dose (250 mg/kg) of pentobarbital. The chest
was opened and the heart was isolated from the aortic root, rinsed
with phosphate-buffered saline (PBS), and the left and right
ventricles were separated. All samples were immediately stored at
−80°C in a liquid nitrogen tank.
ELISA for serum renin and Ang II
levels
ELISA kits (cat. no. csb-e08702r) were used to
measure the serum levels of renin (CUSABIO Biotechnology Co., Ltd.,
Wuhan, China; sensitivity, 1.17 pg/ml) and Ang II (cat. no.
csb-e0449r; CUSABIO Biotechnology Co., Ltd.; sensitivity, 3.9
mU/ml) in rats. The reagents were placed at room temperature
(20–25°C) for 15–20 min prior to use, and the procedures were
performed in strict accordance with the manufacturer's protocol. To
minimize errors, the samples were tested twice to obtain the mean
average.
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) detection of myocardial renin,
AT1R and AT2R mRNA levels
Myocardial tissues were obtained and TRIzol (Tiangen
Biotech Co., Ltd., Beijing, China) was used to extract the total
RNA, according to the manufacturer's protocol. Gel electrophoresis
was performed to detect the integrity of the total RNA extracted.
PrimeScript RT reagent kit with gDNA Eraser (Takara Bio, Inc.,
Otsu, Japan) was used for RT to produce cDNA at 42°C for 50 min and
at 85°C for 5 min, according to the manufacturer's protocol. SYBR
Premix Ex Taq II (Tli RNase H Plus) and ROX plus (Takara Bio, Inc.)
were used for amplification of cDNA with the following program:
95°C for 30 sec followed by 45 cycles of 95°C for 5 sec and 60°C
for 40 sec. An ABI 7500 Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc., Waltham, MA, USA) was used for
qPCR. Primers used for qPCR are presented in Table I and the 2−∆∆Cq method
(18) was used to analyze
quantification data and normalize the myocardial mRNA expression
levels of renin, AT1R and AT2R to GAPDH mRNA expression. This
experiment was performed three times.
 | Table I.Primers for reverse
transcription-quantitative polymerase chain reaction analysis of
AT1R, AT2R, renin and GAPDH mRNA expression in rats. |
Table I.
Primers for reverse
transcription-quantitative polymerase chain reaction analysis of
AT1R, AT2R, renin and GAPDH mRNA expression in rats.
|
| Primer
sequence |
|
|---|
|
|
|
|
|---|
| Gene | Forward | Reverse | Fragment length,
bp |
|---|
| AT1R |
5-GTGTTCCTGCTCACGTGTCT-3 |
5-GATGATGCAGGTGACTTTGG-3 | 108 |
| AT2R |
5-GAAGCTCCGCAGTGTGTTTA-3 |
5-TGGCTAGGCTGATTACATGC-3 | 147 |
| Renin |
5-CTGGGAGGCAGTGACCCTCAACATTACCAG-3 |
5-GAGAGCCAGTATGCACAGGTCATCGTTCCT-3 | 372 |
| GAPDH |
5-ACAACTTTGGCATTGTGGAA-3 |
5-GATGCAGGGATGATGTTCTG-3 | 133 |
Western blot analysis of AT1R and AT2R
in the myocardium
Briefly, myocardial tissue was washed three times
with PBS buffer and lysed in radioimmunoprecipitation assay lysis
buffer (Beyotime Institute of Biotechnology, Haimen, China) on ice.
Total protein (50 µg/sample), which was determined using a BCA
assay (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany), was
extracted and separated by 10% SDS-PAGE and transferred onto
polyvinylidene difluoride membranes (EMD Millipore, Billerica, MA,
USA). The membranes were blocked with 5% non-fat milk in
TBS-Tween-20 (10 mM Tris, pH 7.4, 150 mM NaCl and 0.1% Tween-20) at
room temperature for 1 h. The primary antibodies against AT1R (cat.
no. ab18801; 1:500), AT2R (cat. no. ab92445; 1:500) and GAPDH (cat.
no. ab181602; 1:5,000), and horseradish peroxidase-conjugated
anti-rabbit (cat. no. cw0234; 1:200; www.cwbiotech.com.) and anti-rat (cat. no. cw0108;
1:200; www.cwbiotech.com) secondary antibodies,
were added into a 4 ml centrifuge tube. The sealed membrane was
transferred into the box filled with the antibody mixture,
incubated overnight at 4°C and washed three times with a tris
Buffered saline with Tween-20 washing solution. For enhanced
chemiluminescence (ECL Western Blotting Substrate; cat. no. 32019;
Thermo Fisher Scientific, Inc.) and exposure development, the
reaction time was 5 min, and exposure time was 10 min for AT1R, 1
min for AT2R and 5 min for GAPDH. BandScan 4.3 software was used to
scan and determine the gray value of the target protein (Gel-Pro
analyzer 4.0 (Supplier: Media Cybernetics, USA). The expression of
each protein was compared with that of GAPDH and the control group
was compared with the other groups.
Statistical analysis
Data were analyzed using SPSS 17.0 (SPSS, Inc.,
Chicago, IL, USA). Data are presented as the mean ± standard
deviation. Comparisons among multiple subgroups were performed
using two-way analysis of variance, followed by
Student-Newman-Keuls test. P<0.05 was considered to indicate a
statistically significant difference.
Results
Operation parameters among the
groups
No significant differences were observed among the
groups (P>0.05) in terms of weight, chloral hydrate and EP
usage, asphyxia time, CPR time, ROSC time and resuscitation success
rate (Table II). The overall rate
of successful resuscitation from CA in the present study was 51%
(120/236).
| Table II.Comparison of experimental parameters
in each group. |
Table II.
Comparison of experimental parameters
in each group.
|
| Group |
|---|
|
|
|
|---|
| Parameter | Sham | Vehicle | EP | EPO + EP | EPO | P-value |
|---|
| Weight, g | 326.33±10.33 | 321.23±8.89 | 320.07±8.39 |
326.2±11.15 | 322.63±9.80 | 0.635 |
| Chloral hydrate
usage, mg |
122.3±10.92 |
121.17±10.37 | 120.80±8.08 | 124.97±7.62 | 121.03±9.68 | 0.287 |
| EP usage, µg | NA | NA |
5.55±0.59 |
5.90±0.61 | NA | 0.442 |
| Asphyxia time,
sec | NA |
223.50±13.60 |
222.1±11.00 |
221.5±11.5 |
217.77±12.52 | 0.744 |
| CPR time, sec | NA |
296.2±50.10 |
294.83±49.30 |
269.73±48.14 |
290.77±58.42 | 0.589 |
| ROSC time, sec | NA |
536.2±50.10 |
534.83±49.31 |
509.73±48.14 |
530.77±58.42 | 0.526 |
| Resuscitation
success rate, % (proportion) | NA | 41.67 (30/72) | 56.60 (30/53) | 58.82 (30/51) | 50.00 (30/60) | 0.498 |
EPO alleviates post-resuscitation
myocardial dysfunction
Alterations in cardiac function are presented in
Table III. No differences were
observed in the HR among the five groups. Compared with the sham
group, the vehicle group exhibited a lower MAP, LVSP, +LVdP/dt max
and -LVdP/dt max, and higher LVEDP, following ROSC (P<0.05;
Table III), indicating that CA
resuscitation led to myocardial dysfunction. Compared with the
vehicle group, the only significant differences in the EP group
were a higher MAP at 0 h, a higher +LVdP/dt max at 0 and 1 h after
ROSC, and a higher -LVdP/dt max at 0 h after ROSC (P<0.05;
Table III); however, +LVdP/dt
max and -LVdP/dt max values remained lower compared with the sham
group (P<0.05; Table III).
These results indicated that EP addition may not exacerbate the
myocardial dysfunction upon CA resuscitation, and it appeared to
provide protection, although only at the beginning.
| Table III.Alterations in cardiac function
indices in each group. |
Table III.
Alterations in cardiac function
indices in each group.
|
| Group |
|---|
|
|
|
|---|
| Parameter | Sham | Vehicle | EP | EP + EPO | EPO |
|---|
| HR, beats/min |
|
|
|
|
|
|
Baseline |
334.67±34.90 |
334.00±38.38 |
331.17±31.77 |
331.83±33.65 |
330.67±33.49 |
| 0
h |
330.67±37.06 |
348.00±35.35 |
416.17±32.63 |
420.17±33.88 |
369.33±33.24 |
| 1
h |
328.83±33.13 |
322.67±33.76 |
371.50±31.44 |
361.50±34.50 |
336.00±32.15 |
| 2
h |
319.50±32.52 |
311.17±42.98 |
329.83±35.56 |
322.00±35.13 |
325.33±29.86 |
| 4
h |
318.14±27.98 |
320.00±46.75 |
325.17±37.67 |
323.50±26.08 |
324.67±33.60 |
| 6
h |
313.70±28.53 |
322.83±33.70 |
312.67±34.48 |
313.67±33.73 |
312.17±30.84 |
| MAP, mmHg |
|
|
|
|
|
|
Baseline |
98.17±8.07 |
102.33±8.46 |
102.17±6.26 |
100.50±8.30 |
98.50±5.65 |
| 0
h |
94.17±6.91 |
63.67±6.74a |
127.17±8.33a,b |
124.00±6.45a,b |
62.50±3.21a |
| 1
h |
93.67±5.47 |
65.17±3.49a |
69.83±5.67a |
77.33±9.24a,b |
78.83±8.42a,b |
| 2
h |
95.50±4.51 |
66.17±7.47a |
67.17±3.66a |
70.33±8.36a |
82.33±6.28a,b |
| 4
h |
95.17±3.60 |
68.50±4.09a |
69.83±5.88a |
71.17±7.73a |
86.83±8.18a,b |
| 6
h |
95.67±8.24 |
70.00±7.77a |
72.50±5.39a |
72.33±3.98a |
90.33±6.53b,c |
| LVSP, mmHg |
|
|
|
|
|
|
Baseline |
124.17±14.61 |
122.83±9.37 |
125.50±11.53 |
127.67±8.38 |
122.83±12.61 |
| 0
h |
125.00±13.25 |
93.83±6.37a |
103.50±13.13a |
104.50±9.46a |
96.67±13.46a |
| 1
h |
127.17±7.65 |
84.83±5.38a |
85.17±15.94a |
91.17±9.45a |
93.50±11.78a |
| 2
h |
128.17±4.54 |
80.33±4.37a |
81.67±13.57a |
94.33±10.03a,b |
100.67±10.58a–c |
| 4
h |
128.67±8.66 |
74.67±3.98a |
73.83±12.80a |
95.50±12.32a,b |
107.33±11.04a–c |
| 6
h |
126.10±10.51 |
69.50±5.24a |
67.83±15.92a |
99.00±13.43a,b |
107.67±8.89a–c |
| LVEDP, mmHg |
|
|
|
|
|
|
Baseline |
5.30±0.68 |
5.12±0.76 |
5.14±0.69 |
5.12±0.71 |
5.17±0.85 |
| 0
h |
5.29±0.85 |
6.21±0.62 |
6.20±0.69 |
6.26±0.40 |
6.20±0.59 |
| 1
h |
5.26±0.68 |
6.56±0.83a |
6.89±0.86a |
6.65±0.61a |
6.37±0.56a |
| 2
h |
5.24±0.63 |
7.23±0.81a |
7.59±0.65a |
7.21±0.66a |
6.61±0.54a |
| 4
h |
5.25±0.62 |
8.82±0.67a |
9.31±1.04a |
8.23±0.86a |
7.02±0.50a,c |
| 6
h |
5.25±0.66 |
10.96±0.65a |
11.26±1.11a |
9.30±0.75a–c |
7.75±0.56a–c |
| +LVdP/dt max,
mmHg/sec |
|
|
|
|
|
|
Baseline |
9,064.33±672.53 |
9,291.00±501.27 |
9,162.17±337.50 |
9,200.83±471.40 |
9,173.17±390.54 |
| 0
h |
9,169.00±607.90 |
4,067.00±524.70a |
8,088.00±441.94a,b |
8,173.50±456.83a,b |
4,114.50±513.62a |
| 1
h |
9,172.17±483.60 |
3,792.17±520.68a |
4,949.67±298.39a,b |
5,831.17±445.76a,b |
4,643.00±650.51a,b |
| 2
h |
9,183.00±434.36 |
3,614.83±448.41a |
3,888.83±235.35a |
4,657.50±314.39a–c |
5,030.83±610.52a–c |
| 4
h |
9,080.33±397.61 |
3,422.83±422.84a |
3,352.67±233.10a |
4,220.83±349.03a–c |
5,241.33±565.12a–c |
| 6
h |
9,129.85±436.42 |
3,258.33±381.57a |
2,961.50±229.66a |
4,227.00±426.64a–c |
5,392.67±574.19a–c |
| -LVdP/dt max,
mmHg/sec |
|
|
|
|
|
|
Baseline |
6,218.67±339.28 |
6,260.17±313.37 |
6,098.67±423.04 |
6,418.67±430.05 |
6,423.00±422.07 |
| 0
h |
6,122.00±382.32 |
3,491.33±470.50a |
4,146.67±345.36a,b |
4,205.17±577.13a,b |
3,801.17±411.00a |
| 1
h |
6,193.50±330.42 |
3,304.00±377.34a |
3,615.67±458.70a |
3,687.00±438.40a |
3,779.00±585.96a |
| 2
h |
6,183.00±434.36 |
2,991.17±390.47a |
2,884.33±413.70a |
3,331.33±578.78a |
3,480.33±643.92a |
| 4
h |
6,161.33±441.93 |
2,656.83±308.36a |
2,412.50±355.95a |
2,937.50±598.08a |
3,224.83±567.76a |
| 6
h |
6,139.09±490.82 |
2,507.50±407.66a |
2,104.00±177.22a |
2,630.67±686.80a |
2,980.67±628.92a |
Compared with the vehicle group, the EPO group
generally exhibited a higher MAP, LVSP and +LVdP/dt max, and lower
LVEDP, following ROSC (P<0.05; Table III), indicating that EPO
ameliorated the myocardial dysfunction upon CA resuscitation.
However, the long-term effects of EPO were compromised when
combined with EP addition; EPO pretreatment almost restored MAP to
a normal level at 6 h after ROSC (P>0.05, EPO group vs. sham
group; Table III), but almost no
beneficial effect on MAP was observed in the EP + EPO group
compared with the EP or vehicle groups between 2 and 6 h after ROSC
(P>0.05; Table III).
EPO did not reduce the increased serum
levels of renin and Ang II post-resuscitation
Serum renin and Ang II levels prior to surgery were
similar in all five groups (P>0.05; Tables IV and V, respectively); however, levels were
elevated upon CA resuscitation (P<0.01; vehicle group vs. sham
group; Tables IV and V). Furthermore, EP addition further
augmented the serum renin and Ang II levels (P<0.01, EP group
vs. vehicle group; Tables IV and
V, respectively). EPO treatment
did not reduce the increased serum levels of renin and Ang II upon
CA resuscitation (P>0.05; EPO group vs. vehicle group; Tables IV and V, respectively), but EPO did prevent
further increases in serum renin levels induced by EP addition
(P>0.05; EP + EPO group vs. vehicle group; Table IV).
| Table IV.Alterations in the serum levels of
renin in each group. |
Table IV.
Alterations in the serum levels of
renin in each group.
|
| Group |
|---|
|
|
|
|---|
| Time-point | Sham | Vehicle | EP | EPO | EPO + EP |
|---|
| Baseline |
2.64±0.22 |
2.66±0.26 |
2.23±0.48 |
2.54±0.32 |
2.61±0.28 |
| 2 h |
2.62±0.25 |
56.77±6.12a |
81.67±9.85a,b |
52.42±6.50a,c |
59.62±6.66a,c |
| 4 h |
2.67±0.23 |
53.63±4.32a |
73.90±5.65a,b |
46.10±5.96a,c |
59.77±10.91a,c |
| 6 h |
2.91±0.31 |
46.25±2.94a |
58.97±5.07a,b |
31.81±5.00a,c |
52.76±3.34a |
| Table V.Alterations in the serum levels of
Ang II in each group. |
Table V.
Alterations in the serum levels of
Ang II in each group.
|
| Group |
|---|
|
|
|
|---|
| Time-point | Sham | Vehicle | EP | EPO + EP | EPO |
|---|
| Baseline |
6.72±2.43 |
6.68±2.22 |
6.38±3.88 |
6.73±2.35 |
6.69±3.22 |
| 2 h |
6.88±2.25 |
10.13±3.56a |
20.18±4.94a,b |
14.61±2.14a,b |
10.16±2.82a,c |
| 4 h |
6.32±2.42 |
10.12±5.47a |
18.25±6.10a,b |
18.03±4.09a,b |
8.99±1.39a,c |
| 6 h |
5.93±2.33 |
10.09±4.00a |
16.42±4.62a,b |
15.25±4.71a,b |
8.23±3.15a,c |
EPO did not sustain the downregulation
of renin and AT1R expression in myocardial tissues
post-resuscitation
EPO suppressed the increase of renin mRNA in
myocardial tissues until 4 h after ROSC (both P<0.05, EPO group
vs. vehicle group, 2 and 4 h following ROSC; Fig. 2), but the regulation was not
maintained (P>0.05, EPO group vs. vehicle group, 6 h following
ROSC; Fig. 2). Compared with the
sham group, the mRNA levels of renin (Fig. 2) and AT1R (Fig. 3A), in addition to the protein
expression of AT1R (Fig. 3B), were
increased in myocardial tissues of the vehicle group (all
P<0.05), indicating that CA resuscitation may activate the
canonical RAS signaling of renin-AngII-AT1R.
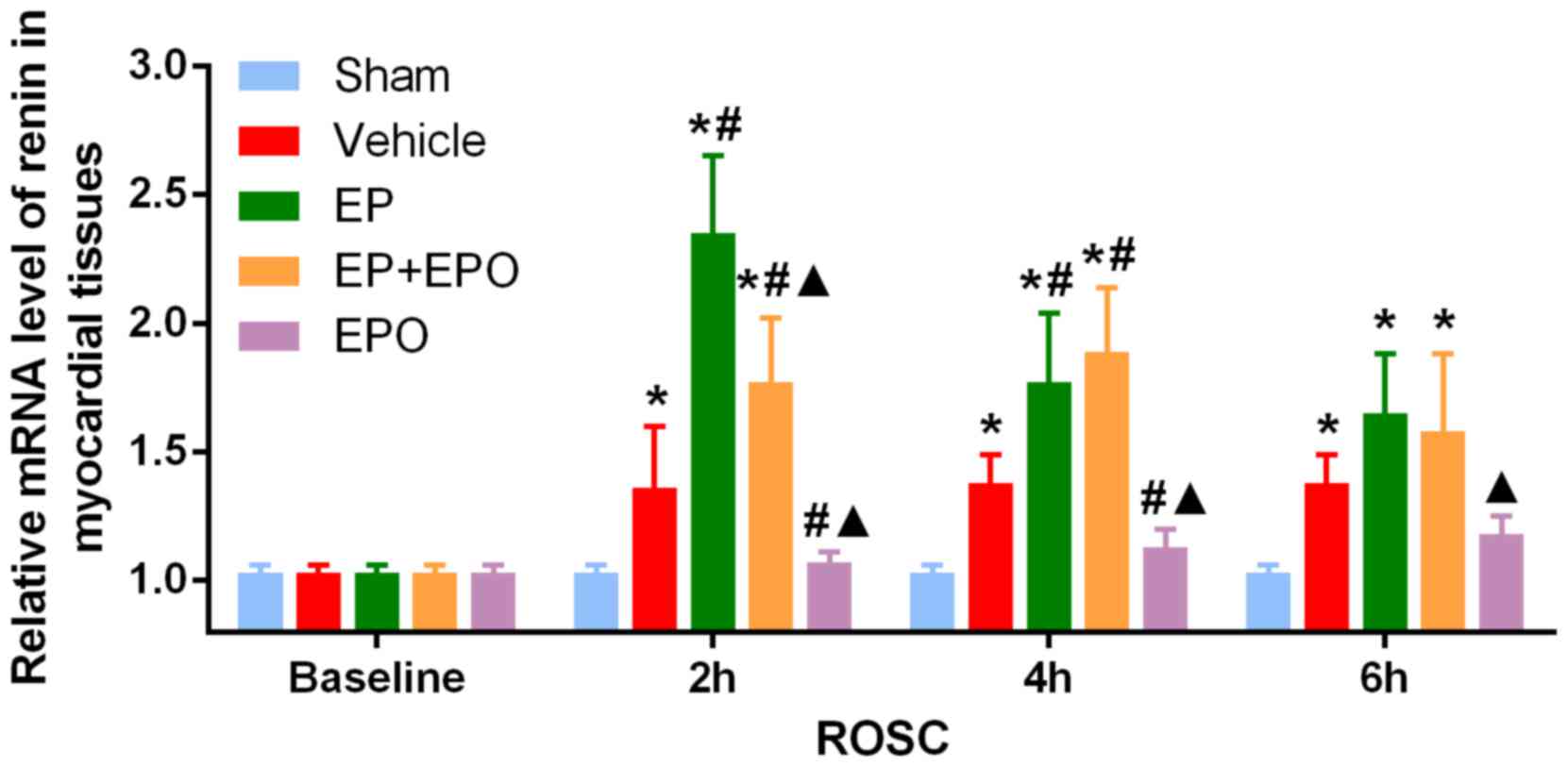 | Figure 2.Alterations in the mRNA level of
renin in myocardial tissues. Sprague-Dawley rats were randomly
divided into five groups with n=30 per group: Sham-operated group,
CA resuscitation group, CA resuscitation + EP group, CA
resuscitation + EPO group and CA resuscitation + EP + EPO group.
Myocardial tissues were obtained from the five groups at baseline
(prior to surgery) and at 2, 4 and 6 h after ROSC, with n=6 per
group at each time-point. The mRNA level of renin in myocardial
tissues was measured by reverse transcription-quantitative
polymerase chain reaction. Comparisons among multiple subgroups
were performed using two-way analysis of variance. *P<0.05 vs.
sham group; #P<0.05 vs. vehicle group;
▲P<0.05 vs. EP group. CA, cardiac arrest; EP,
epinephrine; EPO, erythropoietin; ROSC, return of spontaneous
circulation; Sham group, sham-operated; vehicle group, CA
resuscitation; EP group, CA resuscitation + EP; EPO group, CA
resuscitation + EPO; EP + EPO group, CA resuscitation + EP +
EPO. |
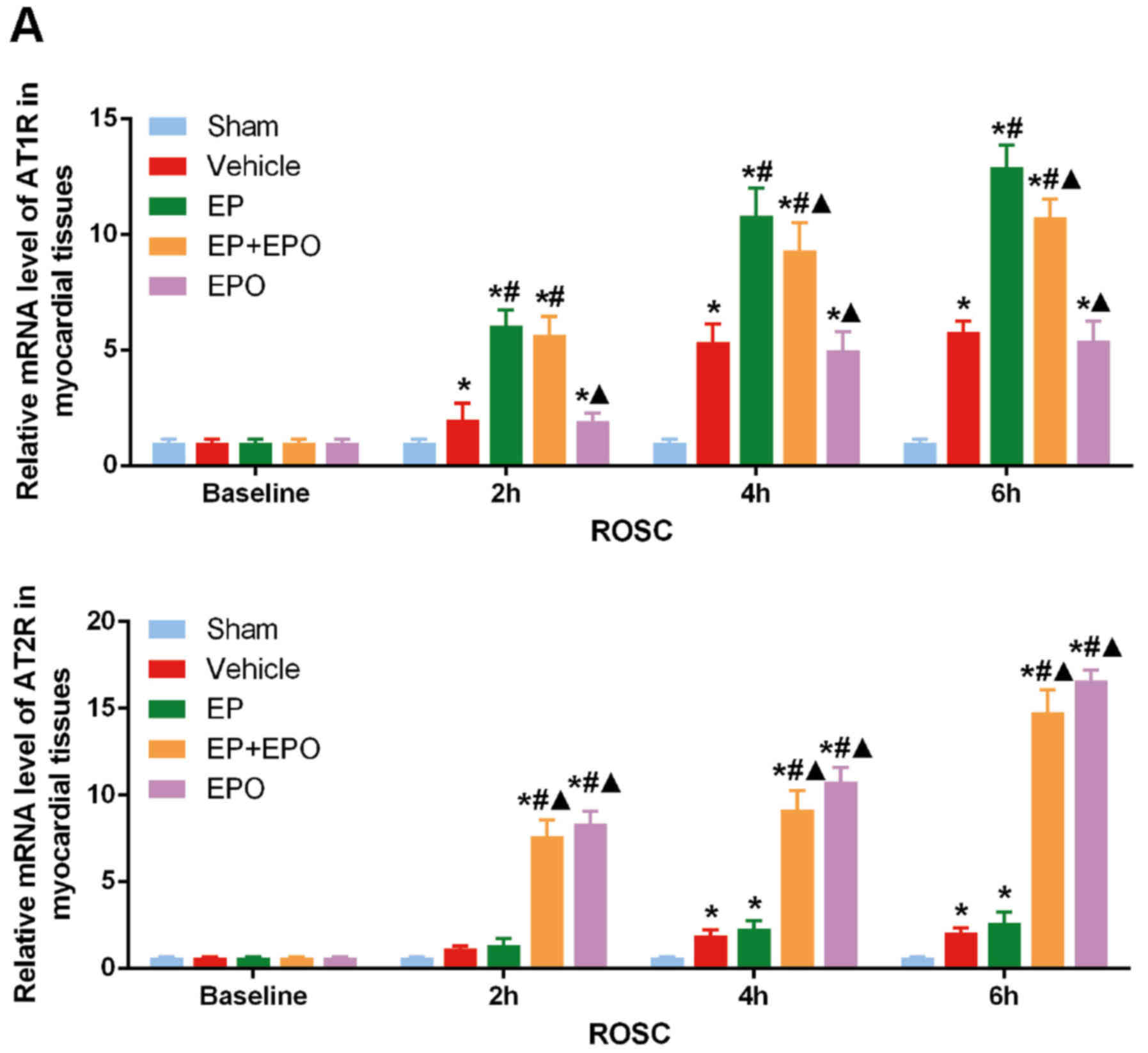 | Figure 3.Alterations in the mRNA and protein
levels of AT1R and AT2R in myocardial tissues. Sprague-Dawley rats
were randomly divided into five groups with n=30 per group:
Sham-operated group, CA resuscitation group, CA resuscitation + EP
group, CA resuscitation + EPO group and CA resuscitation + EP + EPO
group. Myocardial tissues were obtained from the five groups at
baseline (prior to surgery) and at 2, 4 and 6 h after ROSC, with
n=6 per group at each time-point. (A) mRNA levels of AT1R and AT2R
in myocardial tissues at different time-points. (B) Protein
expressions of AT1R and AT2R in myocardial tissues at different
time-points. Representative western blot bands are presented.
Comparisons among multiple subgroups were performed using two-way
analysis of variance. *P<0.05 vs. sham group;
#P<0.05 vs. vehicle group; ▲P<0.05 vs.
EP group. AT1R, angiotensin II receptor type 1; AT2R, angiotensin
II receptor type 2; CA, cardiac arrest; EP, epinephrine; EPO,
erythropoietin; ROSC, return of spontaneous circulation; Sham
group, sham-operated; vehicle group, CA resuscitation; EP group, CA
resuscitation + EP; EPO group, CA resuscitation + EPO; EP + EPO
group, CA resuscitation + EP + EPO. |
The mRNA levels of AT1R were not altered in the EPO
group compared with the vehicle group (Fig. 3A). EPO suppressed the increase of
AT1R protein in myocardial tissues to an extent at 2 h after ROSC
(P<0.05, EPO group vs. vehicle group; Fig. 3B), however, the regulation was not
maintained between 4 and 6 h after ROSC (P>0.05, EPO group vs.
vehicle group; Fig. 3B). The
myocardial expression of AT1R mRNA was reduced in the EP + EPO
group compared with the EP group at 2 h following ROSC (P<0.05;
Fig. 3A), but the difference
disappeared later (both P>0.05, EP+EPO group vs. EP group, 4 and
6 h after ROSC; Fig. 3A). The
myocardial expression of AT1R protein was reduced in the EP + EPO
group compared with the EP group at 4 and 6 h following ROSC
(P<0.05; Fig. 3B), although the
levels in the EP + EPO group remained significantly higher compared
with the vehicle group (P<0.05; Fig. 3B). EP addition further increased
the myocardial expression of renin and AT1R (both P<0.05, EP
group vs. vehicle group; Figs. 2
and 3).
Overall, the results indicated that the effects of
EPO in alleviating post-resuscitation myocardial dysfunction
(Table III) may be mediated
through mechanisms other than direct inhibition of the renin-Ang
II-AT1R signaling pathway.
EPO enhances AT2R expression in
myocardial tissues post-resuscitation
The myocardial expression of AT2R mRNA and protein
was increased upon CA resuscitation (P<0.05, vehicle group vs.
sham group; Fig. 3). The addition
of EP enhanced this increase in AT2R expression significantly at
the protein level at 2 and 4 h after ROSC (P<0.05, EP group vs.
vehicle group; Fig. 3B).
Furthermore, EPO pretreatment in the EPO and EP + EPO groups led to
even higher myocardial AT2R mRNA and protein levels (P<0.05, EPO
group vs. EP and vehicle groups, and EP + EPO group vs. EP and
vehicle groups; Fig. 3).
Taken together, the results indicate that the
activated renin-AngII-AT1R signaling upon CA resuscitation may
contribute to myocardial dysfunction.
Based on the results of the present study, it may be
preliminarily inferred that the mechanisms underlying the effects
of EPO in alleviating post-resuscitation myocardial dysfunction may
be associated with the increase of AT2R expression at mRNA and
protein levels, which may counteract the canonical signaling
mediated by AT1R.
Discussion
Following CA, the interruption of blood flow and
reperfusion injury following resuscitation may lead to intractable
low blood pressure and recurrent malignant ventricular arrhythmia,
causing complex pathophysiological alterations in major organs.
This condition is termed post-resuscitation syndrome and has an
incidence of 30–70% in heart disease, and is also one of the most
important reasons for early mortality following resuscitation
(19,20).
The RAS is thought to have an important role in
post-resuscitation syndrome. This system regulates blood pressure
and consists of endocrine, autocrine and paracrine factors
(8,21). RAS activation, both systemic and
local, is an important step in chronic myocardial remodeling and
one of the decisive factors in the prognosis of myocardial
infarction (22). Furthermore,
clinical trials have demonstrated that RAS inhibitors reduced the
30-day all-cause rehospitalization rate in patients that suffered
heart failure (23). In RAS, Ang I
is converted into Ang II by angiotensin-converting enzyme (ACE).
When Ang II binds to AT1R in the myocardium, the Ang II-AT1R axis
acts as a detrimental effector, causing myocardial cell damage and
development of heart failure in the following manner: Development
of myocardial hypertrophy and increased production of reactive
oxygen free radicals leads to cell mitochondrial damage and
myocardial cell apoptosis (21,24);
promotion of the expression of proinflammatory transcription
factors, nuclear factor-κB and interleukin-6 in cells, which
subsequently promotes inflammatory reactions; through the action of
NADPH oxidase, ultimately increasing the production of reactive
oxygen free radicals, which causes damage to cell mitochondria,
myocardial hypertrophy, and myocardial fibrosis and dysfunction
(25); and through the
phosphatidylinositol 3-kinase/Akt pathway, wherein Akt
phosphorylation is reduced and cardiac hypertrophy and
cardiomyocyte autophagy are promoted, leading to heart failure
(26). Strohmenger et al
(27) demonstrated that
administration of the Ang II antagonist telmisartan during the
port-resuscitation phase in pigs improved myocardial contractility.
Wang et al (4) also
reported that following CA, the use of sildenafil inhibited the
ACE-Ang II-AT1R axis, which reduced myocardial ischemia-reperfusion
injury. In addition, Kaschina et al (28) revealed that indirect stimulation of
AT2R reduced myocardial hypertrophy and fibrosis following
myocardial infarction, thereby improving cardiac function, however,
this was accompanied by hypotension. Furthermore, Xu et al
(29) demonstrated that high AT2R
expression in myocardial cells inhibited myocardial oxidative
stress, reduced myocardial hypertrophy and myocardial fibrosis,
inhibited ventricular remodeling, and improved cardiac function.
Taken together, the results of these studies indicated that
counteracting the Ang II-AT1R axis may have an important role in
ameliorating post-resuscitation myocardial dysfunction, as
supported by an additional previous study (30).
In the present study, lower MAP, LVSP, +LVdP/dt max
and -LVdP/dt max, and higher LVEDP, were observed in rats
post-resuscitation, compared with the sham group (Table III). The serum levels of renin
and Ang II were elevated upon CA resuscitation, as was the
myocardial expression of renin and AT1R, compared with the sham
group. These results indicated that CPR following CA may activate
renin-Ang II-AT1R signaling, which may contribute to myocardial
dysfunction, consistent with previous studies (4,26,28).
EPO has been reported to exert protective effects on
various tissues and organs (31).
The role of EPO in cardiac function protection and reducing
post-resuscitation myocardial dysfunction is well-established
(5,9,11,14,32,33);
however, to the best of our knowledge, its modulation of the RAS
has not been previously investigated. Given the established roles
of the RAS in myocardial ischemia-reperfusion injury following CA,
it may be hypothesized that EPO may alleviate post-resuscitation
myocardial dysfunction by regulating the RAS. The results of the
present study indicated that EPO alleviated post-resuscitation
myocardial dysfunction, as higher MAP, LVSP and +LVdP/dt max, and
lower LVEDP, was observed following ROSC in the EP group, compared
with the vehicle group, which was consistent with previous findings
(32,33).
Within the RAS, EPO does not only mediate AT1R, but
also has effects on AT2R, nitric oxide levels, NAPDH oxidase 4 and
heme oxygenase-1, in addition to other complex signaling pathways.
Patients with congenital AT1R resistance were reported to exhibit
increased EPO secretion (34). The
present study demonstrated that the most noticeable effect of EPO
treatment was the upregulation of AT2R expression in myocardial
tissues, rather than the downregulation of myocardial AT1R or
suppression of renin and Ang II levels, which were not significant
in most cases, with regard to its regulation of RAS upon CA
resuscitation. Therefore, these results indicate that the effects
of EPO in alleviating post-resuscitation myocardial dysfunction may
primarily rely on AT2R activation, which counteracts the canonical
signaling mediated by AT1R. However, AT2R phosphorylation requires
investigation in future studies to reach a firm conclusion
concerning AT2R activation by EPO following CA. In addition,
mechanistic studies using antagonists or knockdown strategies are
required to confirm whether AT2R mediates the effects of EPO
following CA resuscitation. Furthermore, as high AT2R expression in
myocardial cells has been reported to inhibit oxidative stress
(29), the measurement of
oxidative stress markers in myocardial tissues is also required in
future studies. Finally, it should be noted that the long-term
effects of EPO were somewhat compromised when combined with EP
addition (Table III), however
the myocardial expression of AT2R protein was almost identical
between the EPO and EP + EPO groups, which indicates that, in
addition to enhancing the AT2R expression, additional mechanisms
may be involved in mediating the effects of EPO upon CA
resuscitation, which should also be investigated.
EP, which is the resuscitation drug that is
currently recommended in the guidelines, exerts strong β- and
α-adrenergic activation effects (35), which may aggravate myocardial
ischemic injury (36). Therefore,
the present study was designed to include a group with EP addition
as EP may be involved in the mechanisms underlying
post-resuscitation myocardial dysfunction. However, according to
the results of the present study, EP addition did not exacerbate
the myocardial dysfunction post-resuscitation, although it did
increase the production/expression of renin, Ang II and AT1R. One
potential explanation for this discrepancy is that the maximum
effect of renin-Ang II-AT1R signaling is already reached upon CA
resuscitation alone. In addition, the increase in AT2R protein
expression induced by EP addition at 2 and 4 h after ROSC may also
contribute to the counteractive regulation. However, these
hypotheses require further investigation.
The primary limitation of the present study is that
study planning did not include a sham + EPO group. Such a group
would clarify the effect of EPO alone on myocardial dysfunction
upon CA resuscitation. Additional confirmation of EPO effects with
this control group should be performed in future studies.
In conclusion, the results of the current study
indicate that EPO may ameliorate the myocardial dysfunction upon CA
resuscitation, and the underlying mechanisms may include
counteracting the canonical AT1R-mediated RAS signaling as a result
of enhanced AT2R expression, rather than direct inhibition of the
renin-Ang II-AT1R signaling pathway. Further investigation is
required to determine these mechanisms in detail.
Acknowledgements
The present study was supported by the Project of
the Department of Science and Technology, Guizhou province. [grant
nos. (2012)014 and (2016)1411].
References
|
1
|
Jentzer JC, Chonde MD and Dezfulian C:
Myocardial dysfunction and shock after cardiac arrest. Biomed Res
Int. 2015:3147962015. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Bougouin W and Cariou A: Management of
postcardiac arrest myocardial dysfunction. Curr Opin Crit Care.
19:195–201. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rivers EP, Wortsman J, Rady MY, Blake HC,
McGeorge FT and Buderer NM: The effect of the total cumulative
epinephrine dose administered during human CPR on hemodynamic,
oxygen transport, and utilization variables in the
postresuscitation period. Chest. 106:1499–1507. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Wang G, Zhang Q, Yuan W, Wu J and Li C:
Sildenafil protects against myocardial ischemia-reperfusion injury
following cardiac arrest in a porcine model: Possible role of the
renin-angiotensin system. Int J Mol Sci. 16:27015–27031. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Huang CH, Hsu CY, Chen HW, Tsai MS, Cheng
HJ, Chang CH, Lee YT and Chen WJ: Erythropoietin improves the
postresuscitation myocardial dysfunction and survival in the
asphyxia-induced cardiac arrest model. Shock. 28:53–58. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Mollace V, Gliozzi M, Capuano A and Rossi
F: Modulation of RAAS-natriuretic peptides in the treatment of HF:
Old guys and newcomers. Int J Cardiol. 226:126–131. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Inuzuka T, Fujioka Y, Tsuda M, Fujioka M,
Satoh AO, Horiuchi K, Nishide S, Nanbo A, Tanaka S and Ohba Y:
Attenuation of ligand-induced activation of angiotensin II type 1
receptor signaling by the type 2 receptor via protein kinase C. Sci
Rep. 6:216132016. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Kaschina E, Grzesiak A, Li J,
Foryst-Ludwig A, Timm M, Rompe F, Sommerfeld M, Kemnitz UR, Curato
C, Namsolleck P, et al: Angiotensin II type 2 receptor stimulation:
A novel option of therapeutic interference with the
renin-angiotensin system in myocardial infarction? Circulation.
118:2523–2532. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Borovnik-Lesjak V, Whitehouse K, Baetiong
A, Artin B, Radhakrishnan J and Gazmuri RJ: High-dose
erythropoietin during cardiac resuscitation lessens
postresuscitation myocardial stunning in swine. Transl Res.
162:110–121. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Nandra KK, Collino M, Rogazzo M, Fantozzi
R, Patel NS and Thiemermann C: Pharmacological preconditioning with
erythropoietin attenuates the organ injury and dysfunction induced
in a rat model of hemorrhagic shock. Dis Model Mech. 6:701–709.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Sanchis-Gomar F, Garcia-Gimenez JL,
Pareja-Galeano H, Romagnoli M, Perez-Quilis C and Lippi G:
Erythropoietin and the heart: Physiological effects and the
therapeutic perspective. Int J Cardiol. 171:116–125. 2014.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Jie KE, Verhaar MC, Cramer MJ, van der
Putten K, Gaillard CA, Doevendans PA, Koomans HA, Joles JA and
Braam B: Erythropoietin and the cardiorenal syndrome: Cellular
mechanisms on the cardiorenal connectors. Am J Physiol Renal
Physiol. 291:F932–F944. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Rosario R and Epstein M: Relationship
between erythropoietin administration and alterations of
renin-angiotensin-aldosterone. J Renin Angiotensin Aldosterone
Syst. 7:135–138. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Lundby C, Thomsen JJ, Boushel R, Koskolou
M, Warberg J, Calbet JA and Robach P: Erythropoietin treatment
elevates haemoglobin concentration by increasing red cell volume
and depressing plasma volume. J Physiol. 578:309–314. 2007.
View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Cho A and Seok SH: Ethical guidelines for
use of experimental animals in biomedical research. J Bacteriol
Virol. 43:18–26. 2013. View Article : Google Scholar
|
|
16
|
de Prost N, Ricard JD, Saumon G and
Dreyfuss D: Ventilator-induced lung injury: Historical perspectives
and clinical implications. Ann Intensive Care. 1:282011. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Wang P, Yao L, Zhou LL, Liu YS, Chen MD,
Wu HD, Chang RM, Li Y, Zhou MG, Fang XS, et al: Carbon monoxide
improves neurologic outcomes by mitochondrial biogenesis after
global cerebral ischemia induced by cardiac arrest in rats. Int J
Biol Sci. 12:1000–1009. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Mongardon N, Dumas F, Ricome S, Grimaldi
D, Hissem T, Pène F and Cariou A: Postcardiac arrest syndrome: From
immediate resuscitation to long-term outcome. Ann Intensive Care.
1:452011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Lemiale V, Dumas F, Mongardon N,
Giovanetti O, Charpentier J, Chiche JD, Carli P, Mira JP, Nolan J
and Cariou A: Intensive care unit mortality after cardiac arrest:
The relative contribution of shock and brain injury in a large
cohort. Intensive Care Med. 39:1972–1980. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Zablocki D and Sadoshima J: Angiotensin II
and oxidative stress in the failing heart. Antioxid Redox Signal.
19:1095–1109. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Zreikat HH, Harpe SE, Slattum PW, Mays DP,
Essah PA and Cheang KI: Effect of Renin-Angiotensin system
inhibition on cardiovascular events in older hypertensive patients
with metabolic syndrome. Metabolism. 63:392–399. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Sanam K, Bhatia V, Bajaj NS, Gaba S,
Morgan CJ, Fonarow GC, Butler J, Deedwania P, Prabhu SD, Wu WC, et
al: Renin-Angiotensin system inhibition and lower 30-day all-cause
readmission in medicare beneficiaries with heart failure. Am J Med.
129:1067–1073. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Wang X, Yuan B, Dong W, Yang B, Yang Y,
Lin X and Gong G: Humid heat exposure induced oxidative stress and
apoptosis in cardiomyocytes through the angiotensin II signaling
pathway. Heart Vessels. 30:396–405. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Zhang YH: Neuronal nitric oxide synthase
in hypertension - an update. Clin Hypertens. 22:202016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lin L, Liu X, Xu J, Weng L, Ren J, Ge J
and Zou Y: High-density lipoprotein inhibits mechanical
stress-induced cardiomyocyte autophagy and cardiac hypertrophy
through angiotensin II type 1 receptor-mediated PI3K/Akt pathway. J
Cell Mol Med. 19:1929–1938. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Strohmenger HU, Lindner KH, Wienen W and
Vogt J: Effects of the AT1-selective angiotensin II antagonist,
telmisartan, on hemodynamics and ventricular function after
cardiopulmonary resuscitation in pigs. Resuscitation. 35:61–68.
1997. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Kaschina E, Lauer D, Schmerler P, Unger T
and Steckelings UM: AT2 receptors targeting cardiac protection
post-myocardial infarction. Curr Hypertens Rep. 16:4412014.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Xu J, Sun Y, Carretero OA, Zhu L, Harding
P, Shesely EG, Dai X, Rhaleb NE, Peterson E and Yang XP: Effects of
cardiac overexpression of the angiotensin II type 2 receptor on
remodeling and dysfunction in mice post-myocardial infarction.
Hypertension. 63:1251–1259. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Xiao HL, Li CS, Zhao LX, Yang J, Tong N,
An L and Liu QT: Captopril improves postresuscitation hemodynamics
protective against pulmonary embolism by activating the ACE2/Ang-
(1–7)/Mas axis. Naunyn Schmiedebergs Arch Pharmacol. 389:1159–1169.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Sutherland BA, Minnerup J, Balami JS, Arba
F, Buchan AM and Kleinschnitz C: Neuroprotection for ischaemic
stroke: Translation from the bench to the bedside. Int J Stroke.
7:407–418. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Ahmet I, Tae HJ, Juhaszova M, Riordon DR,
Boheler KR, Sollott SJ, Brines M, Cerami A, Lakatta EG and Talan
MI: A small nonerythropoietic helix B surface peptide based upon
erythropoietin structure is cardioprotective against ischemic
myocardial damage. Mol Med. 17:194–200. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Najjar SS, Rao SV, Melloni C, Raman SV,
Povsic TJ, Melton L, Barsness GW, Prather K, Heitner JF, Kilaru R,
et al: Intravenous erythropoietin in patients with ST-segment
elevation myocardial infarction: REVEAL: A randomized controlled
trial. JAMA. 305:1863–1872. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Calò LA, Davis PA, Maiolino G, Pagnin E,
Ravarotto V, Naso E, Carraro G and Naso A: Assessing the
relationship of angiotensin II Type 1 receptors with erythropoietin
in a human model of endogenous angiotensin II Type 1 receptor
antagonism. Cardiorenal Med. 6:16–24. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Ahles A and Engelhardt S: Polymorphic
variants of adrenoceptors: Pharmacology, physiology, and role in
disease. Pharmacol Rev. 66:598–637. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Broadley KJ and Penson PE: The roles of
alpha- and beta-adrenoceptor stimulation in myocardial ischaemia.
Auton Autacoid Pharmacol. 24:87–93. 2004. View Article : Google Scholar : PubMed/NCBI
|