Introduction
Mesenchymal stem cells (MSCs) are multipotent cells
derived from the connective tissues of various organs, they serve
critical roles in tissue regeneration and immunotherapy due to
their self-renewal capacity, multilineage differentiation potential
and immunosuppressive properties (1,2). A
previous study demonstrated that a particular subgroup of MSCs
exists in human dental tissues, including dental pulp stem cells
(DPSCs) and stem cells from human exfoliated deciduous teeth (SHED)
(3).
DPSCs can be separated from enzymatically
disaggregated adult human dental pulp tissue (4). Previous studies have revealed that
DPSCs are highly clonogenic and rapidly proliferative, exhibit
self-renewal and multiple differentiation capabilities (5), and have the potential for use in
tissue regeneration and immunotherapy (6,7).
SHED are derived from exfoliated deciduous teeth in the mixed
dentition stages of children; they are a population of postnatal
stem cells with the ability differentiate into various cell types
(8). SHED are considered to be
immature MSCs that are obtained from naturally exfoliated deciduous
teeth, which may offer a unique, readily accessible and
non-invasive stem cell resource with limited ethical and legal
concerns (9,10). Compared with DPSCs, SHED have been
reported to exhibit a higher proliferation rate, differentiation
potential and increased mineralization capacity in vivo, but
also failed to regenerate a dentin-pulp-like complex (8,11).
Based on the results of previous studies,
researchers are increasingly inclined to select DPSCs and SHED as a
source of stem cells for tissue regeneration and engineering, and
for cellular-based therapies (6,7).
In vitro expansion is necessary to obtain an adequate number
of stem cells for use in tissue engineering and cell therapy
strategies (12). However, MSCs
exhibit certain alterations to their characteristics during
long-term in vitro culture, including spontaneous malignant
transformation, arrested growth, reduced differentiation capacity
and shortened telomeres (13–15).
Additionally, previous studies have indicated that the replicative
senescence of MSCs is a continuous process during which MSCs
exhibit an abnormal morphology, reduced expression of certain
surface markers and arrested proliferation (16). These alterations affect the quality
and efficacy of MSCs and ultimately hinder their practical
application in clinical therapy. Thus, the analysis of SHED and
DPSCs characteristics in in vitro cellular senescence is
vital for basic research and quality control in cellular therapy.
Additionally, the alterations to characteristics and the
differences between SHED and DPSCs in long-term cultivation are yet
to be elucidated.
The present study investigated the effects of
long-term in vitro expansion on the basic properties and
gene expression of SHED and DPSCs. The results revealed that their
capacities for differentiation, proliferation and migration were
decreased at passage 20 (P20), in comparison with passage 4 (P4).
However, senescent SHED and DPSCs retained MSC-specific surface
antigen markers. Furthermore, it was identified that the expression
levels of certain markers associated with senescence were distinct
at relatively advanced passages of SHED and DPSCs. The current
study demonstrated that certain physiological and functional
alterations occur during long-term culture and thus may provide
guidance for the selection of suitable stem cells for therapeutic
application, as well as insight into the various pathways of SHED
and DPSCs in cellular senescence in vitro.
Materials and methods
Cell culture
Healthy and without dental caries exfoliated
deciduous incisors were obtained from 30 children aged 6–7 years
old and human impacted third molars were collected from 30 adults
aged 18–25 years old (sex distribution of both groups, 1:1) from
April 2015 to October 2015, all of whom had provided informed
consent under the approved guidelines set by the Ethics Committee
at the Affiliated Stomatology Hospital of Tongji University
(Shanghai, China). Ethical approval was obtained from the Ethics
Committee at the Affiliated Stomatology Hospital of Tongji
University (approval no. 2015-010). Tooth surfaces were cleaned and
the pulp tissue was gently separated from a remnant or from the
mechanically fractured crown and root, following which the tissues
were rinsed in PBS and minced into fragments of 0.5–1
mm3, which were subsequently uniformly placed in 6-well
plates supplemented with Dulbecco's modified Eagle's medium (DMEM;
cat. no. 11965-092; Gibco; Thermo Fisher Scientific, Inc., Waltham,
MA, USA) containing 10% fetal bovine serum (FBS; cat. no. 16000044;
Gibco; Thermo Fisher Scientific, Inc.), and 100 U/ml penicillin,
100 µg/ml streptomycin (cat. no. 15140-122; Gibco; Thermo Fisher
Scientific, Inc.), as previously reported (17), and incubated at 37°C in 5%
CO2. The culture medium was replaced every 3 days and
the cells were observed every 24 h using an inverted phase-contrast
microscope, magnification, ×100 (Nikon Corp., Tokyo, Japan). When
90% confluence was achieved, cells were harvested using 0.25%
trypsin (Gibco; Thermo Fisher Scientific, Inc.), subcultured and
conventionally cryopreserved in liquid nitrogen. Cells were
cultured to P20 for subsequent experiments and each was repeated at
least three times. Stem cells were collected from at least three
different donors.
Flow cytometry analysis of cell
surface antigens
SHED and DPSCs were harvested at P4 and P20, washed
twice with PBS, passed through a 40 µm cell strainer (BD Falcon; BD
Biosciences, Franklin Lakes, NJ, USA) and then resuspended in PBS
at a concentration of 1×107 cells/ml. The cells were
subsequently incubated in the dark for 30 min on ice with various
antibodies including: Anti-CD90-fluorescein isothiocyanate (FITC),
anti-CD105-peridinin-chlorophyll-protein complex-cyanine 5.5,
anti-CD73-allophycocyanin, anti-CD44-phycoerythrin (PE),
anti-CD34-PE, anti-CD11b-PE, anti-CD19-PE, anti-CD45-PE and
anti-human leukocyte antigen D-related (HLA-DR)-PE (all 1:50
dilution), according to the manufacturer's protocols. All of these
antibodies were included in a Human MSC Analysis kit that was
purchased from BD Biosciences (cat. no. 562245). Non-labeled cells
and isotypes were used as negative controls. After being washed
twice with PBS, the cells were analyzed with a flow cytometer (BD
Biosciences) and a total of 10,000 events were acquired for each
sample and data analysis was performed using FlowJo software
version 7.6 (BD Biosciences).
Cell proliferation assay
P4 and P20 SHED and DPSCs were seeded into 96-well
plates at a density of 3×103 cells/well with DMEM
containing 10% FBS and 100 U/ml penicillin, 100 µg/ml streptomycin,
incubated at 37°C in 5% CO2. Cell proliferation was
determined using a Cell Counting kit-8 assay performed at days 0,
1, 3 and 5, according to the manufacturer's protocol (cat. no.
ZP328; Beijing Zoman Biotechnology Co., Ltd., Beijing, China).
In vitro differentiation
To evaluate cell differentiation, P4/20 SHED and
DPSCs were seeded at a density of 4.2×103
cells/cm2 (for osteogenic differentiation) and
2.1×104 cells/cm2 (for adipogenic
differentiation) in 12/6-well plates with α-minimum essential
medium (α-MEM; R&D Systems, Inc., Minneapolis, MN, USA)
containing 10% FBS and 100 U/ml penicillin, 100 µg/ml streptomycin.
The cells were subsequently incubated overnight at 37°C and 5%
CO2. After reaching the 70% confluence (for osteogenic
differentiation) and 100% confluence (for adipogenic
differentiation), the medium was replaced with osteogenic or
adipogenic differentiation medium to induce osteogenesis and
adipogenesis. The osteogenic differentiation medium consistuted
α-MEM supplemented with 10% FBS and 5% osteogenic supplement, and
the adipogenic differentiation medium consisting of α-MEM
supplemented with 10% FBS and 1% adipogenic supplement (cat. no.
SC006; R&D Systems, Inc.). The differentiation medium was
replaced every 3 days. The cells were cultured for 14 days to
induce adipogenic differentiation or for 21 days to induce
osteogenic differentiation, following which the osteocytes,
adipocytes and control group cells were harvested. The harvested
cells were suspended in 1 ml TRI Reagent®
(Sigma-Aldrich; Merck KGaA, Darmstadt, Germany) for the detection
of mRNA expression, and the cells were fixed in 4% paraformaldehyde
for 30 min at room temperature, washed twice in PBS and stained
with Alizarin Red or Oil Red O at room temperature for 30 min
(Sigma-Aldrich; Merck KGaA), siained cells were observed using an
inverted phase-contrast microscope (magnification, ×100; Nikon
Corporation, Tokyo, Japan), to assess the formation of mineralized
nodules and the accumulation of lipid vacuoles, respectively.
Stained Oil Red O was quantified by dissolving in 100% isopropanol
and the optical density (OD) of the solution at 500 nm was measured
by a microplate reader (Thermo Fisher Scientific, Inc.) (18). Stained Alizarin Red was extracted
using 10% cetylpyridinium chloride buffer and the OD value of the
solution was measured at 550 nm (19).
Reverse transcription-quantitative PCR
(RT-qPCR)
Total RNA was extracted from cells using TRI
Reagent. cDNA was prepared using a TransScript First-Strand cDNA
Synthesis SuperMix kit (Beijing TransGen Biotech Co., Ltd.,
Beijing, China). The RT reaction was set at an initial denaturation
step at 42°C for 15 min, followed by 85°C for 5 sec. Following
first strand cDNA synthesis, qPCR was performed on an ABI Prism
7500 Sequence Detection System (Applied Biosystems; Thermo Fisher
Scientific, Inc.) with a SuperReal PreMix Plus (SYBR-Green) kit
(Tiangen Biotech Co., Ltd., Beijing, China). The reaction mixture
was heated to 95°C for 15 min, followed by amplification that
consisted of 40 cycles of denaturation at 95°C for 10 sec, and
annealing and extension at 60°C for 32 sec, according to the
manufacturer's protocol. The relative alterations in gene
expression were calculated using the 2−∆∆Cq method
(20) with β-actin as a reference
gene. The primer sequences that were utilized are listed in
Table I.
 | Table I.Primer sequences used in reverse
transcription-quantitative polymerase chain reaction analysis. |
Table I.
Primer sequences used in reverse
transcription-quantitative polymerase chain reaction analysis.
| Gene name | Forward primer
sequences | Reverse primer
sequences |
|---|
| ALP |
5′-CCCAAGAATAAAACTGATGTG-3′ |
5′-CTTCCAGGTGTCAACGAG-3′ |
| Runx2 |
5′-GAATGCCTCTGCTGTTATG-3′ |
5′-ACTCTTGCCTCGTCCACT-3′ |
| PPARγ2 |
5′-GGTTGACACAGAGATGCC-3′ |
5′-TGGAGTAGAAATGCTGGAGA-3′ |
| p16Ink4a |
5′-ACTTCAGGGGTGCCACATTC-3′ |
5′-CGACCCTGTCCCTCAAATCC-3′ |
| p21 |
5′-GCGACTGTGATGCGCTAATG-3′ |
5′-GAAGGTAGAGCTTGGGCAGG-3′ |
| p53 |
5′-TGCTCAAGACTGGCGCTAAA-3′ |
5′-CAATCCAGGGAAGCGTGTCA-3′ |
| β-actin |
5′-CTACCTCATGAAGATCCTCACCGA-3′ |
5′-TTCTCCTTAATGTCACGCACGATT-3′ |
Cell migration assay
A migration assay was conducted using Transwell
inserts with an 8 µm pore size (Corning Incorporated, Corning, NY,
USA) according to the manufacturer's protocol. Cells were seeded at
a density of 2×104 cells/well into the upper chambers
with serum-free DMEM while the lower chambers contained DMEM
supplemented with 10% FBS. Chambers were subsequently incubated for
24 h at 37°C and 5% CO2. Migrated cells on the lower
membranes were fixed with anhydrous methanol for 30 min, wash the
cells twice with PBS and stained with 0.1% crystal violet for 30
min at room temperature. They were then imaged using an upright
fluorescent microscope (Nikon Corporation), magnification, ×40 and
counted in five independent, randomly selected fields of view.
Cell apoptosis assay
Apoptotic cells were detected using an Annexin
V-FITC Apoptosis Detection kit (cat. no. ZP327; Beijing Zoman
Biotechnology Co., Ltd.), according to the manufacturer's protocol.
Cells were harvested and resuspended at a concentration of
1×106 cells/ml, the cell suspension (500 µl) was
centrifuged at 426 × g for 5 min at room temperature, washed twice
with cold PBS, then resuspended in 500 µl binding buffer, and
incubated with FITC-conjugated Annexin V (5 µl) and propidium
iodide solution (10 µl) for 15 min at room temperature in the dark,
followed by flow cytometer analysis and data analysis was performed
using FlowJo software version 7.6 (BD Biosciences).
Senescence-associated β-galactosidase
(SA-β-gal) assay
Cellular senescence was assessed via SA-β-gal
staining (21), which was
performed using the Senescence Cells Histochemical Staining kit
(cat. no. CS0030; Sigma-Aldrich; Merck KGaA), according to the
manufacturer's protocol. Cells were seeded into 12-well plates at a
density of 2×105 cells/well and cultured in DMEM
containing 10% FBS and 100 U/ml penicillin, 100 µg/ml streptomycin.
Positive staining was evaluated following overnight incubation at
37°C in a CO2-free atmosphere. The number of
blue-stained cells and the total number of cells were counted from
five distinct fields under a phase-contrast microscope and
subsequently calculate the percentage of blue-stained cells
(senescent cells) in the total number of counted cells.
Western blot analysis
SHED and DPSCs were collected and lysed with RIPA
lysis buffer containing phosphatase and protease inhibitor cocktail
(Beyotime Institute of Biotechnology, Beijing, China). Protein
concentrations were determined by the Bicinchoninic Acid protein
assay kit (Pierce; Thermo Fisher Scientific, Inc.). Total proteins
(20 µg) were separated using 10% SDS-PAGE and transferred to
nitrocellulose membranes. Membranes were subsequently blocked with
5% skimmed milk powder in PBS-Tween-20 (PBST; 0.1% Tween-20) for 2
h at room temperature and gently agitated with the following
primary antibodies overnight at 4°C: Rabbit
anti-p16Ink4a (cat. no. 10883-1-AP; dilution, 1:1,000;
ProteinTech Group, Inc., Chicago, IL, USA), rabbit anti-p21 (cat.
no. 10355-1-AP; dilution, 1:1,000; ProteinTech Group, Inc.), rabbit
anti-p53 (cat. no. 10442-1-AP; dilution, 1:2,000; ProteinTech
Group, Inc.) and rabbit anti-GAPDH (cat. no. G9545; dilution,
1:10,000; Sigma-Aldrich; Merck KGaA). Subsequently, the membranes
were washed in PBST and incubated with goat anti-rabbit horseradish
peroxidase-conjugated secondary antibodies (cat. no. 31460;
dilution, 1:10,000; Invitrogen; Thermo Fisher Scientific, Inc.) for
1 h at room temperature. Proteins were detected using an ECL
detection system (Pierce; Thermo Fisher Scientific, Inc.).
Statistical analysis
All the results were performed at least 3 times and
data are presented as the mean ± standard error of the mean. All
statistical analyses were conducted using SPSS software version
20.0 (IBM Corp., Armonk, NY, USA). Comparisons of normally
distributed data were performed using one-way analysis of variance
followed by Tukey's post-hoc test (for multiple comparisons) or
Student's t-tests (for two samples). Group comparisons of skewed
distributed data were analyzed using the Kruskal-Wallis H test and
variations of statistical significance between groups were further
subjected to post-hoc pairwise analysis by applying
Dunn's-Bonferroni tests. P<0.05 was considered to indicate a
statistically significant difference.
Results
Morphology and long-term growth
kinetics of SHED and DPSCs
Following isolation, the majority of SHED and DPSCs
exhibited a spindle shape, small size and low granularity during
continued culture to P4 (Fig. 1A).
Long-term growth (to P20) led to previously observed (in P4) and
typical morphological alterations, as SHED and DPSCs were notably
larger with irregular and elongated shapes (Fig. 1B). SHED exhibited a significantly
higher proliferation rate in comparison with DPSCs in vitro
at an early passage (P4). However, the rapid growth kinetics of
SHED cells significantly decreased by P20 and no differences were
identified between SHED and DPSCs at P20 (Fig. 1C). These results indicate that SHED
and DPSCs lose their proliferative potential with consecutive cell
passaging.
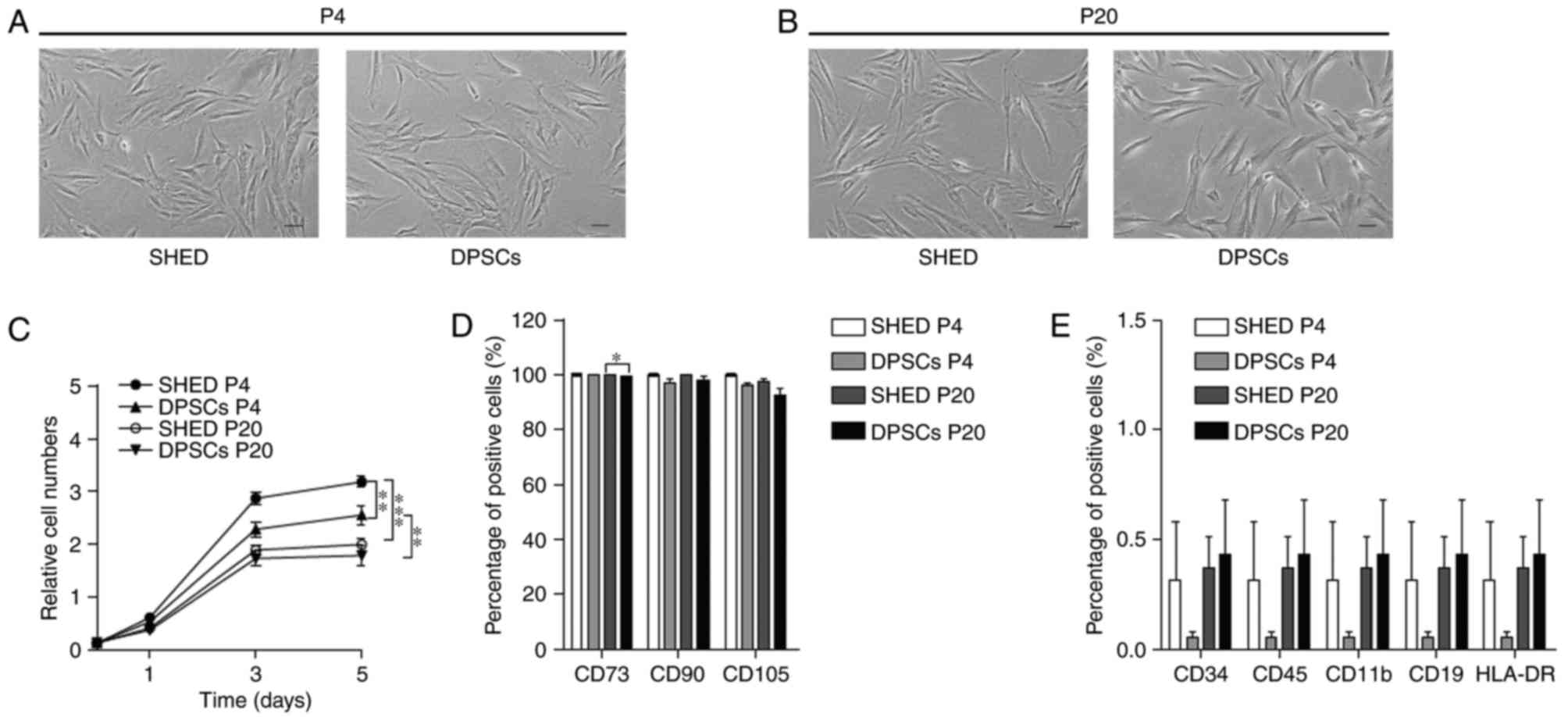 | Figure 1.SHED and DPSCs morphology,
proliferation and immunophenotype. Representative morphological
features of SHED and DPSCs at (A) P4 and (B) P20. Scale bars, 100
µm. (C) Proliferation curves of SHED and DPSCs at P4 and P20. (D)
Statistical analysis of the percentage of CD73-, CD90- or
CD105-positive cells in SHED and DPSCs groups at P4 and P20. (E)
Statistical analysis of the percentage of CD34-, CD11b-, CD19-,
CD45- and HLA-DR-positive cells of the SHED and DPSCs groups at P4
and P20. Data are presented as the mean ± standard error of the
mean (n=3). *P<0.05, **P<0.01 and ***P<0.001, as
indicated. SHED, stem cells from human exfoliated deciduous teeth;
DPSCs, dental pulp stem cells; P4/20, passage 4/20; HLA-DR, human
leukocyte antigen D-related. |
Immunophenotype of SHED and DPSCs
To assess the effects of continuous expansion on the
immunophenotype of SHED and DPSCs, stem cell surface antigens were
assessed using flow cytometry. The results demonstrated that cells
maintained the characteristic immunophenotype of MSCs, with high
rates of positive CD73, CD90 and CD105 expression, and low
expression of CD34, CD11b, CD19, CD45 and HLA-DR during long-term
expansion (Fig. 1D and E).
However, the percentage of CD73-positive DPSCs was lower at P20, in
comparison with SHED at P20 (Fig.
1D). These results indicated that SHED and DPSCs were able to
maintain their specific immunophenotypes during long-term expansion
and that SHED exhibited more properties of stem cells.
Biological characteristics of SHED and
DPSCs
The present study investigated whether the
differentiation potential of cells was influenced by long-term
cultivation in vitro. Oil Red O staining demonstrated that
the presence of lipid vesicles was higher in SHED compared with
DPSCs at P4 and that the adipogenic differentiation potential was
decreased in SHED and DPSCs at P20 in comparison with at P4.
However, no marked differences were observed between the two groups
following consecutive passage in vitro (Fig. 2A-E). The Alizarin Red-positive
condensed nodules of SHED at an early passage were larger and
denser compared with DPSCs. At a later passage, DPSCs still
exhibited reduced formations of mineralized nodules compared with
SHED, but Alizarin Red staining in SHED cells at P20 was also
significantly reduced compared with SHED cells at P4 (Fig. 2F-J). Therefore, the propensity for
adipogenic and osteogenic differentiation in DPSCs and SHED cells
was attenuated with long-term cultivation. Furthermore, RT-qPCR
analysis of differentiation-associated gene expression levels,
including the osteoblast marker genes alkaline phosphatase and
runt-related transcription factor 2, and the adipocyte-specific
transcript, peroxisome proliferator-activated receptor γ2, verified
this conclusion; mRNA expression levels of these markers were all
reduced in DPSCs and SHED cells at P20 compared with P4 (Fig. 3).
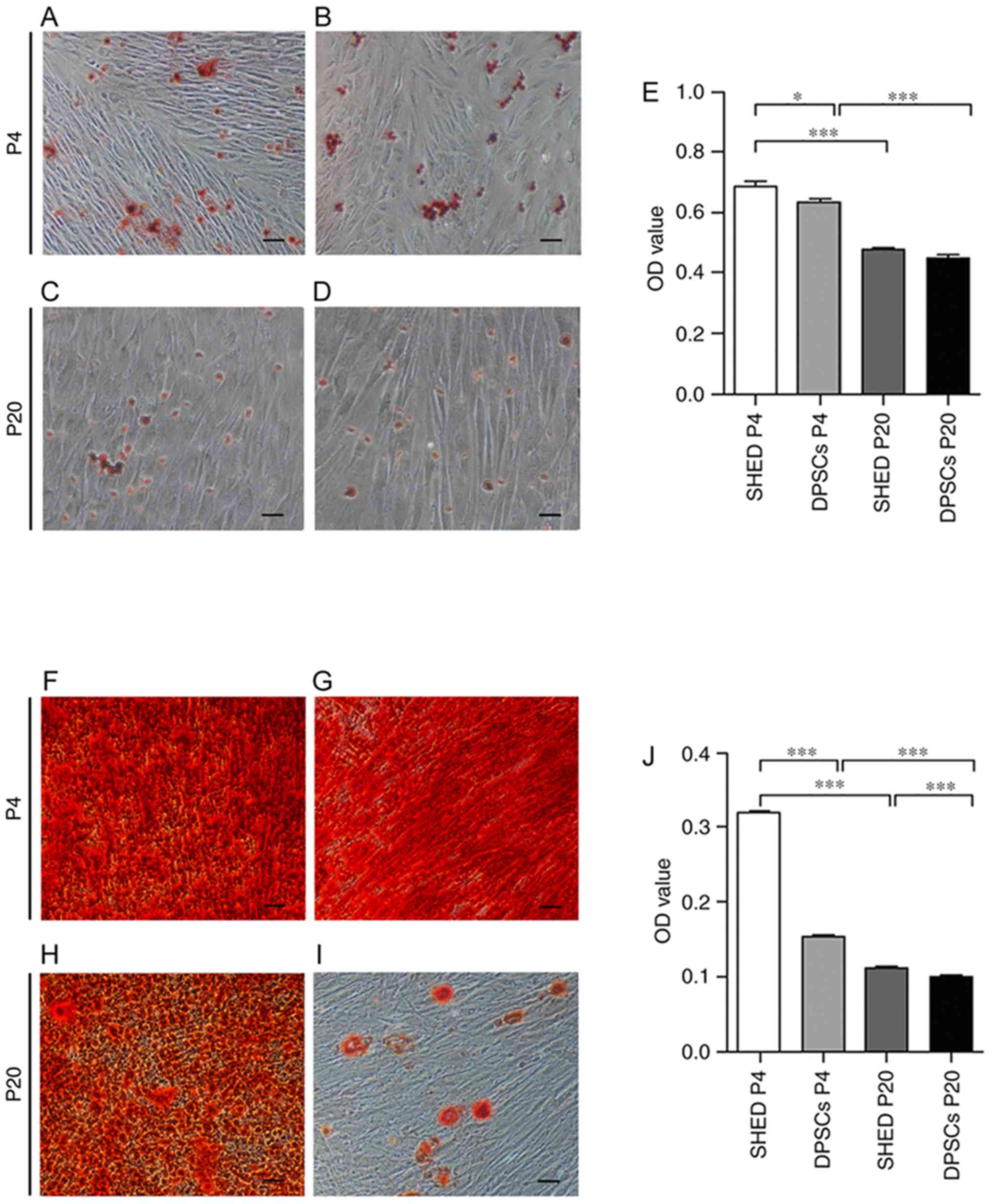 | Figure 2.In vitro differentiation of
SHED and DPSCs. Adipogenesis was assessed by Oil Red O staining of
(A) SHED at P4, (B) DPSCs at P4, (C) SHED at P20 and (D) DPSCs at
P20. Scale bars, 100 µm. (E) Oil Red O staining quantification of
adipogenic differentiation of SHED and DPSCs at P4 and P20).
Osteogenesis was assessed by Alizarin Red staining of (F) SHED at
P4, (G) DPSCs at P4, (H) SHED at P20 and (I) DPSCs at P20. Scale
bars, 100 µm. (J) Alizarin Red staining quantification of
osteogenic differentiation of SHED and DPSCs at P4 and P20. Data
are presented as the mean ± standard error of the mean (n=4).
*P<0.05 and ***P<0.001, as indicated. SHED, stem cells from
human exfoliated deciduous teeth; DPSCs, dental pulp stem cells;
P4/20, passage 4/20; OD, optical density. |
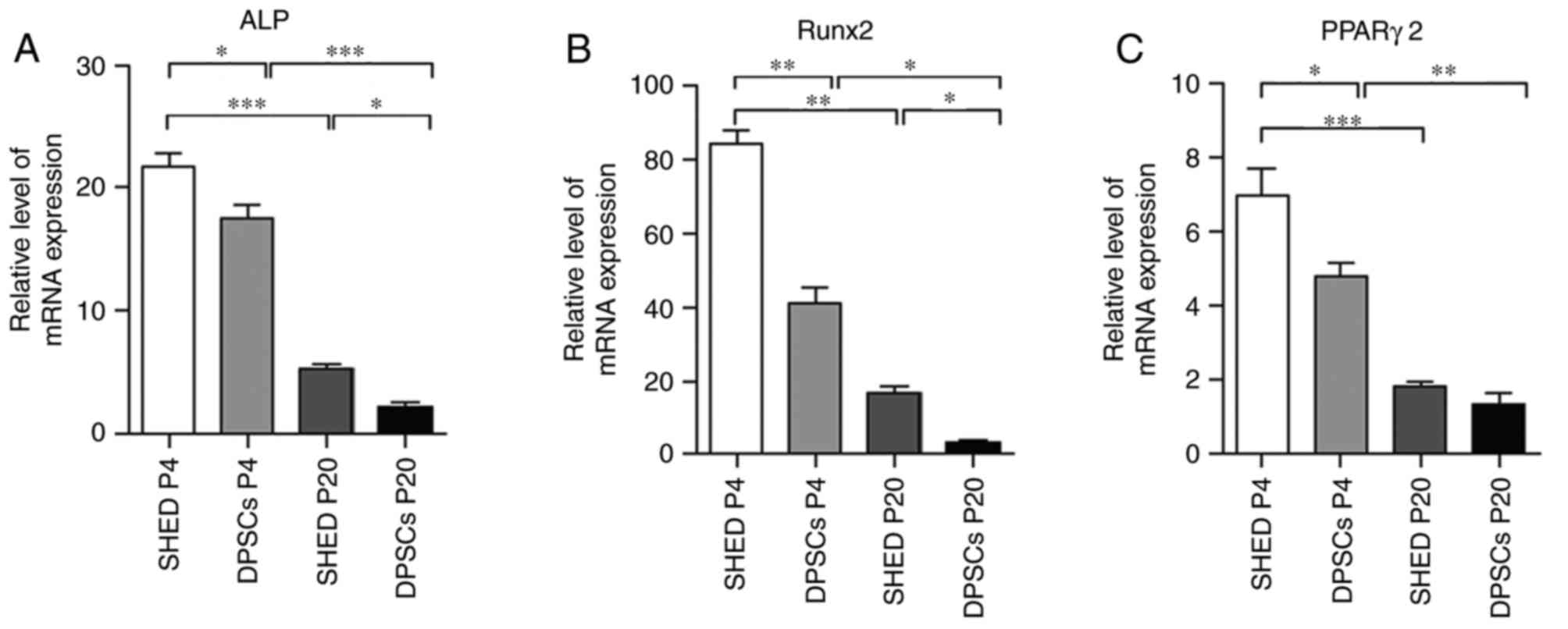 | Figure 3.Quantification of osteogenic and
adipogenic markers. Reverse transcription-quantitative polymerase
chain reaction was performed to determine the mRNA expression
levels of (A) ALP, (B) Runx2 and (C) PPARγ2 in SHED and DPSCs at P4
and P20 following in vitro differentiation. ALP and Runx2
were considered to be osteogenic markers, while PPARγ2 was
considered to indicate adipogenic differentiation. Data are
presented as the mean ± standard error of the mean (n=3).
*P<0.05, **P<0.01 and ***P<0.001, as indicated. ALP,
alkaline phosphatase; Runx2, runt-related transcription factor 2;
PPARγ2, peroxisome proliferator-activated receptor γ2; SHED, stem
cells from human exfoliated deciduous teeth; DPSCs, dental pulp
stem cells; P4/20, passage 4/20. |
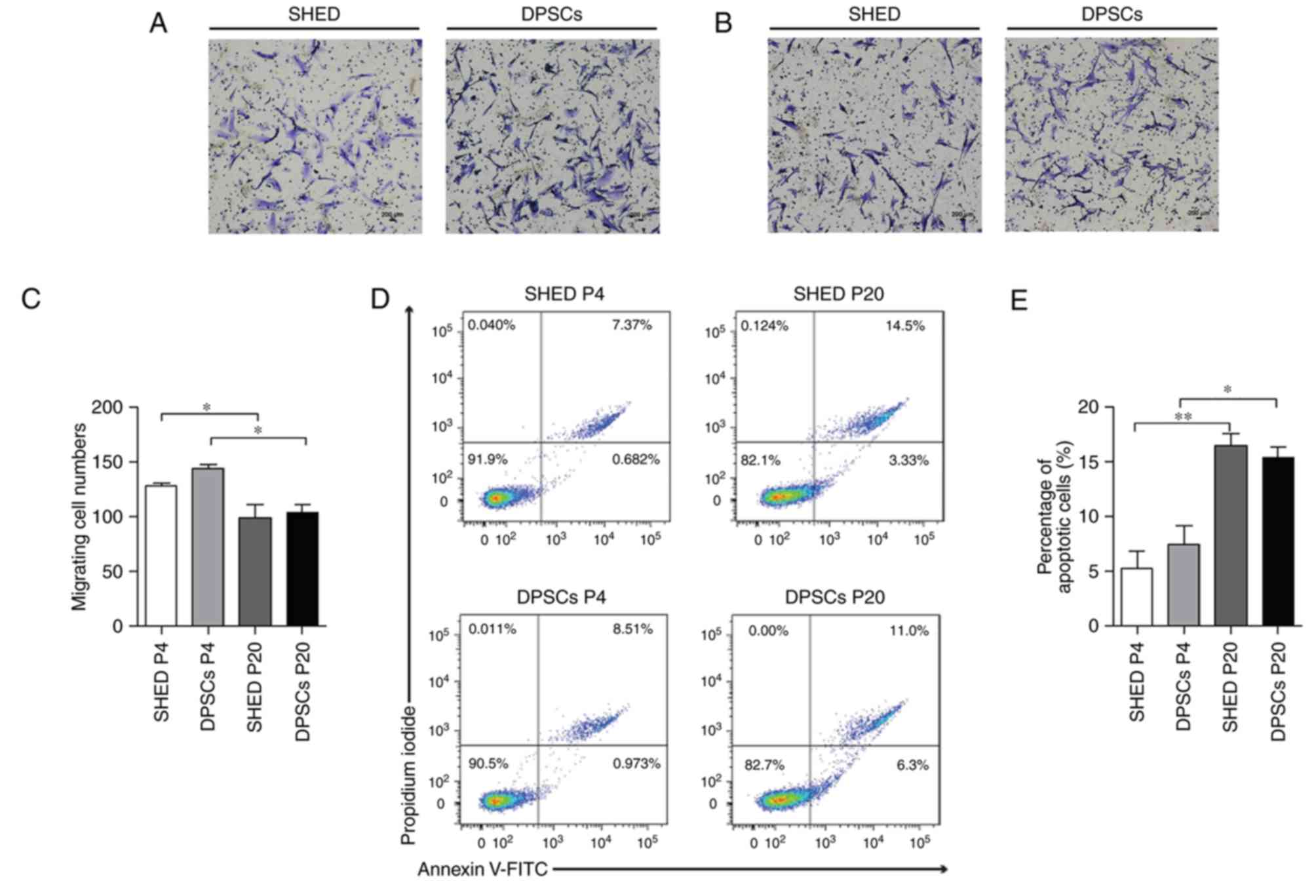
The motility of SHED and DPSCs was subsequently
assessed. It was demonstrated that the migratory abilities of DPSCs
and SHED cells significantly decreased with an increase in passage
number (Fig. 4A-C). However, the
quantification of migratory cell numbers exhibited no marked
difference between SHED and DPSCs at the same passage number
(Fig. 4A-C). Cellular senescence
is associated with DNA damage accumulation and usually leads to
cell apoptosis (22). The present
study therefore assessed the number of apoptotic cells using flow
cytometry. The results demonstrated that long-term cultivation was
associated with a progressive increase in the number of apoptotic
cells in both DPSCs and SHED cells (Fig. 4D and E). Furthermore, the
percentage of apoptotic cells in the DPSC group was similar to that
in the SHED group, irrespective of passage number (Fig. 4D and E).
Expression of senescence molecular
markers in vitro
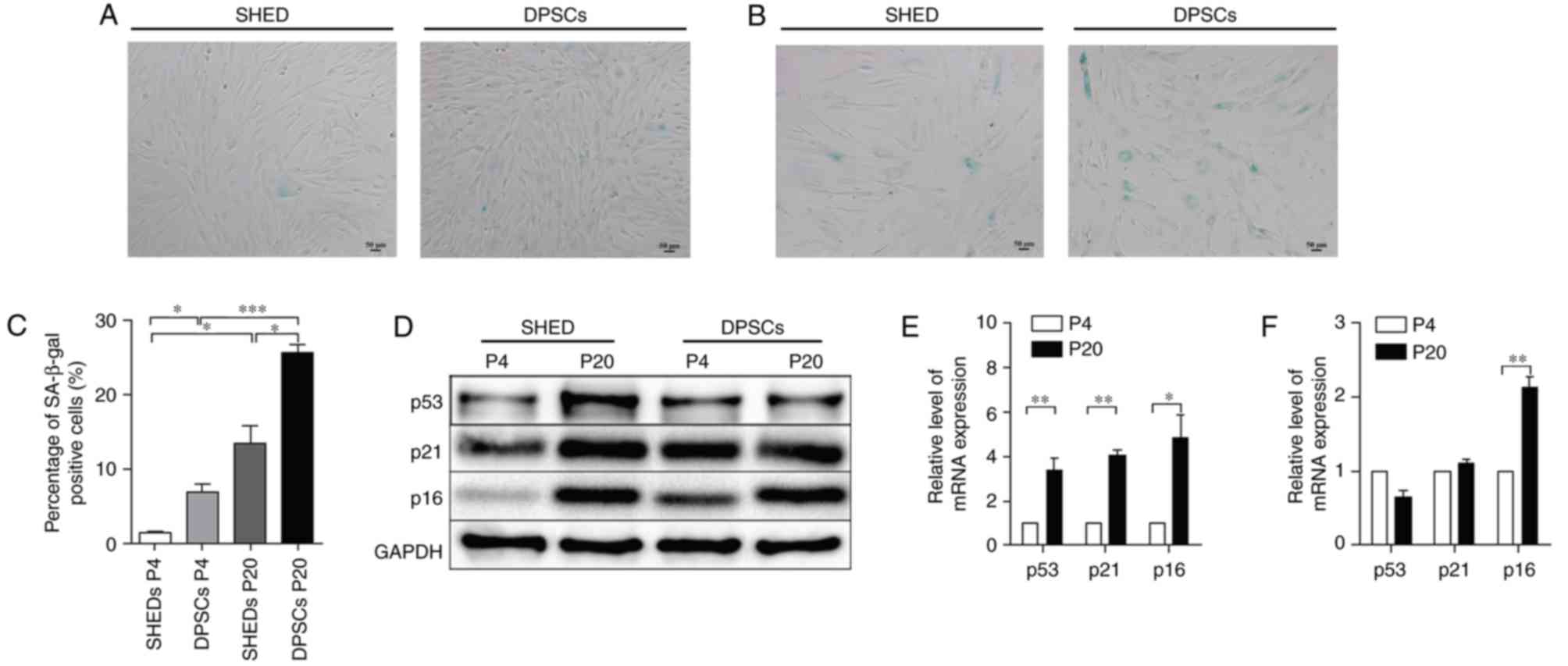
To determine whether the long-term culture of cells
to P20 caused replicative senescence in SHED and DPSCs, P4/20 SHED
and DPSCs were stained with SA-β-gal, a verified cell senescence
marker (23). The results
demonstrated that long-term culture induced marked SA-β-gal
expression in SHED and DPSCs (Fig. 5A
and B). The percentage of SA-β-gal-positive SHED significantly
increased with passage number, from 1.31±0.25% at P4 to 13.44±2.29%
at P20, while in DPSCs, SA-β-gal activity significantly increased
from 6.96±1.02 to 25.62±1.10% at P4 and P20, respectively (Fig. 5C). The results indicated that DPSCs
exhibited a higher predisposition towards senescence during
long-term culture, compared with SHED.
The expression of typical senescence markers p53,
p21 and p16Ink4a in SHED and DPSCs at each passage
number were subsequently assessed to confirm these results at the
mRNA and protein level. The results showed that consecutive
passaging led to a marked increase in the mRNA and protein
expression of p53, p21 and p16Ink4a in SHED cells
(Fig. 5D-F). However, the mRNA and
protein levels of p53 and p21 were not significantly different
between P4 and P20 in DPSCs, while the expression of the senescence
marker p16Ink4a was significantly increased by P20
(Fig. 5D-F). These results
demonstrated that SHED and DPSCs may undergo distinct pathways
during cellular senescence and that p16Ink4a appears to
serve a more prominent role in the senescence of DPSCs.
Discussion
DPSCs and SHED have potential advantages for tissue
regenerative engineering and therapeutic applications due to their
multilineage differentiation potential and immunomodulatory
properties (3). Cell therapy
protocols require 10–400 million human MSCs per treatment and
consequently, MSCs must be expanded in vitro prior to
clinical application (24). Over
five decades ago, Hayflick (25)
demonstrated that all primary human cells exhibit a finite
proliferative capacity in culture and, following a limited number
of cell divisions, enter cellular senescence. Further studies have
reported that MSCs undergo considerable alterations during in
vitro expansion, including spontaneous transformation, reduced
capacity for trafficking and homing, epigenetic and gene expression
alterations, and morphological and multipotent differentiation
potential alterations (26–28).
However, no marked indications of culture degeneration or
spontaneous differentiation have been demonstrated in SHED during
long-term culture up to P19 (29).
Thus, it remains unclear whether SHED and DPSCs undergo
considerable characteristic alterations during in vitro
culture up to P20. In particular, the molecular mechanisms
underlying the phenotypical changes in SHED and DPSCs during
culture expansion remain poorly understood.
A previous study demonstrated that SHED and DPSCs at
early passage numbers exhibited differences in growth and
differentiation (11). However,
differences in their characteristics and gene expression profiles
at later passage numbers are yet to be elucidated. The present
study cultured primary SHED and DPSCs and consecutively expanded
them in vitro. It was demonstrated that SHED exhibited a
significantly higher proliferation rate, compared with DPSCs at P4,
which is similar to the results of a previous study (11). At P20, both SHED and DPSCs
exhibited a progressive loss of proliferative capacity and
morphological alterations, despite maintaining their specific
immunophenotype, as previously observed in other types of MSCs,
such as bone marrow MSC (30).
DPSCs became larger in size, with a more granulated cytoplasm. In
addition, marginal decreases in the expression levels of CD73 were
exhibited following long-term sequential cell passaging in
vitro in DPSCs compared with SHED.
A previous study demonstrated that human MSCs
exhibited reduced differentiation potential as a result of
prolonged culture in vitro (15). However, a previous study also
demonstrated that adipogenic differentiation potential decreases at
later passages, whereas the propensity for osteogenic
differentiation increases in replicative senescence, suggesting
that long-term culture impacts the differentiation potential of
MSCs (16). The present study
indicated that the adipogenic and osteogenic differentiation
potential was reduced with an extended expansion period and that
the two groups exhibited different capacities for differentiation.
This may therefore provide guidance for the selection of cell
sources for tissue regenerative medicine. A previous study also
demonstrated that MSCs, which exhibit higher cell migratory
capacities, are more appropriate for tissue regeneration in terms
of cell homing ability (31). The
current study therefore compared the migratory ability of SHED and
DPSCs during prolonged in vitro expansion. The results
demonstrated that cell migratory capacity was decreased at P20
compared with P4 in each group. However, no significant differences
were identified between the cell migratory abilities of SHED and
DPSCs at early or at later passage stages. In addition, SHED showed
a higher proliferation rate and differentiation capability compared
with in DPSCs in vitro, consequently, these results indicate
that SHED may be an accessible, suitable and potential alternative
source for regenerative medicine and therapeutic application.
Cell senescence is an irreversible process that is
typically accompanied by the upregulation of senescence regulators,
including p53/p21 and p16Ink4a, and the increased
activity of SA-β-gal (32). The
results of the present study indicated that the percentage of
SA-β-gal positive SHED and DPSCs increased with passage number. At
P20, the number of SA-β-gal-positive SHED was reduced in comparison
with DPSCs, indicating that a higher number of senescent cells were
detected in DPSCs under standard culture conditions during
long-term expansion, compared with SHED cells. However, no
significant differences were identified in the number of apoptotic
cells between the two groups at P20. This counterintuitive
observation indicated that DPSCs may initiate an adaptive response
to adopt a compromised state to avoid being eliminated during
severe cellular senescence in vitro. This phenomenon was
similar to that exhibited by neural stem cells (23). p53, p21 and/or p16Ink4a
are key molecules that contribute to cell cycle arrest and cellular
senescence (33). The current
study assessed the expression of p53, p21 and p16Ink4a
at each passage number in the two groups. The results demonstrated
that the mRNA and protein expression of p53, p21 and
p16Ink4a markedly increased at later passage numbers in
SHED, while only p16Ink4a was significantly increased in
DPSCs at a later passage, with a decrease observed for p53 and only
a marginal increase in p21 levels at a later passage in DPSCs. The
reason for this observation may be that as the number of
growth-arrested cells increases with passage, most cells become
senescent and culture growth flattens, leading to decrease in the
number of DPSCs at P20. In addition, DPSCs may adapt a compromised
state to prevent persistent DNA damage, indeed, similar
observations have been noted in a previous study (24). These distinctions indicated that
various distinct pathways may be involved in SHED and DPSCs
cellular senescence during long-term culture in vitro and
that p16Ink4a may be a primary gene associated with the
cellular senescence of DPSCs.
The mechanisms of SHED and DPSCs entry into
senescence during long-term culture in vitro remain
inadequately understood. A previous study demonstrated that
telomeric DNA is subjected to gradual erosion following the
accumulation of MSCs due to consecutive expansion (34). Telomere erosion leads to the
exposure of uncapped, free double-stranded chromosome ends, which
initiates a persistent DNA damage response, and under these
conditions, the recruitment of ataxia telangiectasia mutated, which
is a damage sensor to uncapped telomeres, subsequently resulting in
the stabilization of tumor suppressor p53, enhanced expression of
the p53 transcriptional target, p21, or activated
p16Ink4a (22).
p16Ink4a has been reported to block the CDK4- and
CDK6-mediated inactivation of retinoblastoma to prevent cell cycle
progression and drive cell entry into senescence (32–34).
Further studies investigating the mechanism involved in the in
vitro cellular senescence of SHED and DPSCs are therefore
required.
In conclusion, the present study indicated that SHED
and DPSCs exhibit distinct biological characteristics and gene
expression profiles, and different pathways may be activated in
each of these cell types during long-term in vitro
cultivation. Furthermore, the results provide insight into the
appropriate selection of passaged SHED and DPSCs for use in
cell-based therapies.
Acknowledgements
Not applicable.
Funding
The present study was supported by the Science and
Technology Commission of Shanghai (grant no. 14411963600).
Availability of data and materials
The datasets used and/or analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
HW contributed to the conception and design,
collection and assembly of data, data analysis and interpretation
and writing of the manuscript. QZ, TY, YQ, MF, XY, LQ and QL
contributed to the study design, data analysis and interpretation.
YZ and SL participated in the conception and design of the study,
data analysis and interpretation, financial acquisition, manuscript
writing and final approval of manuscript. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Ethical approval was obtained from the Ethics
Committee at the Affiliated Stomatology Hospital of Tongji
University (approval no. 2015-010). All individuals that
participated in the study provided informed consent under the
approved guidelines set by the Ethics Committee at the Affiliated
Stomatology Hospital of Tongji University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
DPSCs
|
dental pulp stem cells
|
|
SHED
|
stem cells from human exfoliated
deciduous teeth
|
|
SA-β-gal
|
senescence-associated
β-galactosidase
|
|
P4
|
passage 4
|
|
P20
|
passage 20
|
References
|
1
|
Salem HK and Thiemermann C: Mesenchymal
stromal cells: Current understanding and clinical status. Stem
Cells. 28:585–596. 2010.PubMed/NCBI
|
|
2
|
Reddy BY, Xu DS and Hantash BM:
Mesenchymal stem cells as immunomodulator therapies for
immune-mediated systemic dermatoses. Stem Cells Dev. 21:352–362.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Liu J, Yu F, Sun Y, Jiang B, Zhang W, Yang
J, Xu GT, Liang A and Liu S: Concise reviews: Characteristics and
potential applications of human dental tissue-derived mesenchymal
stem cells. Stem Cells. 33:627–638. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Gronthos S, Mankani M, Brahim J, Robey PG
and Shi S: Postnatal human dental pulp stem cells (DPSCs) in vitro
and in vivo. Proc Natl Acad Sci USA. 97:pp. 13625–13630. 2000;
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Gronthos S, Brahim J, Li W, Fisher LW,
Cherman N, Boyde A, DenBesten P, Robey PG and Shi S: Stem cell
properties of human dental pulp stem cells. J Dent Res. 81:531–535.
2002. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bakopoulou A and About I: Stem cells of
dental origin: Current research trends and key milestones towards
clinical application. Stem Cells Int. 2016:42098912016. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Li Z, Jiang CM, An S, Cheng Q, Huang YF,
Wang YT, Gou YC, Xiao L, Yu WJ and Wang J: Immunomodulatory
properties of dental tissue-derived mesenchymal stem cells. Oral
Dis. 20:25–34. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Miura M, Gronthos S, Zhao M, Lu B, Fisher
LW, Robey PG and Shi S: SHED: Stem cells from human exfoliated
deciduous teeth. Proc Natl Acad Sci USA. 100:pp. 5807–5812. 2003;
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Kashyap R: SHED-basic structure for stem
cell research. J Clin Diagn Res. 9:ZE07–ZE09. 2015.PubMed/NCBI
|
|
10
|
Brar GS and Toor RS: Dental stem cells:
Dentinogenic, osteogenic, and neurogenic differentiation and its
clinical cell based therapies. Indian J Dent Res. 23:393–397. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang X, Sha XJ, Li GH, Yang FS, Ji K, Wen
LY, Liu SY, Chen L, Ding Y and Xuan K: Comparative characterization
of stem cells from human exfoliated deciduous teeth and dental pulp
stem cells. Arch Oral Biol. 57:1231–1240. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Banfi A, Muraglia A, Dozin B,
Mastrogiacomo M, Cancedda R and Quarto R: Proliferation kinetics
and differentiation potential of ex vivo expanded human bone marrow
stromal cells: Implications for their use in cell therapy. Exp
Hematol. 28:707–715. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Røsland GV, Svendsen A, Torsvik A, Sobala
E, McCormack E, Immervoll H, Mysliwietz J, Tonn JC, Goldbrunner R,
Lønning PE, et al: Long-term cultures of bone marrow-derived human
mesenchymal stem cells frequently undergo spontaneous malignant
transformation. Cancer Res. 69:5331–5339. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Baxter MA, Wynn RF, Jowitt SN, Wraith JE,
Fairbairn LJ and Bellantuono I: Study of telomere length reveals
rapid aging of human marrow stromal cells following in vitro
expansion. Stem Cells. 22:675–682. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Bonab MM, Alimoghaddam K, Talebian F,
Ghaffari SH, Ghavamzadeh A and Nikbin B: Aging of mesenchymal stem
cell in vitro. BMC Cell Biol. 7:142006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Wagner W, Horn P, Castoldi M, Diehlmann A,
Bork S, Saffrich R, Benes V, Blake J, Pfister S, Eckstein V and Ho
AD: Replicative senescence of mesenchymal stem cells: A continuous
and organized process. PLoS One. 3:e22132008. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Bakopoulou A, Leyhausen G, Volk J,
Tsiftsoglou A, Garefis P, Koidis P and Geurtsen W: Assessment of
the impact of two different isolation methods on the
osteo/odontogenic differentiation potential of human dental stem
cells derived from deciduous teeth. Calcif Tissue Int. 88:130–141.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Naderi N, Wilde C, Haque T, Francis W,
Seifalian AM, Thornton CA, Xia Z and Whitaker IS: Adipogenic
differentiation of adipose-derived stem cells in 3-dimensional
spheroid cultures (microtissue): Implications for the
reconstructive surgeon. J Plast Reconstr Aesthet Surg.
67:1726–1734. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Bakopoulou A, Leyhausen G, Volk J,
Tsiftsoglou A, Garefis P, Koidis P and Geurtsen W: Comparative
analysis of in vitro osteo/odontogenic differentiation potential of
human dental pulp stem cells (DPSCs) and stem cells from the apical
papilla (SCAP). Arch Oral Biol. 56:709–721. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Dimri GP, Lee X, Basile G, Acosta M, Scott
G, Roskelley C, Medrano EE, Linskens M, Rubelj I, Pereira-Smith O,
et al: A biomarker that identifies senescent human cells in culture
and in aging skin in vivo. Proc Natl Acad Sci USA. 92:pp.
9363–9367. 1995; View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Childs BG, Durik M, Baker DJ and van
Deursen JM: Cellular senescence in aging and age-related disease:
From mechanisms to therapy. Nat Med. 21:1424–1435. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Dong CM, Wang XL, Wang GM, Zhang WJ, Zhu
L, Gao S, Yang DJ, Qin Y, Liang QJ, Chen YL, et al: A
stress-induced cellular aging model with postnatal neural stem
cells. Cell Death Dis. 5:e11162014. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Estrada JC, Torres Y, Benguría A, Dopazo
A, Roche E, Carrera-Quintanar L, Pérez RA, Enríquez JA, Torres R,
Ramírez JC, et al: Human mesenchymal stem cell-replicative
senescence and oxidative stress are closely linked to aneuploidy.
Cell Death Dis. 4:e6912013. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Hayflick L: The Limited in vitro lifetime
of human diploid cell strains. Exp Cell Res. 37:614–636. 1965.
View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Li Z, Liu C, Xie Z, Song P, Zhao RC, Guo
L, Liu Z and Wu Y: Epigenetic dysregulation in mesenchymal stem
cell aging and spontaneous differentiation. PLoS One. 6:e205262011.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Wu Y and Zhao RC: The role of chemokines
in mesenchymal stem cell homing to myocardium. Stem Cell Rev.
8:243–250. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Philippe B, Luc S, Valerie PB, Jerome R,
Alessandra BR and Louis C: Culture and use of mesenchymal stromal
cells in phase I and II clinical trials. Stem Cells Int.
2010:5035932010. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Suchánek J, Visek B, Soukup T, El-Din
Mohamed SK, Ivancaková R, Mokrỳ J, Aboul-Ezz EH and Omran A: Stem
cells from human exfoliated deciduous teeth-isolation, long term
cultivation and phenotypical analysis. Acta Medica (Hradec
Kralove). 53:93–99. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Madeira A, da Silva CL, dos Santos F,
Camafeita E, Cabral JM and Sa-Correia I: Human mesenchymal stem
cell expression program upon extended ex-vivo cultivation, as
revealed by 2-DE-based quantitative proteomics. PLoS One.
7:e435232012. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Andersen RK, Zaher W, Larsen KH, Ditzel N,
Drews K, Wruck W, Adjaye J, Abdallah BM and Kassem M: Association
between in vivo bone formation and ex vivo migratory capacity of
human bone marrow stromal cells. Stem Cell Res Ther. 6:1962015.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Kuilman T, Michaloglou C, Mooi WJ and
Peeper DS: The essence of senescence. Genes Dev. 24:2463–2479.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
van Deursen JM: The role of senescent
cells in ageing. Nature. 509:439–446. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Ksiazek K: A comprehensive review on
mesenchymal stem cell growth and senescence. Rejuvenation Res.
12:105–116. 2009. View Article : Google Scholar : PubMed/NCBI
|