Introduction
Cervical cancer, the fourth most common cancer in
women worldwide, with relatively high numbers occurring in
developing countries, including China, has the major etiological
factor which is persistent infection of oncogenic Human
papillomavirus (HPV) (1,2).
The HPV genome is a small double-stranded circular
DNA molecule of 8,000 base pairs (bp) which contains three regions:
i) 4,000 bp region encoding 6 early proteins participating in viral
replication and cell transformation, including 3 regulatory
proteins (E1, E2 and E4) and 3 oncoproteins (E5, E6 and E7); ii)
3,000 bp region encoding the structural proteins of the virus,
including major capsid protein L1 and the minor capsid protein L2;
and iii) 1,000 bp long control region (LCR) that contains the
origin of viral DNA replication and transcriptional regulatory
elements (3).
Any two HPV strains may be classified into types,
subtypes or variants according to the difference rate between their
L1 sequences that is the difference rate >10%, between 2 and 10%
or <2% (4). Currently, >200
HPV types have been fully characterized (http://www.hpvcenter.se/html/refclones.html), and
the International Agency for Research on Cancer defined 12 HPV
types as oncogenic or potentially carcinogenic groups (5). Except for HPV51 (belongs to a-5) and
HPV-56 (belongs to a-6), others of these carcinogenic types are
phylogenetically clustered with a-7 (HPV-18, HPV-39, HPV-45,
HPV-59) or a-9 (HPV-16, HPV-31, HPV-33, HPV-35, HPV-52, HPV-58)
(6).
HPV-56 is an oncogenic HPV type, which was initially
described in the United States in 1989 (7). Globally, HPV-56 accounts for <1%
of cervical cancer cases (1)
However, compared with the rest of the world, the prevalence of
HPV-56 is remarkably high in Asia, particularly in China (8,9).
According to a recent study from China's largest KingMed Laboratory
(Guangzhou, Guangdong, China), from January 2011 to June 2014, the
detection rate of HPV-56 among the 51,345 samples was ranked fourth
below the three most highly detected types, HPV-52, HPV-16, and
HPV-58 (10). Additionally,
another previous study on the prevalence and distribution of HPV in
37 cities in China revealed that HPV-56 infection rate is 6.09%,
ranking fifth from 2,580 high risk HPV-positive samples (11). These findings are similar to those
observed in southwest China in the past two years (12).
Previous studies suggested that variants of the same
HPV type are biologically distinct and may result in differing
pathogenic risks (13–15). The viral LCR region as the primary
recognition site of the transcription factor, variations in this
region may affect virus replication rates and the transcriptional
activity (16). When considering
the E6 and E7 oncoproteins, one or more amino acid changes may
potentially alter cellular immortalization, transformation and
carcinogenesis (17,18). Changes in the L1 amino acid
sequence, which contains sequences important for conformation of
viral neutralization-relevant epitopes, may affect viral
antigenicity or influence the effectiveness of viral infection
(19). Currently, the information
available on sequence variation in HPV-16, HPV-18, HPV-52, and
HPV-58 is relatively comprehensive (20–23).
However, data on HPV-56 variants is limited and sequence variations
of HPV-56 in the Asian population remain to be elucidated (24,25).
Therefore, the aim of the present study was to detect sequence
variations within LCR, E6, E7, and L1 of HPV-56 in women from
southwest China. The current study may improve the understanding of
the viral persistence, transmission and oncogenic potential.
Furthermore, identifying novel variants of HPV-56 may assist in the
design of vaccines and development of diagnostic probes for
specific populations.
Materials and methods
Ethics statement
The present study was approved by the Education and
Research Committee and the Ethics Committee of Sichuan University
(Chengdu, China). Prior to sample collection, written informed
consent was obtained from the patients and study subject privacy
was carefully protected.
Sample collection
From January 2014 to January 2016, 7,707 cervical
swabs were collected from female patients (age range, 18–65 years)
that received HPV infection screening at the following maternity
hospitals in Sichuan, China: Sichuan Reproductive Health Research
Center Affiliated Hospital, The Angel Women's and Children's
Hospital, The Chengdu Western Hospital Maternity Unit, The Peoples'
Hospital of Pengzhou, Jinjiang Maternity and Child Health Hospital,
Chengdu Zongnan Gynecology Hospital and Chengdu Songziniao
Sterility Hospital. Samples were placed in preservative buffer
(Yaneng Bioscience Co., Ltd., Shenzhen, China) and stored at
−20°C.
HPV DNA detection and typing
Following sample collection, the viral genomic DNA
was extracted and genotyped using the human papillomavirus
genotyping kit for 23 types (Yaneng Bioscience Co., Ltd.) according
to the manufacturer's protocol. This kit is based on a reverse dot
blot principle and is able to classify 23 HPV genotypes, including
the 18 high-risk, the probable high-risk types (16, 18, 31, 33,35,
39, 45, 51, 52, 53, 56, 58, 59, 66, 68, 73, 83, and MM4), and the 5
low-risk types (6, 11, 42, 43, and 44). A total of 101 of HPV-56
positive samples DNA were detected and subsequently stored at
−20°C.
PCR amplification and variant
identification
The LCR fragment was amplified using specific
primers as previously described (24). The complete genes of E6, E7 and L1
primers were designed using Primer Primier version 5.0 (Premier
Biosoft, Palo Alto CA, USA; http://www.premierbiosoft.com/) (26) based on the HPV-56 GenBank reference
sequences X74483 (accession no. X74483; www.ncbi.nlm.nih.gov/nuccore/X74483) and listed in
Table I. The primers were
synthesized by Sangon Biotech Co., Ltd., (Shanghai, China).
 | Table I.Primers used for the molecular
characterization of human papillomavirus-56 E6, E7 and L1 long
control regions. |
Table I.
Primers used for the molecular
characterization of human papillomavirus-56 E6, E7 and L1 long
control regions.
| Gene | Direction | Sequence 5′-3′ | Primer
position | Product size,
bp | Annealing
Temperature, °C |
|---|
| LCR | Forward |
TGTGTCATTATTGTGGCTTTTGTTTTGT | 7323 | 603 | 57 |
|
| Reverse |
AGCTGCCTTTTATATGTACCGTTTTC | 81 |
|
|
| E6 | Forward |
ATTGGGAGTGACCGAAAAGG | 26 | 803 | 58 |
|
| Reverse |
ACAACACGCAGGTCCTCTTT | 828 |
|
|
| E7 | Forward |
GCTACAGATGTCAAAGTCCG | 444 | 542 | 57.5 |
|
| Reverse |
GCCTCTACTTCAAACCATCC | 985 |
|
|
| L1-1 | Forward |
GCCCCTTTAGGTAATGTGTGG | 5418 | 943 | 59 |
|
| Reverse |
CCACATAGAATCACCATAGGCAT | 6360 |
|
|
| L1-2 | Forward |
ATGATAGACACAGGATTTGGCG | 6217 | 948 | 58 |
|
| Reverse |
ATACAACACACAAACACAGTTACAC | 7164 |
|
|
Each 25 µl PCR reaction contained 3 µl of extracted
DNA (10–100 ng), 2.5 mM 10X PCR buffer (Mg2+) (Beijing
TransGen Biotech Co., Ltd., Beijing, China), 2.5 mM dNTPs (Beijing
TransGen Biotech Co., Ltd.), 2U Taq DNA polymerase (Sangon Biotech
Co., Ltd.) and 90 pmol of each primer (Sangon Biotech Co., Ltd.).
The PCR conditions were as follows: Initial denaturation at 95°C
for 5 min, followed by 35 amplification cycles, with each cycle
including a 45 sec denaturation step at 94°C, 1 min annealing step
at a primer specific temperature, and a 1 min elongation step at
72°C, the process was completed with a final 10 min extension step
at 72°C. The annealing temperatures for the PCR primer sequences of
the selected genes are presented in Table I.
Following PCR amplification, electrophoresis was
performed to separate the products, using 2% agarose gel (Sangon
Biotech Co., Ltd.) stained by GeneGreen (Tiangen Biotech Co., Ltd.,
Beijing, China) nucleic acid dye and the products were detected
under ultra violet light. Target products were sequenced by Sangon
Biotech Co., Ltd. (Shanghai, China) and the data were confirmed by
repeating the PCR amplification and sequence analysis at least
twice.
The sequences were subsequently analyzed by NCBI
Blast (blast.ncbi.nlm.nih.gov/Blast.cgi) and DNAMAN version
5.2.2 (Lynnon Biosoft LLC, San Ramon, CA, USA). HPV-56 nucleotide
positions were numbered according to the reference sequence X74483.
Only when the LCR-E6-E7-L1 were amplified and sequenced
simultaneously, was the resulting sequence selected for subsequent
analyses.
Phylogenetic trees analysis
Phylogenetic trees of HPV-56 LCR-E6-E7 and L1 were
constructed with the maximum-likelihood trees method by Molecular
Evolutionary Genetics Analysis version 6 software using Kimura's
two-parameter model, respectively. The reference viral sequences
EF177181, EF177180, EF177179, EF177178, KU298915, KU298916,
KU298918, KU298917, KU298919, EF177176, EF177177, collected from
the GenBank sequence database were used to construct the distinct
phylogenetic branches. The tree topology was evaluated by using
bootstrap resampled 1,000 times as previously described (27). Numbers above the branches indicate
the bootstrap values that are >70%.
Sequence analysis
The secondary structure was predicted with the
PSIPRED server (bioinf.cs.ucl.ac.uk/psipred/), which provides a simple
and accurate secondary structure prediction method as previously
described (28). The prediction of
the damaging effect of missense mutation to protein structure and
function was performed using PolyPhen-2 software (genetics.bwh.harvard.edu/pph2/)
(29).
The MATCH 1.0 server (www.gene-regulation.com/pub/programs.html#match)
(30) was used to search within
the LCR of HPV-56 for potential binding sites for cellular and
viral transcriptional factors. Various transcriptional factors were
identified, including AP-1, E2, GRE, NF-1, Oct-1, TATA, YY1, C/EBP,
Sp1, SRY, AML-1a and c-Myc/c-Max.
Selective pressure analysis
To estimate for positive selection at particular
sites of the HPV-56 E6-E7 and L1 gene sequences, the codeml
program in the Phylogenetic Analyses by Maximum Likelihood (PAML)
version 4.8 package was used to perform likelihood ratio tests to
infer nonsynonymous and synonymous nucleotide divergence for coding
regions by the method of Nei and Gojobor (31).
Results
Characteristics of HPV-56 prevalence
in southwest China in the span of 2 years
During the period of January 2014 to January 2016,
the overall prevalence of detectable HPV infection was 31.27%
(2,407/7,707) and 21.42% (1,651/7,707) samples were high-risk HPV
type and 9.85% (759/7,707) were low-risk HPV type. From the
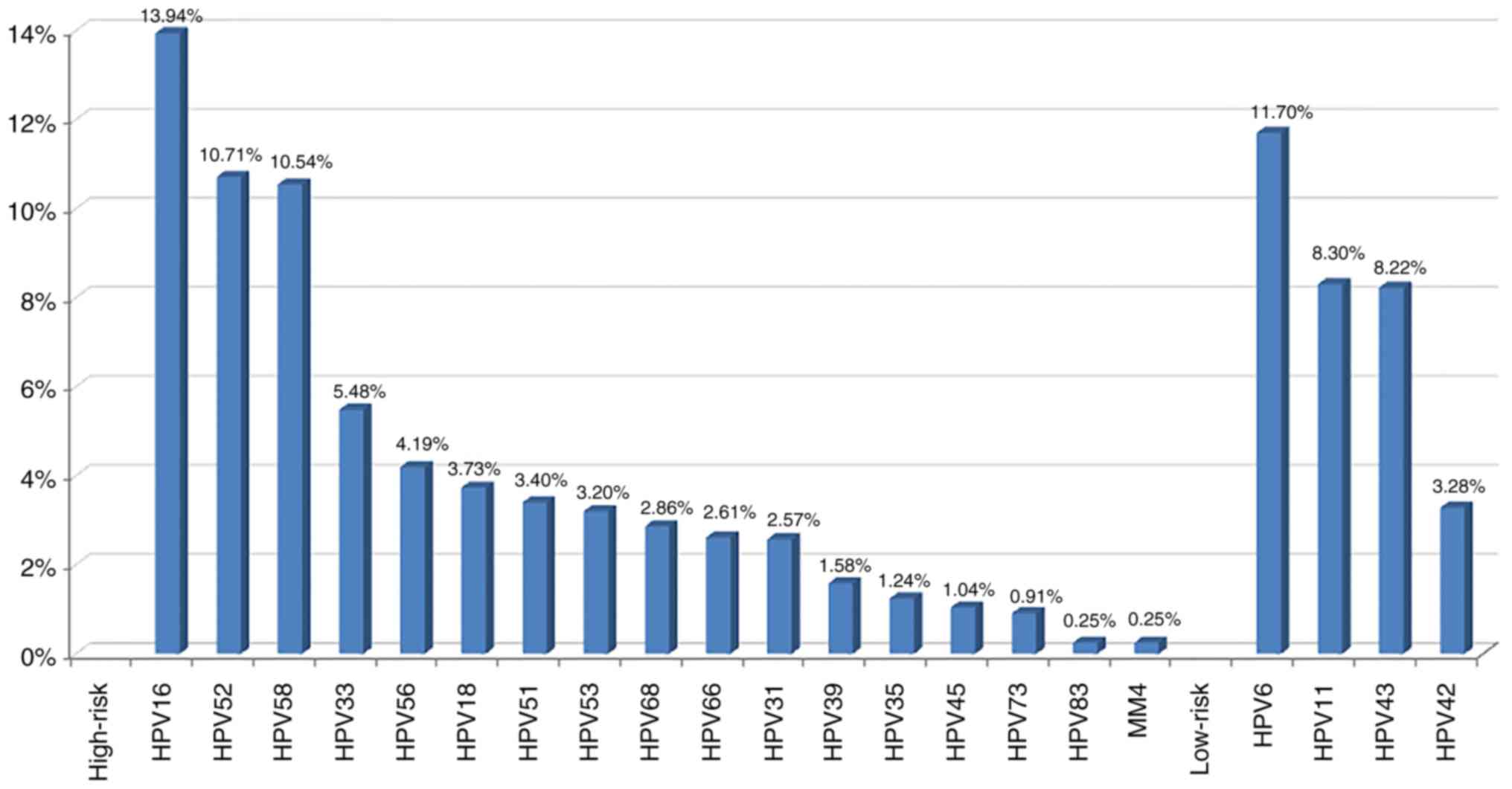
HPV-positive women involved in the present study, the most commonly
detected high-risk HPV genotype was HPV-16 (336, 13.94%), followed
by HPV-52 (258, 10.71%), HPV-58 (254, 10.54%) and HPV-33 (132,
5.48%). HPV-56 ranked fifth (101, 4.19%). Additional high-risk HPV
genotypes were listed in descending order of priority as follows:
HPV-18, −51, −53, −68, −66, −31, −39, −35, −45, −73, −83 and MM4
(Fig. 1).
HPV-56 LCR-E6-E7 sequence
variation
From the 101 samples which tested positive for
HPV-56, 75 (75.25%) LCR-E6-E7-L1 sequences were amplified by PCR
for variant analysis. The remaining 26 (25.75%) sequences were not
obtained due to unsuccessful PCR.
All 75 HPV-56 LCR-E6-E7 isolates showed nucleotide
variation when compared with the HPV-56 reference sequence
(GenBank: X74483). The isolates were divided into 23 different
groups denoted as 56SE01-56SE23. And these sequences were published
with GenBank accession codes from KX645742 to KX645764.
From the HPV-56 LCR sequences, 18 point
substitutions were detected, 11 of which were located in putative
transcriptional factors binding sites and four substitutions
(T7485C, T7581C, A7628C and T7800G) were present in all isolates.
In addition, a 42 bp deletion and a 19 bp deletion were observed,
leading to loss of a putative binding site for the YY1
transcriptional factor (Table
II).
| Table II.Nucleotide sequence variations at
long control regions-E6-E7 of 23 HPV-56 isolates. |
Table II.
Nucleotide sequence variations at
long control regions-E6-E7 of 23 HPV-56 isolates.
A total of 18 single nucleotide changes occurred in
the E6-E7 sequence, with 11/18 (61.11%) being non-synonymous
substitutions and 7/18 (38.89%) being synonymous mutations. The
sequence variability of E6 was lower than that of E7. The average
probability of a nucleotide sequence deviation from the prototype
was 6.2 substitutions per 1,000 bp for E6 and 6.4 substitutions per
1,000 bp for E7 (Table II). Three
amino acid mutations occurred in sequences encoding the α-helix,
and five mutations (four non-synonymous) were observed in sequences
encoding the β-sheet (Table II).
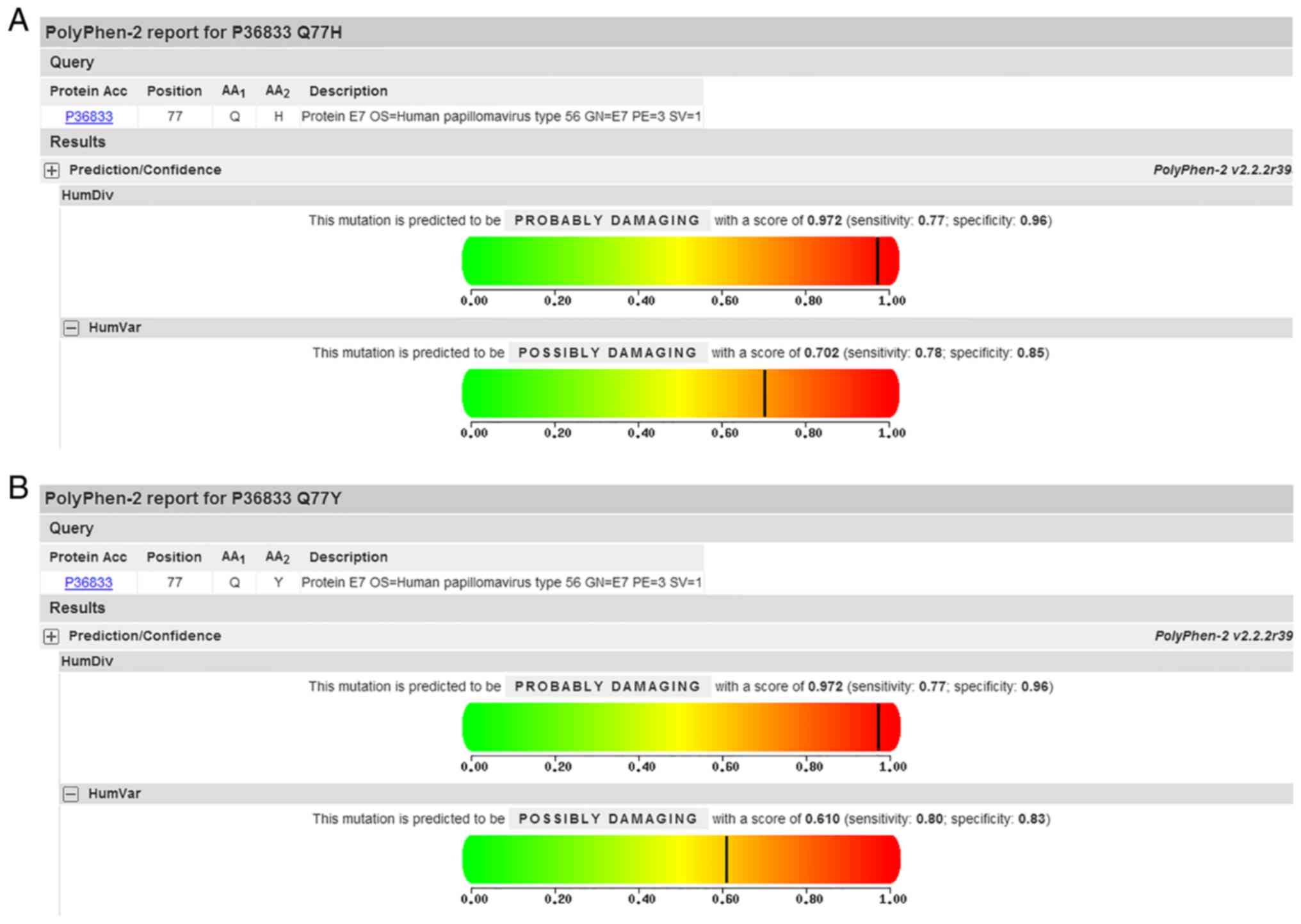
Additionally, PolyPhen-2 analysis predicted a Q77H/Y mutation of E7
to be ‘possibly damaging’ with a score of 0.702/0.610 on the HumVar
model (Fig. 2). Mutations
generating a frame shift or a premature stop codon were not
observed.
Phylogenetic analysis of HPV-56
LCR-E6-E7 sequences
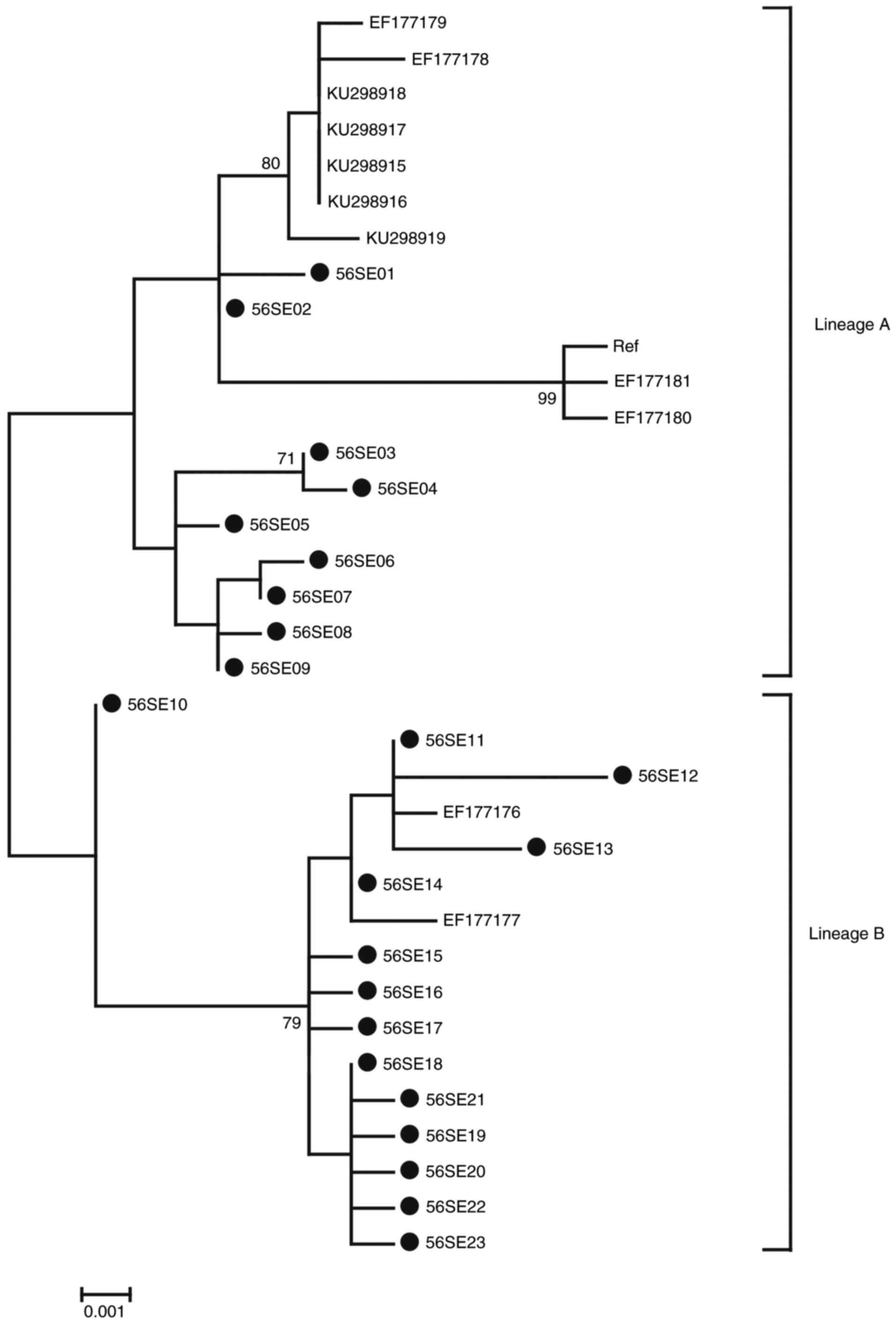
Phylogenetic analysis based on HPV-56 LCR-E6-E7
1,259 bp nucleotide sequences was performed including 23 isolates
in the current study, accompanied by 12 reference sequences. Based
on whole genome study and the topology, two distinct branches were
formed (Fig. 3) (32). A total of 9 isolates in the current
study (56SE01-S6SE09) were classified into the A variant lineage.
The remaining 14 isolates (56SE10-S6SE23) were classified into the
B variant lineage.
HPV-56 L1 sequence variation
The nucleotide variation rate of HPV-56 L1 was 100%
among the 75 HPV-56 isolates when compared with the HPV-56
reference sequence (GenBank: X74483). The L1 isolates were divided
into 16 different groups denoted as 56HL01-56HL16 (Table III). And these sequences were
published in GenBank with the accession codes KX645765-645780.
| Table III.Nucleotide sequence variations at L1
of 16 HPV-56 isolates. |
Table III.
Nucleotide sequence variations at L1
of 16 HPV-56 isolates.
|
|
L1 |
|
|---|
|
|
|
|
|---|
|
| 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 5 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 6 | 7 |
|
|---|
|
| 5 | 5 | 5 | 6 | 6 | 6 | 8 | 8 | 8 | 8 | 8 | 9 | 4 | 4 | 4 | 4 | 5 | 6 | 7 | 8 | 8 | 8 | 8 | 0 |
|
|---|
|
| 2 | 4 | 5 | 2 | 3 | 8 | 0 | 1 | 3 | 4 | 6 | 3 | 2 | 2 | 5 | 6 | 8 | 0 | 3 | 2 | 4 | 6 | 8 | 2 |
|
|---|
| Isolates | 9 | 7 | 7 | 0 | 5 | 0 | 6 | 8 | 8 | 5 | 7 | 2 | 4 | 7 | 4 | 9 | 6 | 7 | 3 | 6 | 5 | 8 | 0 | 0 | n, 75 |
|---|
| Ref | C | G | A | A | A | A | G | A | A | G | A | A | A | G | G | T | T | A | T | C | A | A | G | A |
|
| 56HL01 | – | – | – | – | – | – | – | – | – | – | G | – | – | A | T | – | G | – | – | – | – | – | – | – | 14 |
| 56HL02 | – | – | – | – | – | – | – | – | – | – | G | – | – | A | T | – | G | – | – | – | – | C | – | – | 1 |
| 56HL03 | – | – | – | – | – | – | – | – | T | – | G | – | – | – | T | – | – | – | – | – | – | C | – | – | 1 |
| 56HL04 | – | – | – | – | – | – | – | – | – | – | G | – | – | – | T | – | – | – | – | – | – | C | – | – | 4 |
| 56HL05 | – | – | – | – | – | – | – | – | – | – | G | – | – | – | T | – | – | – | – | – | – | T | – | – | 2 |
| 56HL06 | – | – | – | – | – | – | – | – | – | – | G | – | – | – | T | – | – | – | – | – | – | – | – | – | 2 |
| 56HL07 | T | – | – | C | – | – | A | – | – | – | – | – | – | – | T | – | – | G | – | – | – | – | – | – | 1 |
| 56HL08 | T | – | – | C | – | – | – | – | – | – | – | – | – | – | T | – | – | G | – | – | – | – | – | – | 1 |
| 56HL09 | – | – | – | – | – | G | – | – | – | A | – | G | – | – | T | G | – | – | – | – | – | – | – | – | 4 |
| 56HL10 | – | – | – | – | – | G | – | – | – | A | – | G | – | – | T | G | – | – | – | T | – | – | – | – | 2 |
| 56HL11 | – | A | G | – | – | G | – | – | – | – | – | G | – | – | T | G | – | – | – | T | – | – | A | – | 1 |
| 56HL12 | – | – | – | – | – | G | – | – | – | A | – | G | – | – | T | G | – | – | – | T | – | – | A | G | 28 |
| 56HL13 | – | – | G | – | – | G | – | – | – | A | – | G | – | – | T | G | – | – | – | T | – | – | A | G | 1 |
| 56HL14 | – | – | – | – | G | G | – | – | – | A | – | G | G | – | T | G | – | – | – | T | C | – | A | G | 1 |
| 56HL15 | – | – | – | – | – | G | – | G | – | A | – | G | – | – | T | G | – | – | – | T | – | – | A | G | 11 |
| 56HL16 | – | – | – | – | – | G | – | G | – | A | – | G | – | – | T | G | – | – | C | T | – | – | A | G | 1 |
|
|
| P13L | C19Y | – | E43D | – | – | – | – | K116M | – | N126D | – | – | – | – | – | – | – | – | – | – | – | – | K510R |
|
|
| Second
structure | – | S | S | – | S | S | S | S | – | – | – | – | – | – | – | S | S | – | – | – | – | – | – | – |
|
A total of 24 single nucleotide changes were
identified in the 1,605 bp L1 sequence, 6 (33.33%) substitutions
were non-synonymous mutations and 18 (66.67%) substitutions were
synonymous mutations and G64554T was present in all samples
analyzed (Table III). The
average probability of a nucleotide sequence deviation from
prototype was 4.04 substitutions per 1,000 bp for L1. No
non-synonymous mutation was observed in the HPV-56 L1 sequences
encoding the a-helix. 8 mutations (one non-synonymous) were
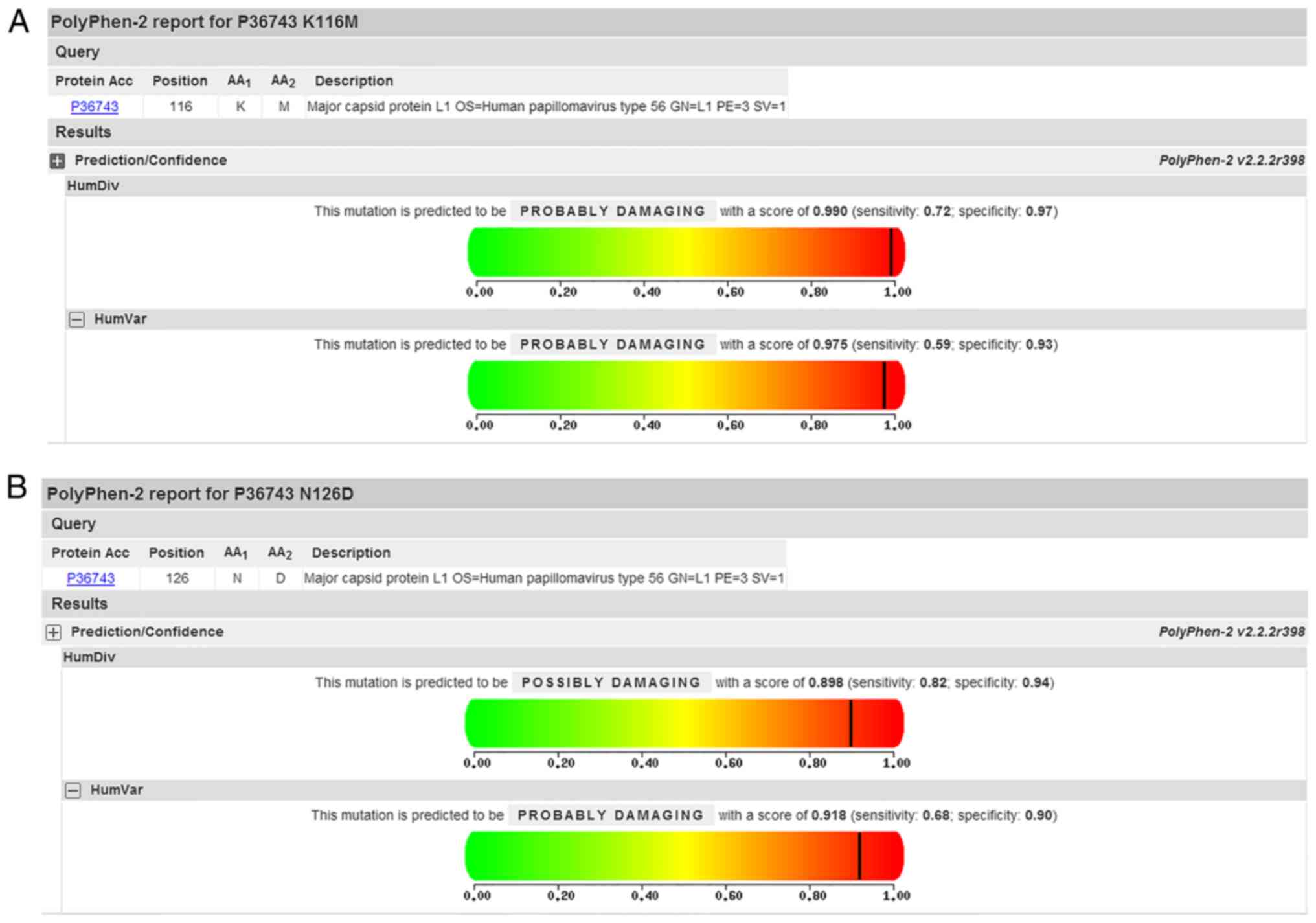
observed in the sequences encoding the b sheet (Table III). In addition, PolyPhen-2
analysis predicted that K116M and N126D mutations of L1 were
‘probably damaging’ on HumVar model with the score of 0.975 and
0.918, respectively (Fig. 4). No
frame shift and premature stop caused by mutations were
observed.
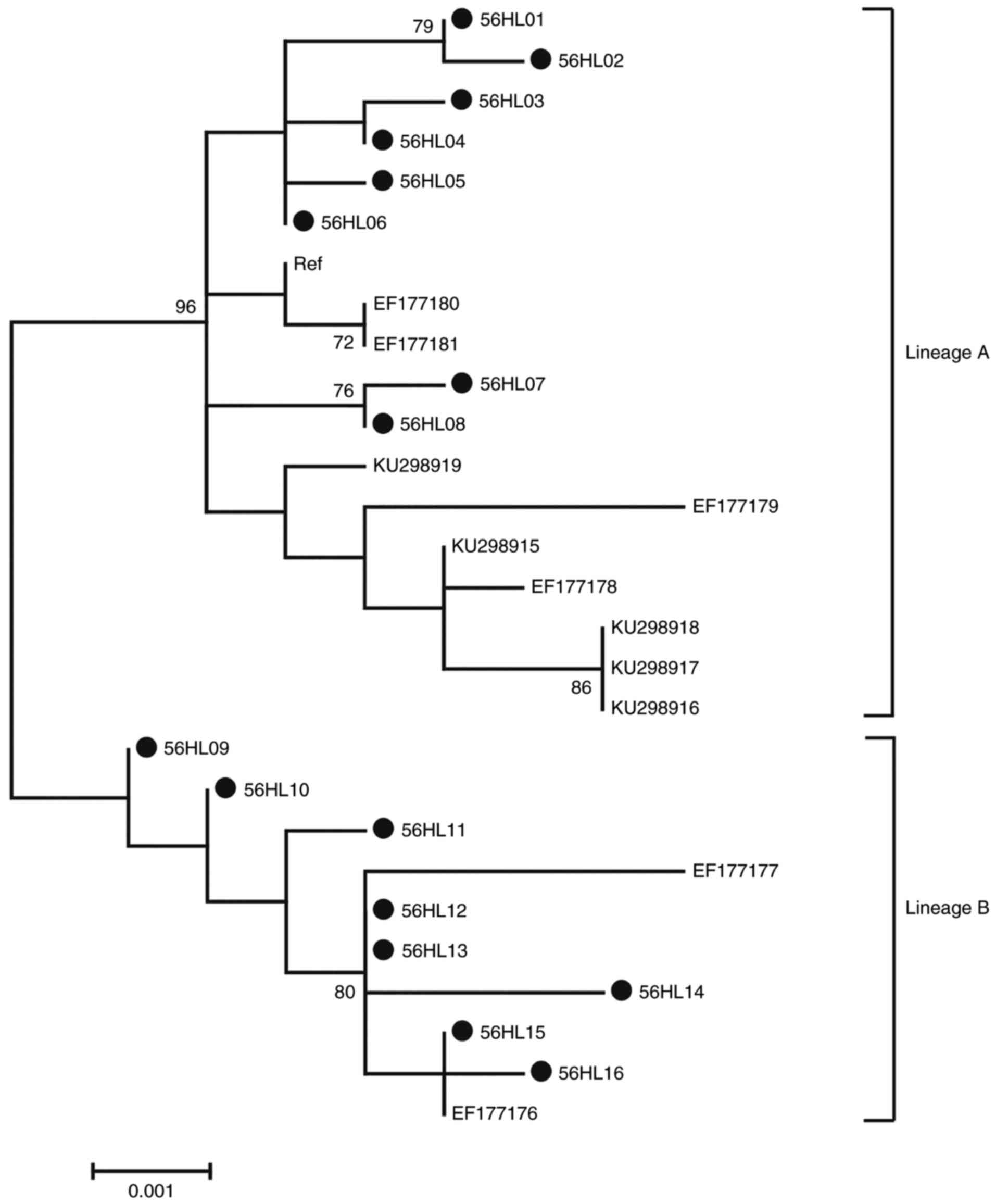
Phylogenetic analysis of HPV-56 L1
sequences
Phylogenetic analysis based on HPV-56 L1 1,605 bp
nucleotide sequences was performed including 16 isolates in the
current study, accompanied by 12 reference sequences. Based on
whole genome study and the topology, 2 distinct branches were
formed (Fig. 5) (32). A total of 8 isolates in the current
study (56HL01-S6HL08) were classified into the A variant lineage.
The remaining 8 isolates (56HL09-S6HL18) were classified into the B
variant lineage.
Selective pressure analysis of HPV 56
E6-E7 and L1 sequences
PAML software was used to estimate the selective
pressure of HPV-56. Positive selection for E6-E7 and L1 was
identified and has been presented in Tables IV and V, respectively. In addition, there was no
evidence of negative selection in the sequence alignment of these
genes (P<0.1).
| Table IV.Site-specific tests for positive
selection on human papillomavirus-56 E6-E7. |
Table IV.
Site-specific tests for positive
selection on human papillomavirus-56 E6-E7.
| Models | InL | Parameters
estimates | 2Δl | E6 positively
selected sites | E7 positively
selected sites |
|---|
| M7 | −1340.45 | p=0.005;
q=0.05 | n/a | NA | NA |
| M8 | −1318.69 | p0=0.9;
p=2.24; q=99; (p1=0.05); ω=15.63 | 43.52;
P<0.01 | 14Sb; 54Kb; 60Db | 10Db; 12Va; 67Ea; 77Qb |
| Table V.Site-specific tests for positive
selection on human papillomavirus-56 L1. |
Table V.
Site-specific tests for positive
selection on human papillomavirus-56 L1.
| Models | InL | Estimates of
parameters | 2Δl | L1 positively
selected sites |
|---|
| M7 | −2542.50 | p=0.005;
q=0.049 | n/a | NA |
| M8 | −2531.74 |
p0=0.986; p=0.005; q=4.034;
(p1=0.014); ω=10.537 | 21.51;
P<0.01 | 510Ka |
Discussion
When considering the association between HPV and
cervical cancer, HPV-16 and HPV-18 are the most frequent worldwide,
with HPV-56 only accounting for a small proportion of the
infections worldwide, with no available commercial vaccines
(33). However, the prevalence of
high-risk HPV types differs greatly among different countries and
regions. Previous studies have revealed a high detection rate of
the HPV-56 strain among Chinese women (9–11).
The purpose of the present study was to determine a ranking of HPV
type prevalence and to demonstrate the importance and significance
of HPV-56 in southwest China.
Previous studies have demonstrated that specific
intratypic HPV genome variations may affect the virus infectivity,
pathogenicity, oncogenicity, viral particle assembly and host
immune response (15,34). In the current study, the genetic
variability of LCR, E6, E7 and L1 genes of HPV-56 in southwest
China were analyzed. Additionally, phylogenetic trees were
constructed for all variant strains and selection pressures of the
E6, E7, and L1 genes were estimated.
The binding affinity of the cellular and viral
transcriptional factor may be influenced by nucleotide variation
within LCR (35). Some of the
mutations identified in the LCR in the present study are embedded
in the putative binding sites for YY1, SRY, NF-1, TATA and C/EBP
transcription factors. These mutations may affect the binding
affinity of transcriptional factor and the transcription of the
downstream E6 and E7 oncogenes; however, it remains to be
determined by in vitro studies. When compared with a
previous study, changes at positions 7,485, 7,504, 7,546, 7,582,
7,589, 7,678, 7,741, 7,781 and 7,796 of LCR, to the best of our
knowledge, were described for the first time in the present study,
and a new 19 bp deletion was not described previously (14).
The early expressing proteins E6 and E7 of high-risk
HPV are the primary oncoproteins involved in human epithelial cell
immortalization and transformation due to their ability to
inactivate p53 and pRb proteins, respectively (36,37).
In addition, amino acid mutation of E6/E7 genes may influence host
immunologic responses (38).
Therefore, studies on the genetic variability in HPV E6/E7 may
provide important basic data for future research on viral molecular
mechanisms and contribute to the design of therapeutic vaccines,
which target these proteins (39).
In the current study, 11/18 amino acid mutations identified in
E6/E7 were non-synonymous, except for A141C, T158C and A263C, all
the remaining mutations were described, to the best of our
knowledge, for the first time in the current study. S14R and K54N
of E6 amino acid substitutions were apparently associated and were
simultaneously found in 47 isolates. It is of note that the present
study identified that the E7 variation frequency is higher than E6
variations, similar to findings in the HPV-58 type (40). Variants of the Q77H/Y changes of E7
was analyzed using PolyPhen-2 with results of ‘possibly damaging’,
at the same time, the variant occurred in the E7 β-sheet, which may
influence E7 structure.
Overall, the L1 major capsid protein may be used to
inform the design of prophylactic vaccines and main components
virus-like particles (VLPs) that induce high levels of neutralizing
antibodies (19). Additionally,
conformational epitopes of HPV are primarily located in
hypervariable immuno-dominant regions (BC, DE, EF, FG and HI
loops). Therefore, previous studies demonstrated that polymorphisms
in the L1 gene may have a critical role in the structure of the
capsid protein, VLPs assembly and recognition epitopes (41–43).
The nucleotide changes detected at positions 6,826 and 6,880 of
HPV-56 L1 gene were previously described (25); however, to the best of our
knowledge the remaining mutations at other positions are described
for the first time in the present study. Variants of K116M and
N126D were analyzed using PolyPhen-2 and the mutation was
identified as ‘probably damaging’. Additionally, C19Y is located in
the sequences encoding the b sheet, those variants may affect L1
structure.
In terms of phylogenetics, the current known HPV
types have high diversity. According to the maximum-likelihood
trees topology constructed by the current study and the reference
strains, the strains are distributed in two clusters, not in one
single cluster (32). Selective
pressure analysis revealed that the majority of the identified HPV
amino acid changes were of positive selection, which indicated that
these mutations were beneficial for HPV to accommodate the
environment.
In summary, the current study reported the
prevalence of HPV-56 and sequence variations in the LCR, E6, E7 and
L1 genes of HPV-56 isolates from southwest China. Some of the
variations identified in LCR are located within binding sites of
transcriptional factors. In addition, the majority of nucleotide
changes were described for the first time by the current study. To
the best of our knowledge, this was the first comprehensive study
to assess genetic variation of HPV-56 in southwest China. The
present study provided important basic data for further research on
viral molecular mechanisms and future development of diagnostic
probes and therapeutic vaccines.
Acknowledgements
The authors would like to thank the following
hospitals for the sample collection: Sichuan Reproductive Health
Research Center Affiliated Hospital, The Angel Women's and
Children's Hospital, The Chengdu Western Hospital Maternity Unit,
The Peoples' Hospital of Peng Zhou, Jinjiang Maternity and Child
Health Hospital, Chengdu Zongnan Gynecology Hospital and Chengdu
Songziniao Sterility Hospital.
Funding
The present study was funded by the Institute of
Medical Genetics, College of Life Science, Sichuan University,
Chengdu, China.
Availability of data and materials
All data generated or analysed during this study are
included in this published article and GeneBank.
Authors' contributions
YJ and XD conceived and designed the study. YJ, TW,
ZC, JX, XM, MC and HC performed the experiments. ZC and JX analyzed
the data. YJ and TW were major contributors in writing the
manuscript. All authors read and approved the final manuscript.
Ethics approval and consent to
participate
The present study was approved by the Education and
Research Committee and the Ethics Committee of Sichuan University
(Chengdu, China; approval number SCU20100196494).
Consent for publication
Prior to sample collection, written informed consent
was obtained from the patients or their guardians, and
patient/study subject privacy was carefully protected.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
de Sanjose S, Quint WG, Alemany L, Geraets
DT, Klaustermeier JE, Lloveras B, Tous S, Felix A, Bravo LE, Shin
HR, et al: Human papillomavirus genotype attribution in invasive
cervical cancer: A retrospective cross-sectional worldwide study.
Lancet Oncol. 11:1048–1056. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Ferlay J SI, Ervik M, Dikshit R, Eser S,
Mathers C, Rebelo M, Parkin DM, Forman D and Bray F: GLOBOCAN 2012
v1.0Cancer Incidence and Mortality Worldwide: IARC CancerBase. No.
11. IARC; Lyon: 2013, http://publications.iarc.fr/Databases/Iarc-Cancerbases/Globocan-2012-Estimated-Cancer-Incidence-Mortality-And-Prevalence-Worldwide-In-2012-V1-0-2012May
30–2012
|
|
3
|
Rampias T, Sasaki C and Psyrri A:
Molecular mechanisms of HPV induced carcinogenesis in head and
neck. Oral Oncol. 50:356–363. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Bernard HU, Burk RD, Chen Z, van Doorslaer
K, zur Hausen H and de Villiers EM: Classification of
papillomaviruses (PVs) based on 189 PV types and proposal of
taxonomic amendments. Virology. 401:70–79. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Bouvard V, Baan R, Straif K, Grosse Y,
Secretan B, El Ghissassi F, Benbrahim-Tallaa L, Guha N, Freeman C,
Galichet L, et al: A review of human carcinogens-Part B: Biological
agents. Lancet Oncol. 10:321–322. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Arbyn M, Tommasino M, Depuydt C and
Dillner J: Are 20 human papillomavirus types causing cervical
cancer? J Pathol. 234:431–435. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Lörincz AT, Quinn AP, Goldsborough MD,
McAllister P and Temple GF: Human papillomavirus type 56: A new
virus detected in cervical cancers. J Gen Virol. 70:3099–3104.
1989. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhai L and Tumban E: Gardasil-9: A global
survey of projected efficacy. Antiviral Res. 130:101–109. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Yang L, Yang H, Wu K, Shi X, Ma S and Sun
Q: Prevalence of HPV and variation of HPV 16/HPV 18 E6/E7 genes in
cervical cancer in women in South West China. J Med Virol.
86:1926–1936. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Zeng Z, Yang H, Li Z, He X, Griffith CC,
Chen X, Guo X, Zheng B, Wu S and Zhao C: Prevalence and genotype
distribution of HPV infection in China: Analysis of 51,345 HPV
genotyping results from China's largest CAP certified laboratory. J
Cancer. 7:1037–1043. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Wang R, Guo XL, Wisman GB, Schuuring E,
Wang WF, Zeng ZY, Zhu H and Wu SW: Nationwide prevalence of human
papillomavirus infection and viral genotype distribution in 37
cities in China. BMC Infect Dis. 15:2572015. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chen Z, Wang Q, Ding X, Li Q, Zhong R and
Ren H: Characteristics of HPV prevalence in Sichuan Province,
China. Int J Gynecol Obstet. 131:277–280. 2015. View Article : Google Scholar
|
|
13
|
Lee K, Magalhaes I, Clavel C, Briolat J,
Birembaut P, Tommasino M and Zehbe I: Human papillomavirus 16 E6,
L1, L2 and E2 gene variants in cervical lesion progression. Virus
Res. 131:106–110. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Cento V, Rahmatalla N, Ciccozzi M, Perno
CF and Ciotti M: Intratype variations of HPV 31 and 58 in Italian
women with abnormal cervical cytology. J Med Virol. 83:1752–1761.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Xi LF, Schiffman M, Koutsky LA, Hughes JP,
Hulbert A, Shen Z, Galloway DA and Kiviat NB: Variant-specific
persistence of infections with human papillomavirus Types 31, 33,
45, 56 and 58 and risk of cervical intraepithelial neoplasia. Int J
Cancer. 139:1098–1105. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Hubert WG: Variant upstream regulatory
region sequences differentially regulate human papillomavirus type
16 DNA replication throughout the viral life cycle. J Virol.
79:5914–5922. 2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Vande Pol SB and Klingelhutz AJ:
Papillomavirus E6 oncoproteins. Virology. 445:115–137. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Roman A and Munger K: The papillomavirus
E7 proteins. Virology. 445:138–168. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Buck CB, Day PM and Trus BL: The
papillomavirus major capsid protein L1. Virology. 445:169–174.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Chan PK, Luk AC, Park JS, Smith-McCune KK,
Palefsky JM, Konno R, Giovannelli L, Coutlée F, Hibbitts S, Chu TY,
et al: Identification of human papillomavirus type 58 lineages and
the distribution worldwide. J Infect Dis. 203:1565–1573. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sun Z, Lu Z, Liu J, Wang G, Zhou W, Yang
L, Liu C and Ruan Q: Genomic polymorphism of human papillomavirus
type 52 in women from Northeast China. Int J Mol Sci.
13:14962–14972. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Hang D, Yin Y, Han J, Jiang J, Ma H, Xie
S, Feng X, Zhang K, Hu Z, Shen H, et al: Analysis of human
papillomavirus 16 variants and risk for cervical cancer in Chinese
population. Virology. 488:156–161. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Shen M, Ding X, Li T, Chen G and Zhou X:
Sequence variation analysis of HPV-18 Isolates in Southwest China.
PLoS One. 8:e566142013. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Prado JC, Calleja-Macias IE, Bernard HU,
Kalantari M, Macay SA, Allan B, Williamson AL, Chung LP, Collins
RJ, Zuna RE, et al: Worldwide genomic diversity of the human
papillomaviruses-53, 56, and 66, a group of high-risk HPVs
unrelated to HPV-16 and HPV-18. Virology. 340:95–104. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Wyant PS, Cerqueira DM, Moraes DS, Leite
JP, Martins CR, de Macedo Brígido M and Raiol T: Phylogeny and
polymorphism in the long control region, E6, and L1 of human
papillomavirus types 53, 56, and 66 in central Brazil. Int J
Gynecol Cancer. 21:222–229. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Clarke KR and Gorley RN: Primer v5: User
Manual\TutorialPRIMER-E. Plymouth: pp. 912001
|
|
27
|
Tamura K, Stecher G, Peterson D, Filipski
A and Kumar S: MEGA6: Molecular evolutionary genetics analysis
version 6.0. Mol Biol Evol. 30:2725–2729. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Buchan DW, Minneci F, Nugent TC, Bryson K
and Jones DT: Scalable web services for the PSIPRED protein
analysis workbench. Nucleic Acids Res. 41:W349–W357. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Adzhubei IA, Schmidt S, Peshkin L,
Ramensky VE, Gerasimova A, Bork P, Kondrashov AS and Sunyaev SR: A
method and server for predicting damaging missense mutations. Nat
Methods. 7:248–249. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kel AE, Gössling E, Reuter I, Cheremushkin
E, Kel-Margoulis OV and Wingender E: MATCHTM: A tool for searching
transcription factor binding sites in DNA sequences. Nucleic Acids
Res. 31:3576–3579. 2003. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Nei M and Gojoborit T: Simple methods for
estimating the numbers of synonymous and nonsynonymous nucleotide
substitutions. Mol Biol Evol. 3:418–426. 1986.PubMed/NCBI
|
|
32
|
Burk RD, Harari A and Chen Z: Human
papillomavirus genome variants. Virology. 445:232–243. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Schiller JT and Lowy DR: Understanding and
learning from the success of prophylactic human papillomavirus
vaccines. Nat Rev Microbiol. 10:681–692. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Pista A, Oliveira A, Barateiro A, Costa H,
Verdasca N and Paixao MT: Molecular variants of human
papillomavirus type 16 and 18 and risk for cervical neoplasia in
Portugal. J Med Virol. 79:1889–1897. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Bernard HU: Regulatory elements in the
viral genome. Virology. 445:197–204. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Moody CA and Laimins LA: Human
papillomavirus oncoproteins: Pathways to transformation. Nat Rev
Cancer. 10:550–560. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Thomas M, Narayan N, Pim D, Tomaić V,
Massimi P, Nagasaka K, Kranjec C, Gammoh N and Banks L: Human
papillomaviruses, cervical cancer and cell polarity. Oncogene.
27:7018–7030. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Chagas BS, Batista MV, Crovella S, Gurgel
AP, Silva Neto Jda C, Serra IG, Amaral CM, Balbino VQ, Muniz MT and
Freitas AC: Novel E6 and E7 oncogenes variants of human
papillomavirus type 31 in Brazilian women with abnormal cervical
cytology. Infect Genet Evol. 16:13–18. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yao Y, Huang W, Yang X, Sun W, Liu X, Cun
W and Ma Y: HPV-16 E6 and E7 protein T cell epitopes prediction
analysis based on distributions of HLA-A loci across populations:
An in silico approach. Vaccine. 31:2289–2294. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Chan PK, Zhang C, Park JS, Smith-McCune
KK, Palefsky JM, Giovannelli L, Coutlée F, Hibbitts S, Konno R,
Settheetham-Ishida W, et al: Geographical distribution and
oncogenic risk association of human papillomavirus type 58 E6 and
E7 sequence variations. Int J Cancer. 132:2528–2536. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Fleury MJ, Touze A and Coursaget P: Human
papillomavirus type 16 pseudovirions with few point mutations in L1
major capsid protein FG loop could escape actual or future
vaccination for potential use in gene therapy. Mol Biotechnol.
56:479–486. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Sapp M and Day PM: Structure, attachment
and entry of polyoma- and papillomaviruses. Virology. 384:400–409.
2009. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Pande S, Jain N, Prusty BK, Bhambhani S,
Gupta S, Sharma R, Batra S and Das BC: Human papillomavirus type 16
variant analysis of E6, E7, and L1 genes and long control region in
biopsy samples from cervical cancer patients in north India. J Clin
Microbiol. 46:1060–1066. 2008. View Article : Google Scholar : PubMed/NCBI
|