Introduction
More than 70% of hereditary hearing loss is
nonsyndromic and exhibits extremely high genetic heterogeneity.
Nonsyndromic hearing impairment (NSHI) exhibits autosomal recessive
inheritance in ~75–80% of cases, autosomal dominant inheritance in
20–25% of cases, and X-linked inheritance in 1–1.5% of cases
(1). Mutations in at least 30
genes have been identified in patients with autosomal dominant
sensory NSHI (ADNSHI).
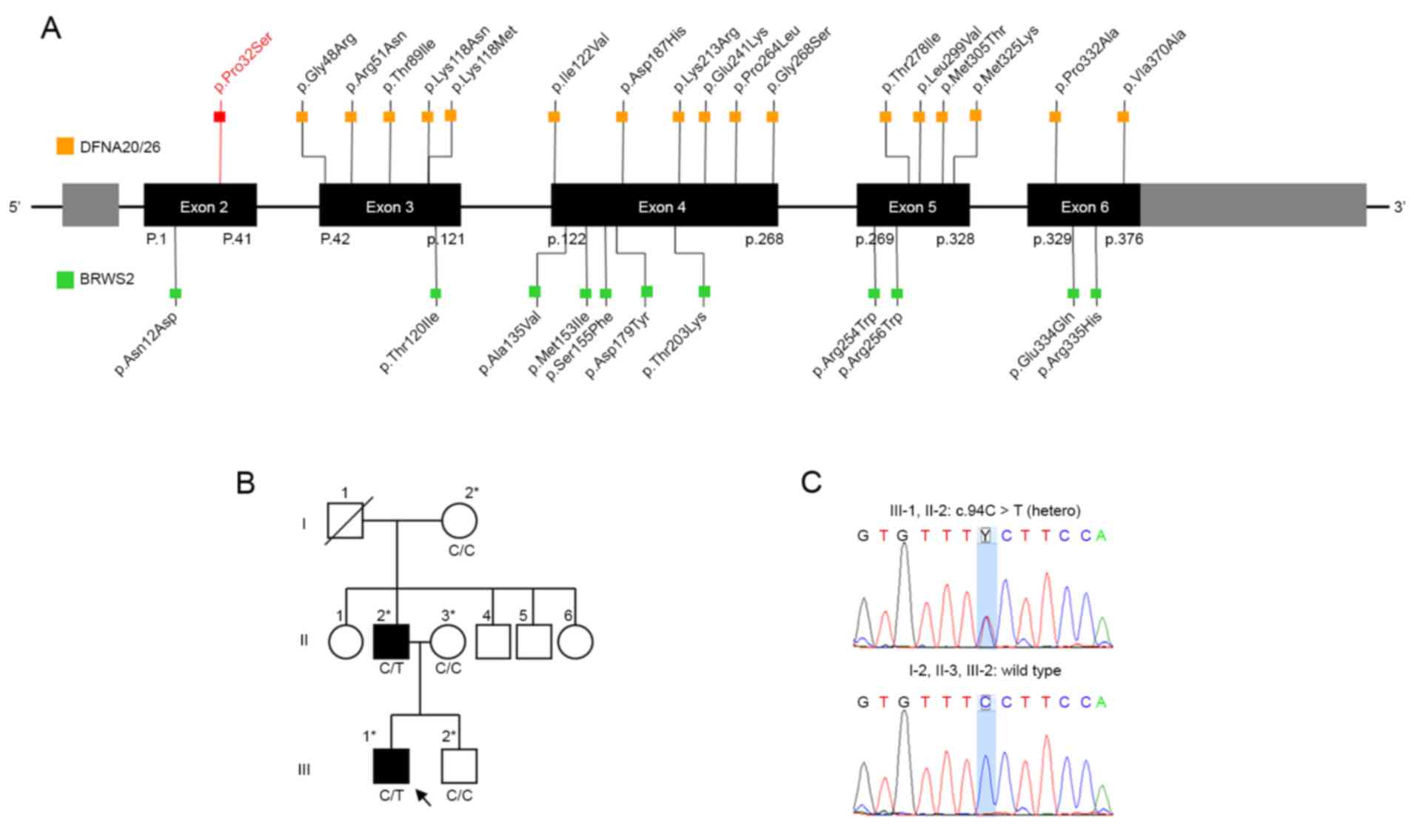
ACTG1 was initially identified as a causative
gene for ADNSHI linked to the DFNA20/26 locus (MIM #604717)
on chromosome 17q25.3 in 2003 (2,3). The
ACTG1 gene encodes a protein called γ-actin, which is part
of the actin protein family. The γ-actin is a cytoskeletal protein,
which makes up the structural framework inside cells. This protein
is particularly abundant in the specialized cells of inner ear
called hair cells, which are essential for normal hearing. Thus
ACTG1 is predicted to be crucial for the shape and function
of the stereocilia of the cochlear hair cells (4–6). To
date, 17 missense mutations in ACTG1 have been reported in
20 families (Fig. 1A; Table I) (2–5,7–15).
The majority of these patients present progressive post-lingual
hearing loss with ages of onset ranging between the first and
fourth decades (13).
 | Table I.ACTG1 variants in autosomal
dominant sensorineural hearing impairment and Baraitser-Winter
cerebrofrontofacial syndrome 2 (BWCFF2). |
Table I.
ACTG1 variants in autosomal
dominant sensorineural hearing impairment and Baraitser-Winter
cerebrofrontofacial syndrome 2 (BWCFF2).
| No. | Author, year | Phenotype | AA change | Exon | Age of onset | N | (Refs.) |
|---|
| 1 | Present study | DFNA20/26 | p.Pro32Ser | 2 | At birth | 1 family |
|
| 2 | Miyagawa et
al, 2015 | DFNA20/26 | p.Gly48Arg | 3 | First decade | 1 family | (7) |
| 3 | Verloes et al,
1993–2016 | DFNA20/26 | p.Arg51Asn | 3 | First decade | 1 family | (14) |
| 4 | Zhu et al,
2003 | DFNA20/26 | p.Thr89Ile | 3 | Third decade | 3 families | (2) |
| 5 | Zhu et al,
2003; Miyagawa et al, 2015 | DFNA20/26 | p.Lys118Asn | 3 | First-third
decade | 1 family | (2,7) |
| 6 | Morín et al,
2009 | DFNA20/26 | p.Lys118Met | 3 | Second-third
decade | 1 family | (4) |
| 7 | Liu et al,
2008 | DFNA20/26 | p.Ile122Val | 4 | First decade | 1 family | (8) |
| 8 | Baek et al,
2012 | DFNA20/26 | p.Asp187His | 4 | First decade (1 year
old) | 1 family | (9) |
| 9 | Yuan et al,
2016 | DFNA20/26 | p.Lys213Arg | 4 | Second decade | 1 family | (5) |
| 10 | Morín et al,
2009; Miyagawa et al, 2015 | DFNA20/26 | p.Glu241Lys | 4 | First decade | 1 family | (4,7) |
| 11 | Zhu et al,
2003 | DFNA20/26 | p.Pro264Leu | 4 | First-second
decade | 1 family | (2) |
| 12 | Mutai et al,
2013 | DFNA20/26 | p.Gly268Ser | 4 | First-fourth
decade | 1 family | (10) |
| 13 | van Wijk et
al, 2003 | DFNA20/26 | p.Thr278Ile | 5 | First-second
decade | 1 family | (3) |
| 14 | Miyagawa et
al, 2015 | DFNA20/26 | p.Leu299Val | 5 | Second decade | 2 families | (7,11) |
| 15 | Park et al,
2013 | DFNA20/26 | p.Met305Thr | 5 | Third-fourth
decade | 1 family | (12) |
| 16 | Vona et al,
2014 | DFNA20/26 | p.Met325Lys | 5 | At birth | 1 family | (13) |
| 17 | Zhu et al,
2003 | DFNA20/26 | p.Pro332Ala | 6 | Second decade | 1 family | (2) |
| 18 | Rendtorff et
al, 2006 | DFNA20/26 | p.Val370Ala | 6 | First-second
decade | 1 family | (15) |
| 1 | Di Donato et
al, 2016 | BWCFF 2 | p.Asn12Asp | 2 |
| 1 patient | (16) |
| 2 | Rivière et
al, 2012 | BWCFF 2 | p.Thr120Ile | 3 |
| 1 patient | (17) |
| 3 | Rivière et
al, 2012; Verloes et al, 2015 | BWCFF 2 | p.Ala135Val | 4 |
| 2 patients | (17,18) |
| 4 | Poirier et
al, 2015 | BWCFF 2 | p.Met153Ile | 4 |
| 1 patient | (19) |
| 5 | Rivière et
al, 2012 | BWCFF 2 | p.Ser155Phe | 4 |
| 3 patients | (17) |
| 6 | Di Donato et
al, 2016 | BWCFF 2 | p.Asp179Tyr | 4 |
| 1 patient | (16) |
| 7 | Rivière et
al, 2012 | BWCFF 2 | p.Thr203Lys | 4 |
| 1 patient | (17) |
| 8 | Di Donato et
al, 2016; Rivière et al, 2012 | BWCFF 2 | p.Arg254Trp | 4 |
| 2 patients | (16,17) |
| 9 | Di Donato et
al, 2016; Rivière et al, 2012 | BWCFF 2 | p.Arg256Trp | 4 |
| 2 patients | (16,17) |
| 10 | Di Donato et
al, 2016 | BWCFF 2 | p.Glu334Gln | 6 |
| 1 patient | (16) |
| 11 | Di Donato et
al, 2016 | BWCFF 2 | p.Arg335His | 6 |
| 1 patient | (16) |
The present study presented a small family
exhibiting autosomal dominant, congenital, sensorineural hearing
loss with an autosomal dominant inherited novel heterozygous C-to-T
transition at nucleotide 94 (p.Pro32Ser) of the ACTG1 gene
identified using the TruSight One sequencing panel. The proband was
the first case of congenital hearing loss identified by newborn
hearing screening. The present study provided a new insight into
the genotype-phenotype correlation for ACTG1 mutations in
ADNSHI.
Case report
A small family with ADNSHI was identified at the
Eulji Medical Center (Seoul, Korea). The pedigree chart is
illustrated in Fig. 1B, and the
results of Sanger sequencing are presented in Fig. 1C.
The Institutional Review Board of Eulji General
Hospital in Seoul, Korea (IRB approval no. 2014-06-007-001)
approved the use of medical records and 2 ml peripheral blood
samples in the present study. Written informed consent for genetic
testing and medical photography was obtained from all subjects
prior to participation.
Patient 1 (proband)
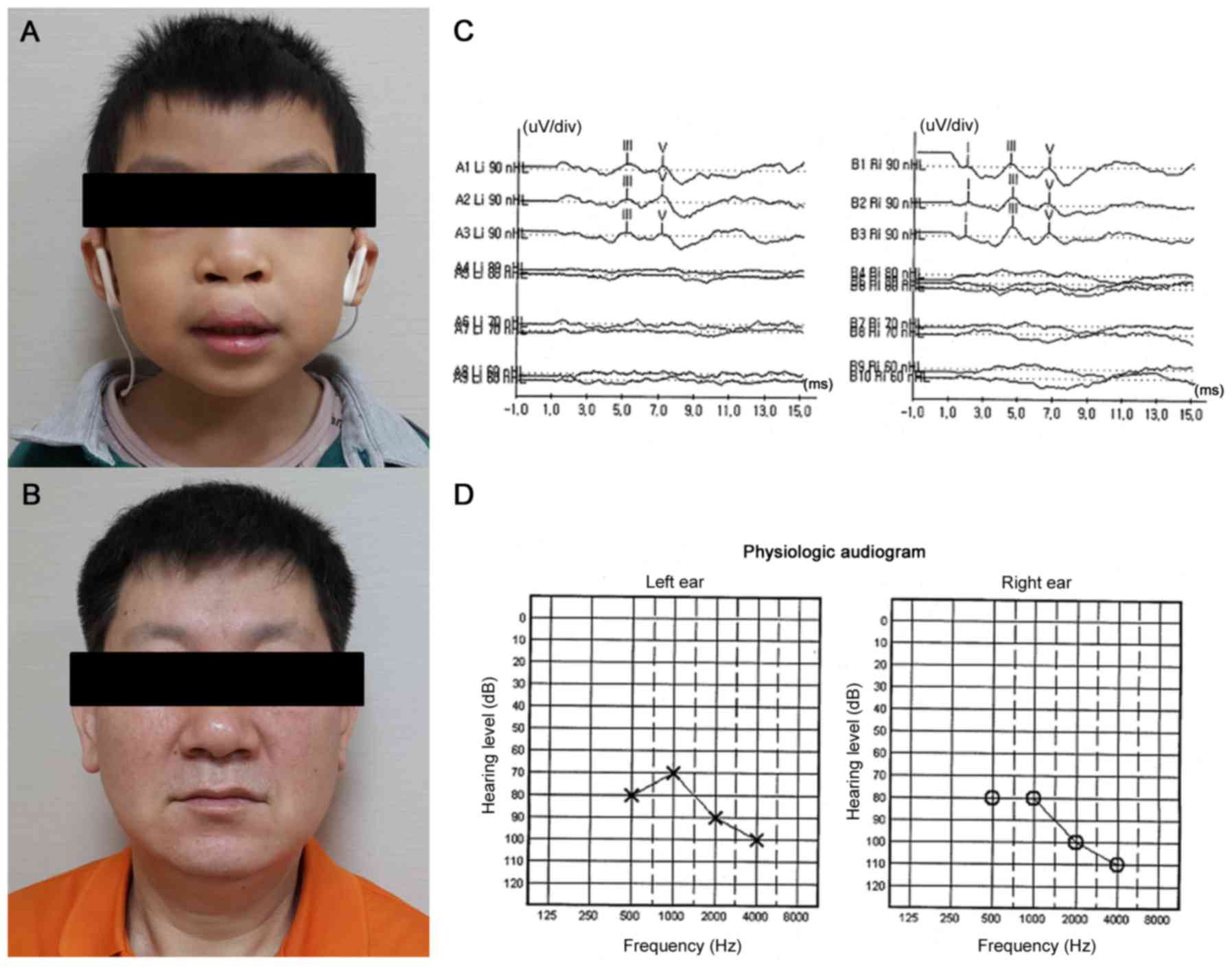
A boy was born at term with a birth weight of 2,770
g (10–25th percentile) and an occipitofrontal circumference of 34.5
cm (75–90th percentile) as the first child of a non-consanguineous
33-year-old Filipina mother and a 49-year-old Korean father
(Fig. 2A and B). He had one
sibling who was healthy. At birth, he had a complete bilateral
cleft lip and palate. He did not pass the initial screening test
with otoacoustic emissions at birth. He exhibited bilateral severe
hearing loss on auditory brainstem response at threshold 90 dB
above normal adult hearing level (nHL; Fig. 2C) and auditory steady-state
response audiometry at threshold 80 dB nHL (Fig. 2D). He underwent surgical repair of
the cleft lip (cheiloplasty) at 4 months and cleft palate
(palatoplasty) at 12 months. He received cochlear implant (CI)
surgery in the right ear at 22 months and in the left ear at 4
years. When he visited our clinic (Nowon Eulji Medical Center,
Eulji University, Seoul, Republic of Korea) at 4 years and 6 months
old, his body weight was 17.8 kg [+0.00 standard deviation (SD)],
height was 99.9 cm (−1.53 SD), and head circumference was 53 cm
(+1.44 SD). On examination, he presented a distinctive face owing
to the reconstructed philtrum and upper lip by cheiloplasty
(Fig. 2A). He had no coloboma or
ptosis in an eye examination performed by an ophthalmologist. There
were no visible abnormalities of the external ear. There were no
skeletal abnormalities. He exhibited no seizures, movement
problems, or other behavioral problems. He revealed developmental
language delay based on the sequenced language scale of infants at
4 years and 5 months of age; expressive language function was at
the 21-month-old level, and receptive language function was at the
23-month-old level. This was attributed to congenital deafness with
relatively delayed cochlear implants. The results of brain magnetic
resonance imaging (MRI) were normal. There were no associated
structural ear abnormalities in temporal bone computer tomography
findings. Electrocardiography and chest radiography results were
normal. Echocardiograms exhibited a very small patent ductus
arteriosus. No evidence of malformation of the kidneys was observed
in an abdominal ultrasound evaluation. The laboratory tests,
including a complete blood count, chemistry panel, lipid profile,
thyroid function test, and urinalysis, were all normal.
Patient 2 (father)
The patient's father first visited our clinic at 53
years of age with his son. He was the second child of healthy
non-consanguineous Korean parents. There was no family history of
neurologic disease, deafness, or developmental delay, with the
exception of his son. At this visit, he exhibited profound
pre-lingual deafness (Fig. 2B). He
did not acquire speech and language. On examination, he had no
specific dysmorphic craniofacial features. Brain MRI was
normal.
Results
Cytogenetic analysis
To identify the causative genetic alterations in the
proband (III-1), cytogenetic and analysis were performed.
Peripheral blood lymphocytes were cultured for 72 h according to
routine cytogenetic protocols. G-banding normal chromosomes (46,
XY) was found in the proband. A genomic microarray of DNA from the
proband's peripheral blood using a 750K High-resolution Genotyping
SNP Microarray (Affymetrix; Thermo Fisher Scientific, Inc.,
Waltham, MA, USA) revealed no abnormalities.
Mutation analysis
Considering the genetic heterogeneity of the disease
and for differential diagnosis, clinical sequencing of regions
containing Mendelian disease-causing genes in the proband (III-1)
was performed for genetic confirmation. Genomic DNA was extracted
from peripheral blood leukocytes using the Wizard Genomic DNA
Purification kit following the manufacturer's protocols (Promega
Corporation, Madison, WI, USA). Library preparation was conducted
using TruSight One Sequencing Panel (Illumina, Inc., San Diego, CA,
USA), which included 125,395 probes targeting a 12-Mb region
spanning 4,813 genes. Massively parallel sequencing was performed
using Illumina MiSeq platform (Illumina, Inc.). Sequence reads were
aligned to hg19 using Burrow-Wheeler Aligner (version 0.7.12).
Duplicate reads were removed by Picard (version 1.96, http://picard.sourceforge.net). Local realignment and
base quality recalibration were conducted using Genome Analysis
Toolkit (GATK version 3.5). Variant calling was performed using
GATK HaplotypeCaller. Variants were annotated using Variant Effect
Predictor and dbNSFP (version 3.0b2a). Sanger sequencing was used
to determine the genotypes of affected and unaffected family
members at candidate loci. From sequencing data of the proband with
the TruSight One Panel, it was expected to discover a heterozygote
ADNSHI-causing mutation in this family. Mutations in known genes
causing autosomal recessive in addition to autosomal dominant
syndromic/nonsyndromic hearing loss were also evaluated in the
exome data of the proband. Two missense mutations were detected:
c.94C>T (p.Pro32Ser) in MYO6 and c.94C>T (p.Pro32Ser)
in ACTG1. The mutation c.94C>T (p.Pro32Ser) in
MYO6 displayed lack of segregation in affected and
unaffected members of this family. The mutation was not observed in
another affected family member (II-2) and was observed in two
unaffected family members (II-3 and III-2). In contrast, the
heterozygous novel missense mutation, c.94C>T (p.Pro32Ser), in
the ACTG1 gene was supported as a disease-causing mutation
based on segregation in the family. Sanger sequencing confirmed
that the affected family members (III-1 and II-2) were heterozygous
for the mutation, while the mutation was not observed in the
unaffected family members (I-2, II-3 and III-2; Fig. 1C). This mutation is absent from
controls in NHLBI GO Exome Sequencing Project (http://evs.gs.washington.edu/EVS/), 1000 Genomes
Project Phase 3 (http://browser.1000genomes.org), Exome Aggregation
Consortium v0.3 (http://exac.broadinstitute.org/), and Korean Reference
Genome Database (http://152.99.75.168/KRGDB/menuPages/introKor.jsp).
Multiple lines of evidence from in silico prediction
indicated that the mutation was potentially deleterious. The
characteristics of the novel missense mutation are summarized in
Table II.
| Table II.Summary of the disease-causing novel
mutation in ACTG1. |
Table II.
Summary of the disease-causing novel
mutation in ACTG1.
| Gene | ACTG1 | (Refs.) |
|---|
| Position
(hg19) | chr17q25
(79,479,287 base pair) |
|
| Exome | 2/7 |
|
| Accession
number | NM_001614.3 |
|
| Nucleotide
change | c.94C>T,
hetero |
|
| Amino acid
change | p.Pro32Ser |
|
|
HGMD® | None | (20) |
| Classification by
ACMG guidelines | Likely
pathogenic | (21) |
| Allele frequency in
controls |
|
|
| dbSNP
142 | No rsID |
|
|
1000Gp3 | None |
|
|
ESP6500 | None |
|
|
ExAC | None |
|
|
KRGdb | None |
|
| Missense
prediction |
|
|
| SIFT_
pred | Deleterious |
|
| LRT_
pred | Unknown |
|
|
PolyPhen2 HDIV_ pred | Probably
damaging |
|
|
Polyphen2_HVAR_ pred | Probably
damaging |
|
|
MutationTaster_pred | Disease
causing |
|
|
MutationAssessor_pred | Medium predicted
functional |
|
|
MetaSVM_pred | Deleterious |
|
|
MetaLR_pred | Deleterious |
|
|
FATHMM_pred | Deleterious |
|
|
PROVEAN_pred | Damaging |
|
|
CADD_phred | 16.96 |
|
| Nucleotide
conservation prediction |
|
|
|
GEFF++_RS | 3.99 |
|
|
PhyloP7way_vertebrate | 0.917 |
|
|
phastCons7way_vertebrate | 0.98 |
|
|
SiPhy_29way_logOdds | 15.0358 |
|
In addition, genes known to serve a role in
craniofacial development including cleft lip and palate were
examined. A missense mutation c.251A>T (p.Glu84Val) in the
MSX1 gene was identified but lacked segregation in this
family, as the mutation was also observed in two unaffected family
members (II-3 and III-2).
Structural modelling of
p.Pro32Ser
The effect of the p.Pro32Ser mutation on the
structure and function of the ACTG1 protein product,
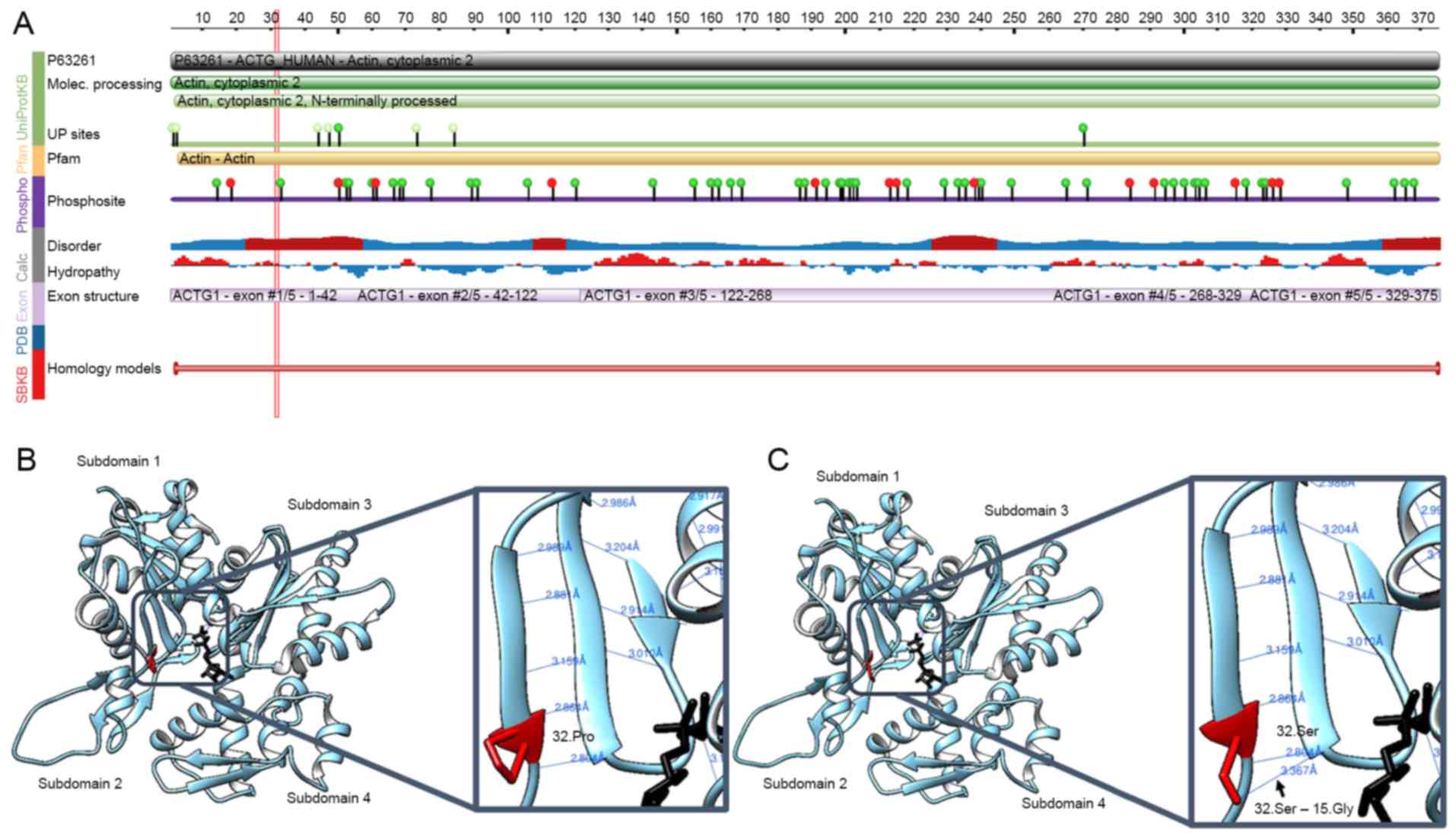
γ-actin, was next assessed. The p.Pro32Ser region of ACTG1
was checked against the protein feature view of the Protein Data
Bank (http://www.wwpdb.org/) entries mapped to
a UniProt Knowledgebase (http://www.uniprot.org) sequence. The native amino
acid at position 32 is hydrophobic and in a predicted potentially
disordered region (Fig. 3A). The
substitution of proline (nonpolar, hydrophobic) by serine
(uncharged polar, hydrophilic) is likely to change the
hydrophobicity of the hydrophobic region and influence hydrophobic
interactions. It may affect protein folding or decrease the
stability of the native protein structure of γ-actin. Pro32Ser is
also close to a phosphorylation site (Fig. 3A). Serine is one of most commonly
phosphorylated amino acids. A substitution by serine could affect
protein phosphorylation and post-translational modifications. In
silico predictions were performed to infer the
three-dimensional structure of γ-actin using UCSF chimera
(https://www.cgl.ucsf.edu/chimera). The
32.Pro in wild-type γ-actin participates in two hydrogen bonds
(distances, 2.804 Å and 2.864 Å), as demonstrated in Fig. 3B. Structural modeling of mutant
32.Ser identified a new hydrogen bond (distance, 3.367 Å) that
interacted with 15.Gly (Fig. 3C).
Hydrogen bonding serves a particularly important role in secondary
structure formation and the determination of three-dimensional
structures. Prolines are flexible amino acids and are often in β
turns with glycine. In a β turn, there is a tight 180° reversal in
the direction of the polypeptide chain, and proline can readily
assume the cis configuration, which facilitates a tight turn.
Accordingly, the Pro32Ser mutation may result in a conformational
change in γ-actin. Molecular modelling analyses therefore predicted
alterations in γ-actin and actin-based structures following this
mutation.
Discussion
The auditory hair cells are located within the
spiral organ of Corti on the thin basilar membrane in the cochlea
of the inner ear. The inner hair cells transform the sound
vibrations in the fluids of the cochlea into electrical signals
that are then relayed via the auditory nerve to the auditory
brainstem and to the auditory cortex. Humans and other mammals are
generally incapable of regrowth of the inner ear cells that convert
sound into neural signals when those cells are damaged by age or
disease. Actins are highly conserved proteins involved in cell
motility and cytoskeletal maintenance. γ-Actin is encoded by the
ACTG1 gene and is identified in the cytoplasm of non-muscle
cells. It is abundant in the auditory hair cells of the cochlea.
Actin filaments are essential for the shape and function of the
stereocilia of hair cells. An alteration in actin filament
regulation caused by actin-binding proteins is a major factor in
deafness caused by ACTG1 mutations (6). The DFNA20/26 phenotype includes
hearing loss with post-lingual onset and an increasing degree of
hearing loss with age (DFNA20/26, MIM# 604717). Impaired actin
structures may be more susceptible to age-dependent degeneration,
resulting in progressive late-onset, post-lingual hearing loss. In
patients with very early-onset hearing loss, suspected hearing loss
is rarely reported (13). The
proband in the present study was the first case of congenital
hearing loss to be identified by a newborn hearing screening test.
His father exhibited pre-lingual profound hearing impairment. It is
hypothesised that congenital and pre-lingual-onset hearing loss is
possible in patients with ADNSHI associated with ACTG1
mutations.
A missense mutation in ACTG1 has also been
identified in patients with Baraitser-Winter cerebrofrontofacial
syndrome 2 (BWCFF2, MIM# 614583), which is a multiple congenital
malformation syndrome including specific facial gestalt, short
status, brain malformation, and sensorineural hearing loss
(16). Eleven mutations in 16
patients have been detected in BWCFF2 to date (Fig. 1A; Table I). All mutations in BWCFF2, in
addition to DFNA20/26, are heterozygous missense mutations. The
spectrum of pathogenic mutations observed in DFNA20/26 does not
overlap with the spectrum of pathogenic mutations observed in
BWCFF2 (Fig. 1A). Mutations
causing DFNA20/26 and BWCFF2 are distributed over the entire exome
without regional clusters, as shown in Fig. 1A. The molecular genetic mechanisms
of ACTG1 heterozygous missense mutations in DFNA20/26 and
BWCFF2 are unclear; this is an important open question for future
research. Up to 83% of patients with BWCFF2 have congenital hearing
loss. Riviere et al (17)
suggested that BWCFF2 represents the severe end of a spectrum of
cytoplasmic actin-associated phenotypes that begins with ADNSHI and
extends to BWCFF2. In the present study, the father (II-2) carried
a de novo mutation in ACTG1 and presented with
non-syndromic pre-lingual deafness. The proband (III-1) had
additional features of cleft lip and cleft palate. While cleft lip
and cleft palate may be identified in various craniofacial
syndromes, environmental influences may also cause or interact with
genetics to produce orofacial clefting. In addition, the proband
exhibited no other minor or major anomalies suggestive of BWCFF2
with the exception of cleft lip and palate.
Advances in the acquisition of clinical and
molecular data may improve molecular genetic diagnoses, including
those based on ACTG1 mutations, and may contribute to
therapeutic decisions. Previous studies have suggested that CI
surgery is a good therapeutic option for patients with ACTG1
mutations, as the etiology involves the cochlea and normal brain
structures; therefore, comparatively improved outcomes are
predicted (7,11). In the present study, the proband
with congenital deafness underwent CI surgery. Although cochlear
implantation was performed late, at 2 and 4 years old, he received
language therapy and has the ability to communicate. The present
findings support the recommendation for early CI surgery in
patients with ACTG1 mutations.
In summary, a novel heterozygous missense mutation
P32S in the ACTG1 gene was identified in a small family with
autosomal dominant nonsyndromic hearing loss. The present findings
expand our understanding of the phenotypes associated with
ACTG1. Specifically, the results of the present study
emphasized that mutations in ACTG1 result in a diverse
spectrum of onset ages, including congenital in addition to
post-lingual onset.
Acknowledgements
This research was supported by the Basic Science
Research Program through the National Research Foundation of Korea
funded by the Ministry of Science, ICT and Future Planning (grant
no. 2014R1A1A1007569).
References
|
1
|
Smith RJH, Shearer AE, Hildebrand MS and
Van Camp G: Deafness and hereditary hearing loss
overviewGeneReviews. Pagon RA, Adam MP, Ardinger HH, et al:
University of Washington; Seattle: 1993–2016
|
|
2
|
Zhu M, Yang T, Wei S, DeWan AT, Morell RJ,
Elfenbein JL, Fisher RA, Leal SM, Smith RJ and Friderici KH:
Mutations in the gamma-actin gene (ACTG1) are associated with
dominant progressive deafness (DFNA20/26). Am J Hum Genet.
73:1082–1091. 2003. View
Article : Google Scholar : PubMed/NCBI
|
|
3
|
van Wijk E, Krieger E, Kemperman MH, De
Leenheer EM, Huygen PL, Cremers CW, Cremers FP and Kremer H: A
mutation in the gamma actin 1 (ACTG1) gene causes autosomal
dominant hearing loss (DFNA20/26). J Med Genet. 40:879–884. 2003.
View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Morín M, Bryan KE, Mayo-Merino F, Goodyear
R, Mencía A, Modamio-Høybjør S, del Castillo I, Cabalka JM,
Richardson G, Moreno F, et al: In vivo and in vitro effects of two
novel gamma-actin (ACTG1) mutations that cause DFNA20/26 hearing
impairment. Hum Mol Genet. 18:3075–3089. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Yuan Y, Gao X, Huang B, Lu J, Wang G, Lin
X, Qu Y and Dai P: Phenotypic Heterogeneity in a DFNA20/26 family
segregating a novel ACTG1 mutation. BMC Genet. 17:332016.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Bryan KE, Wen KK, Zhu M, Rendtorff ND,
Feldkamp M, Tranebjaerg L, Friderici KH and Rubenstein PA: Effects
of human deafness gamma-actin mutations (DFNA20/26) on actin
function. J Biol Chem. 281:20129–20139. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Miyagawa M, Nishio SY, Ichinose A, Iwasaki
S, Murata T, Kitajiri S and Usami S: Mutational spectrum and
clinical features of patients with ACTG1 mutations identified by
massively parallel DNA sequencing. Ann Otol Rhinol Laryngol. 124
Suppl 1:84S–93S. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Liu P, Li H, Ren X, Mao H, Zhu Q, Zhu Z,
Yang R, Yuan W, Liu J, Wang Q and Liu M: Novel ACTG1 mutation
causing autosomal dominant non-syndromic hearing impairment in a
chinese family. J Genet Genomics. 35:553–558. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Baek JI, Oh SK, Kim DB, Choi SY, Kim UK,
Lee KY and Lee SH: Targeted massive parallel sequencing: The
effective detection of novel causative mutations associated with
hearing loss in small families. Orphanet J Rare Dis. 7:602012.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Mutai H, Suzuki N, Shimizu A, Torii C,
Namba K, Morimoto N, Kudoh J, Kaga K, Kosaki K and Matsunaga T:
Diverse spectrum of rare deafness genes underlies early-childhood
hearing loss in Japanese patients: A cross-sectional, multi-center
next-generation sequencing study. Orphanet J Rare Dis. 8:1722013.
View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Miyagawa M, Nishio SY, Ikeda T, Fukushima
K and Usami S: Massively parallel DNA sequencing successfully
identifies new causative mutations in deafness genes in patients
with cochlear implantation and EAS. PLoS One. 8:e757932013.
View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Park G, Gim J, Kim AR, Han KH, Kim HS, Oh
SH, Park T, Park WY and Choi BY: Multiphasic analysis of whole
exome sequencing data identifies a novel mutation of ACTG1 in a
nonsyndromic hearing loss family. BMC Genomics. 14:1912013.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Vona B, Müller T, Nanda I, Neuner C,
Hofrichter MA, Schröder J, Bartsch O, Läßig A, Keilmann A, Schraven
S, et al: Targeted next-generation sequencing of deafness genes in
hearing-impaired individuals uncovers informative mutations. Genet
Med. 16:945–953. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Verloes A, Drunat S, Pilz D and Di Donato
N: Baraitser-Winter cerebrofrontofacial syndromeGeneReviews. Pagon
RA, Adam MP, Ardinger HH, et al: University of Washington; Seattle:
1993–2016
|
|
15
|
Rendtorff ND, Zhu M, Fagerheim T, Antal
TL, Jones M, Teslovich TM, Gillanders EM, Barmada M, Teig E, Trent
JM, et al: A novel missense mutation in ACTG1 causes dominant
deafness in a Norwegian DFNA20/26 family, but ACTG1 mutations are
not frequent among families with hereditary hearing impairment. Eur
J Hum Genet. 14:1097–1105. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Di Donato N, Kuechler A, Velgano S,
Heinritz W, Bodurtha J, Merchant SR, Breningstall G, Ladda R, Sell
S, Altmüller J, et al: Update on the ACTG1-associated
Baraitser-Winter cerebrofrontofacial syndrome. Am J Med Genet A.
170:2644–2651. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Rivière JB, van Bon BW, Hoischen A,
Kholmanskikh SS, O'Roak BJ, Gilissen C, Gijsen S, Sullivan CT,
Christian SL, Abdul-Rahman OA, et al: De novo mutations in the
actin genes ACTB and ACTG1 cause Baraitser-Winter syndrome. Nat
Genet. 44:440–444. 2012. View
Article : Google Scholar : PubMed/NCBI
|
|
18
|
Verloes A, Di Donato N, Masliah-Planchon
J, Jongmans M, Abdul-Raman OA, Albrecht B, Allanson J, Brunner H,
Bertola D, Chassaing N, David A, et al: Baraitser-Winter
cerebrofrontofacial syndrome: Delineation of the spectrum in 42
cases. Eur J Hum Genet. 23:292–301. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Poirier K, Martinovic J, Laquerrière A,
Cavallin M, Fallet-Bianco C, Desguerre I, Valence S,
Grande-Goburghun J, Francannet C, Deleuze JF, et al: Rare ACTG1
variants in fetal microlissencephaly. Eur J Med Genet. 58:416–418.
2015. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Human Gene Mutation Database Professional.
version 2017.1. http://www.hgmd.cf.ac.uk
|
|
21
|
Richards S, Aziz N, Bale S, Bick D, Das S,
Gastier-Foster J, Grody WW, Hegde M, Lyon E, Spector E, et al:
Standards and guidelines for the interpretation of sequence
variants: A joint consensus recommendation of the American college
of medical genetics and genomics and the association for molecular
pathology. Genet Med. 17:405–424. 2015. View Article : Google Scholar : PubMed/NCBI
|