Introduction
Endometriosis is characterized by endometriotic
tissue growth outside the uterine cavity. The pathophysiological
mechanism of this disorder remains unknown (1). It is a common, chronic, benign,
estrogen-dependent, gynecological disorder associated with pelvic
pain and infertility (2). Numerous
studies have indicated that endometriosis is associated with
increased endometrial stromal cell (ESC) survival and invasion,
suggesting a link between a heightened menstrual repair response
and ectopic endometrial implant formation (3–5). ESC
survival and migration is controlled by a complex array of
hormones, growth factors, chemokines and inflammatory mediators,
involving signaling through phosphatidylinositol-4,5-biphosphate
3-kinase/AKT and mitogen-activated protein kinase (MAPK) pathways,
particularly MAPK/c-Jun N-terminal kinase (JNK) signaling (6). Therefore, therapies that inhibit ESC
migration and promote apoptosis may have the potential to
effectively treat endometriosis.
Mitochondria are central to a variety of cellular
physiological processes, including bioenergetic regulation,
cellular oxidation-reduction status maintenance and apoptosis
induction (7). The mitochondrial
network is associated with mitochondrial dynamics and the
morphology ranges from a highly interconnected and elongated to a
highly fragmented and punctated network (8). The primary mitochondrial dynamics
machinery is responsible for mitochondrial membrane fusion and
fission. Changes in mitochondrial fission have been reported as an
early event occurring in cancer cell proliferation, apoptosis,
metabolism, cell motility and migration (9,10).
Mitochondrial fission contributes to the mitochondrial pathway of
apoptosis via the induction of cardiolipin oxidation by
mitochondrial reactive oxygen species (mROS) and mitochondrial
permeability transition pore (mPTP) opening, induced by hexokinase
2 (HK2)/voltage-dependent anion channel 1 (VDAC1) dissociation
(11). ESC apoptosis and migration
are pathogenic factors in endometriosis (12,13).
Cell motility is regulated by the balance of F-actin/G-actin.
Notably, these actin proteins also facilitate the recruitment of
dynamin-related protein 1 (Drp1) to the mitochondria and subsequent
mitochondrial fission (14,15).
Previous study has confirmed that the accumulation of actin
filaments at future fission sites increases the rate of fission,
indicating that mitochondrial fission is associated with the
migration of malignant cells through the influence of actin
homeostasis (16). However, the
ability of mitochondrial fission to modulate the apoptosis and
migration of ESCs remains unknown.
Zearalenone (ZEA) is a non-steroidal mycotoxin
produced by several fungi of the genus Fusarium (17). Accumulating evidence has
demonstrated that ZEA may regulate the cancer cell cycle (18), mitochondrial metabolism (19) and apoptosis (20). However, whether ZEA promotes ESC
apoptosis through mitochondrial integrity modification remains
unknown. Therefore, the role of mitochondrial fission in ESC
apoptosis and migration were investigated in the present study. The
protective mechanism of ZEA on ESCs was also examined. The results
revealed that ZEA induces mitochondrial fission via amplification
of the JNK/Drp1 pathway. Furthermore, increased mitochondrial
fission caused ESC apoptosis by inducing functional and structural
mitochondrial damage. ZEA-mediated mitochondrial fission also
impaired ESC migration through the promotion of F-actin
depolymerization.
Materials and methods
Ethics statement
The present study was conducted in accordance with
the Declaration of Helsinki. The experimental protocol was approved
by the Ethics Committee of the Department of Gynecology and
Obstetrics, Beijing Tongren Hospital (Beijing, China).
Cell culture
The human ESCs was an American Type Culture
Collection (ATCC; Manassas, VA, USA) culture (ATCC-CRL-4003; lot
no. 3857441) purchased from LGC Promochem GmbH, Teddington, UK.
ESCs were cultured according to ATCC recommendations in RPMI medium
(Thermo Fisher Scientific, Inc., Waltham, MA, USA) supplemented
with 10% fetal bovine serum (HyClone; GE Healthcare Life Sciences,
Logan, UT, USA) (21), 1%
L-glutamine and 0.5% gentamycin (Sigma-Aldrich; Merck KGaA,
Darmstadt, Germany) at 37°C in 5% CO2. Increasing
concentrations of ZEA (1, 5, 10 and 20 µM; Z2125; Sigma-Aldrich;
Merck KGaA, Darmstadt, Germany) were applied to ESCs for 24 h once
70–80% confluence was reached (22).
Effects of ZEA on cell viability
To analyze the effects of various ZEA concentrations
(1–20 µM) on ESC viability, an MTT assay was performed. ESCs were
seeded into a 96-well plate (103 cells/well) (23) and 20 µl MTT (5 mg/ml PBS; pH 7.4;
Sigma-Aldrich; Merck KGaA) was subsequently added to the medium for
4 h. The supernatant was discarded and 100 µl dimethyl sulfoxide
was added to each well for 10 min. The optical density was measured
at 490 nm (24,25).
Cell proliferation, migration and
scratch assay
Cell proliferation was measured with a cell counting
kit-8 (CCK-8) (Beyotime Institute of Biotechnology, Beijing, China)
assay. Cell suspension (200 µl) was seeded in 96-well cell culture
plates at a density of 1,000 cells/well and incubated at 37°C for
1–4 days, as previously described (26). Cell migration was analyzed using a
Transwell chamber assay (1×105 cells; pore size, 8 µm)
with a polycarbonate membrane, as previously described (27,28).
For the scratch assay, cells were cultured in
serum-free medium for 24 h and were subsequently scratched with
pipette tips, as described previously (29). Wound healing was observed for 48 h
and images were captured using a light microscope (Olympus DX51;
Olympus Corporation, Tokyo, Japan) every 24 h.
Western blot analysis
Cells were lysed with radioimmunoprecipitation assay
buffer (Beyotime Institute of Biotechnology) supplemented with
phenylmethylsufonyl fluoride (Beyotime Institute of Biotechnology).
Protein concentration was determined using a bicinchoninic acid
protein assay. Protein (50 µg) was separated by 10% SDS-PAGE and
transferred to polyvinylidene difluoride membranes. The membranes
were subsequently blocked with 5% non-fat milk for 1 h at room
temperature prior to incubation with the following primary
antibodies: GAPDH (1:1,000; 5174; CST Biological Reagents Co.,
Ltd., Shanghai, China), caspase-3 (1:2,000; 9662; CST Biological
Reagents Co., Ltd.), X-linked inhibitor of apoptosis (X–IAP;
1:1,000; 14334; CST Biological Reagents Co., Ltd.), phosphorylated
(p-)JNK (1:1,000; 9225; CST Biological Reagents Co., Ltd.), p-Drp1
(1:500; ab193216; Abcam, Cambridge, MA, USA), B-cell lymphoma 2
(Bcl-2; 1:1,000; 15071; CST Biological Reagents Co., Ltd.)
associated X protein (Bax; 1:2,000; ab32503), γ-actin (1:1,000;
ab123034) and F-actin (1:1,000; ab205; all Abcam) overnight at 4°C
(30). The membranes were washed
in Tris-buffered saline with Tween-20 for 15 min and were
subsequently incubated with horseradish peroxidase-conjugated
secondary antibody (1:1,000; sc-2004/sc-2005; Santa Cruz
Biotechnology, Inc., Dallas, TX, USA) for 1 h at room temperature.
Blots were visualized with an enhanced chemiluminescence substrate
kit (Thermo Fisher Scientific, Inc.) (31,32).
The bands were scanned and quantified by Quantity One (version
4.6.2; Bio-Rad Laboratories, Inc., Hercules, CA, USA).
Detection of lactate dehydrogenase
(LDH) release, mitochondrial membrane potential (ΔΨm), adenosine
triphosphate (ATP) production and mPTP opening
LDH released into the medium from injured cells was
detected with an LDH cytotoxicity kit (Roche Diagnostics,
Indianapolis, IN, USA). ΔΨm was analyzed with a JC-1 kit (Beyotime
Institute of Biotechnology, Shanghai, China), according to the
manufacturer's protocol (33,34).
Cellular ATP was detected using a firefly luciferase-based ATP
assay kit (Beyotime Institute of Biotechnology). mPTP opening was
visualized as a rapid dissipation of tetramethylrhodamine ethyl
ester fluorescence, as described previously (35).
Terminal deoxynucleotidyl transferase
dUTP nick end labelling (TUNEL) staining
A TUNEL assay was performed using a one-step TUNEL
kit (Beyotime Institute of Biotechnology, Haimen, China), according
to the manufacturer's protocol. Briefly, cells were fixed for 1 h
in 4% (w/v) paraformaldehyde at room temperature. Following
specific labelling, the cells were exposed to DAPI (5 mg/ml) with
PBS in the dark for 5 min at room temperature. TUNEL-positive cells
were defined as those with fluorescein-dUTP staining present. Then
20 different fields were randomly selected under magnification, ×40
to count the number of apoptotic cells by confocal microscopy
(FluoView 1000; Olympus Corporation) (36).
Determination of caspase-3 and
caspase-9 activity
Caspase-3 and caspase-9 activities were detected
with their respective activity assay kits (C1115 and C1157;
Beyotime Institute of Biotechnology), according to the
manufacturer's protocol. The assay was repeated three times
(37).
Immunocytochemistry
To determine cytochrome c (cyt-c)
localization and mitochondrial division, immunocytochemistry was
performed (38). Cells (1,000
cells/well) were fixed in 4% paraformaldehyde in PBS for 15 min at
room temperature, washed with PBS, and then permeabilized with 0.1%
Triton X-100 in PBS for 5 min. Subsequently, the cells were blocked
for 1 h with PBS containing 5% bovine serum albumin (Sigma-Aldrich)
at room temperature incubated with primary antibodies against
cyt-c (1:500; 12963; CST Biological Reagents Co., Ltd.) and
translocase of outer mitochondrial membrane 20 (Tom20; 1:500;
42406; CST Biological Reagents Co., Ltd.) for 1 h at room
temperature. Mitochondrial fission was observed via Tom20. F-actin
was stained with rhodamin-phalloidin (1:100; Molecular Probes;
Thermo Fisher Scientific, Inc.) in the dark for 15 min at room
temperature. DAPI (1:100, Sigma-Aldrich; Merck KGaA) was used to
stain the nuclei in the dark for 5 min at room temperature. Images
of the immunostained cells were captured using a fluorescence
microscope through a 50× objective (VANOX-S; Olympus
Corporation).
ATP production and respiratory chain
complex activities assays
The cellular ATP levels were measured using a
firefly luciferase-based ATP assay kit (Beyotime, Shanghai, China).
Complex I, II, and V activity was measured according to previous
studies (7). Mitochondrial
respiratory function was measured polarographically at 30°C using a
Biological Oxygen Monitor System (Hansatech Instruments, King's
Lynn, UK) and a Clarktype oxygen electrode (Hansatech DW1, Norfolk,
UK).
Statistical analysis
All analyses were performed with SPSS software
version 20.0 (SPSS Inc.; IBM Corp., Armonk, NY, USA). All
experiments were repeated three times. All results are expressed as
the mean ± standard deviation and statistical significance for each
variable was estimated by a one-way analysis of variance followed
by Tukey's test for the post hoc analysis. P<0.05 was considered
to indicate a statistically significant difference.
Results
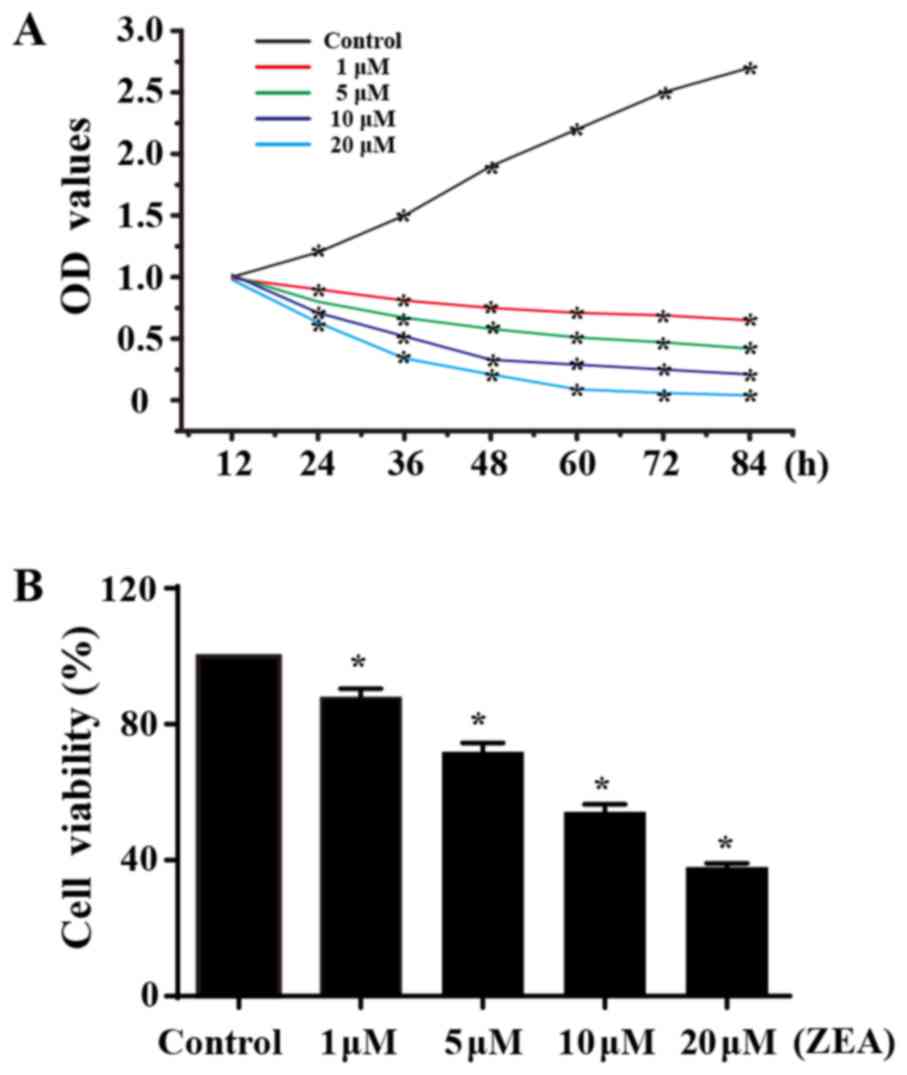
ZEA reduces ESC viability and
growth
The MTT assay revealed a dose-dependent decrease in
cell viability following treatment with ZEA for 24 h (Fig. 1B), indicating that ZEA has a toxic
effect on ESC viability. The CCK-8 assay was performed to assess
the growth capacity of ESCs treated with ZEA. Compared with the
control group, ZEA treatment significantly interfered with the
proliferative capacity of ESCs. However, no statistical difference
was noted between the groups in the first 12 h of treatment. The
growth capacity of ESCs reduced as the ZEA concentration increased
(Fig. 1A), suggesting that ZEA
reduced ESC proliferation.
ZEA induces ESC apoptosis through
mitochondrial fission
As no significant difference was observed in ESC
proliferation following 12 h ZEA treatment, ESCs were treated with
ZEA for 24 h in the subsequent experiments to exclude the influence
of proliferation on cell numbers (Fig.
2). A TUNEL assay was performed to detect the apoptotic rate of
ESCs treated with ZEA. The results demonstrated that ZEA
significantly increased the apoptotic rate in ESCs compared with
the rate observed in the control group (Fig. 2A and B). Approximately 8.35±2.73%
of ESCs were TUNEL-positive in the control group. The percentage of
TUNEL-positive cells was significantly increased by ZEA in a
concentration-dependent manner: 1 µM/l, 19.35±4.64%; 5 µM/l,
36.58±3.29%; 10 µM/l, 58.37±4.26%; and 20 µM/l, 64.21±4.57%
(Fig. 2B). These data indicate
that ZEA had a lethal impact on ESCs. Caspase-3 activity was
investigated as its activation represents an essential and final
step in the apoptotic process, leading to the induction of DNA
breakage and subsequent cellular apoptosis (7). Significantly increased caspase-3
activity was observed in the ZEA-treated cells compared with the
level of activity in the control group (Fig. 2C). The largest pro-apoptotic effect
was detected in the of 20 µM/l ZEA group. Thus, this concentration
was used for subsequent experiments.
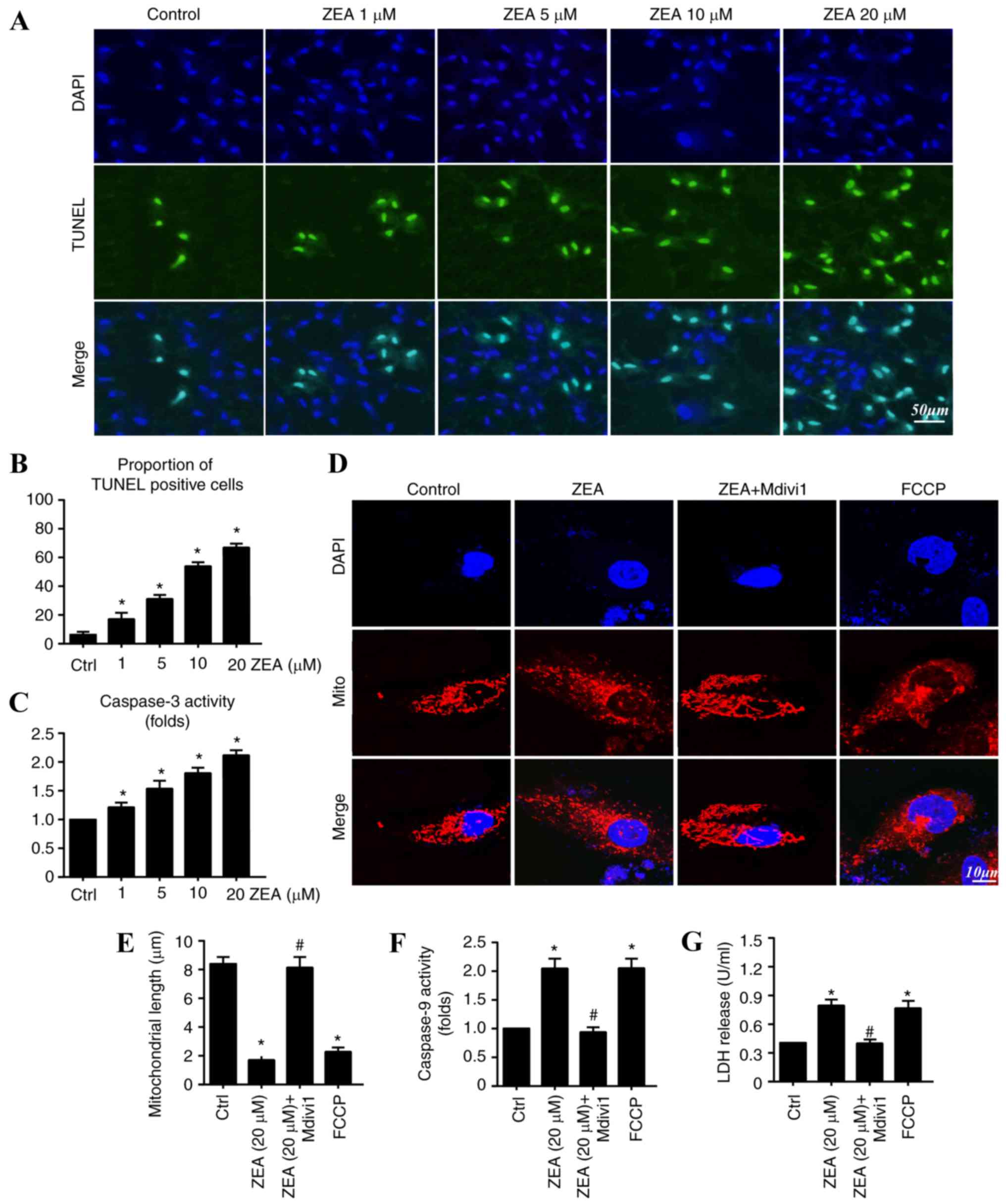 | Figure 2.ZEA induces ESC apoptosis by
increasing mitochondrial fission. (A) A TUNEL assay was performed
to detect the apoptotic rate of ESCs treated with ZEA. Cells were
visualized with a confocal microscope. Scale bar, 50 µm. (B) Cells
were counted and the proportion of TUNEL-positive cells was
calculated. (C) Changes in caspase-3 activity were detected with an
assay kit (D) Mitochondrial fission was observed using Tom20. Scale
bar, 10 µm. (E) Mitochondrial length was calculated. (F) Changes in
caspase-9 activity were detected with an assay kit. (G) LDH
released into the medium from injured cells was detected using an
LDH cytotoxicity kit. *P<0.05 vs. Ctrl; #P<0.05
vs. ZEA group. ZEA, zearalenone; ESC, endometrial stromal cell;
TUNEL, terminal deoxynucleotidyl transferase dUTP nick end
labelling; Tom20, translocase of outer mitochondrial membrane; LDH,
lactate dehydrogenase; FCCP, carbonyl
cyanide-p-trifluoromethoxyphenylhydrazone; Mdivi1, mitochondrial
fission inhibitor; Ctrl, control. |
Several studies have identified that mitochondrial
fission is a critical process in determining cellular survival and
apoptosis (39,40). Changes in mitochondrial morphology
were investigated to examine the role of mitochondrial fission in
ZEA-mediated cellular apoptosis. The mitochondrial fission inducer,
carbonyl cyanide-p-trifluoromethoxyphenylhydrazone (FCCP), and the
inhibitor, Mdivi1, were used. Compared with the control group, the
ZEA-treated group had a marked increase in the level of fragmented
mitochondria, with evidence of rounder mitochondria and increased
debris (Fig. 2D). The
mitochondrial length was also significantly reduced following
treatment with ZEA compared with the length in the control group
(Fig. 2E). Mdivi1 application with
ZEA markedly blocked the mitochondrial fragmentation mediated by
ZEA. FCCP was used as the positive control group and induced
substantial amounts of mitochondrial debris. These results indicate
that ZEA promoted mitochondrial fission.
To establish whether ZEA induced ESC apoptosis by
mitochondrial fission induction, LDH release and caspase-9 activity
assays were conducted with and without Mdivi1. Inhibition of
mitochondrial fission with Mdivi1 significantly reduced the
concentration of LDH detected in the medium compared with the level
in the ZEA group (Fig. 2G).
Similarly, increased caspase-9 activity in the ZEA treatment group
was significantly reduced with Mdivi1 application. Notably, FCCP
not only promoted LDH release but also enhanced caspase-9 activity.
Similar results were observed in the ZEA group (Fig. 2F). Taken together, these data
suggest that ZEA induced ESC apoptosis through the upregulation of
mitochondrial fission.
Mitochondrial fission contributes to
mitochondrial dysfunction and structural damage
Changes in mitochondrial function and structure were
examined to elucidate the mechanism by which mitochondrial fission
activates cellular apoptosis (11). The primary function of the
mitochondria is to produce adequate ATP to fuel cell metabolism
(41,42). Significantly reduced ATP production
was detected in the ZEA treatment group when compared with the
control group, demonstrating that ZEA treatment reduces the ability
of mitochondria to generate sufficient ATP (Fig. 3A). Mdivi1 was demonstrated to
reverse the ZEA-mediated decrease in ATP, indicating that
ZEA-induced mitochondrial fission is responsible for mitochondrial
energy disorder. This functional change was induced by the
ZEA-induced structural damage. ΔΨm is fundamental to ATP production
(43). Over time, the
mitochondrial membrane potential dissipates following ZEA treatment
(Fig. 3B and C) and these changes
were reversed with the addition of Mdivi1 (Fig. 3D and E). ΔΨm collapse is a marker
of increased mitochondrial membrane permeability, which may lead to
the leakage of mitochondrial proteins from the mitochondria into
the cytoplasm (44). The addition
of ZEA resulted in increased mitochondrial release of cyt-c,
with evidence of dispersion into the cytoplasm and nucleus
(Fig. 3F). Furthermore, ZEA
treatment promoted excessive mPTP opening and decreased electron
transport chain complex function (ETCx; Fig. 3G and H). Mdivi1 was observed to
partially suppress cyt-c diffusion and mPTP opening.
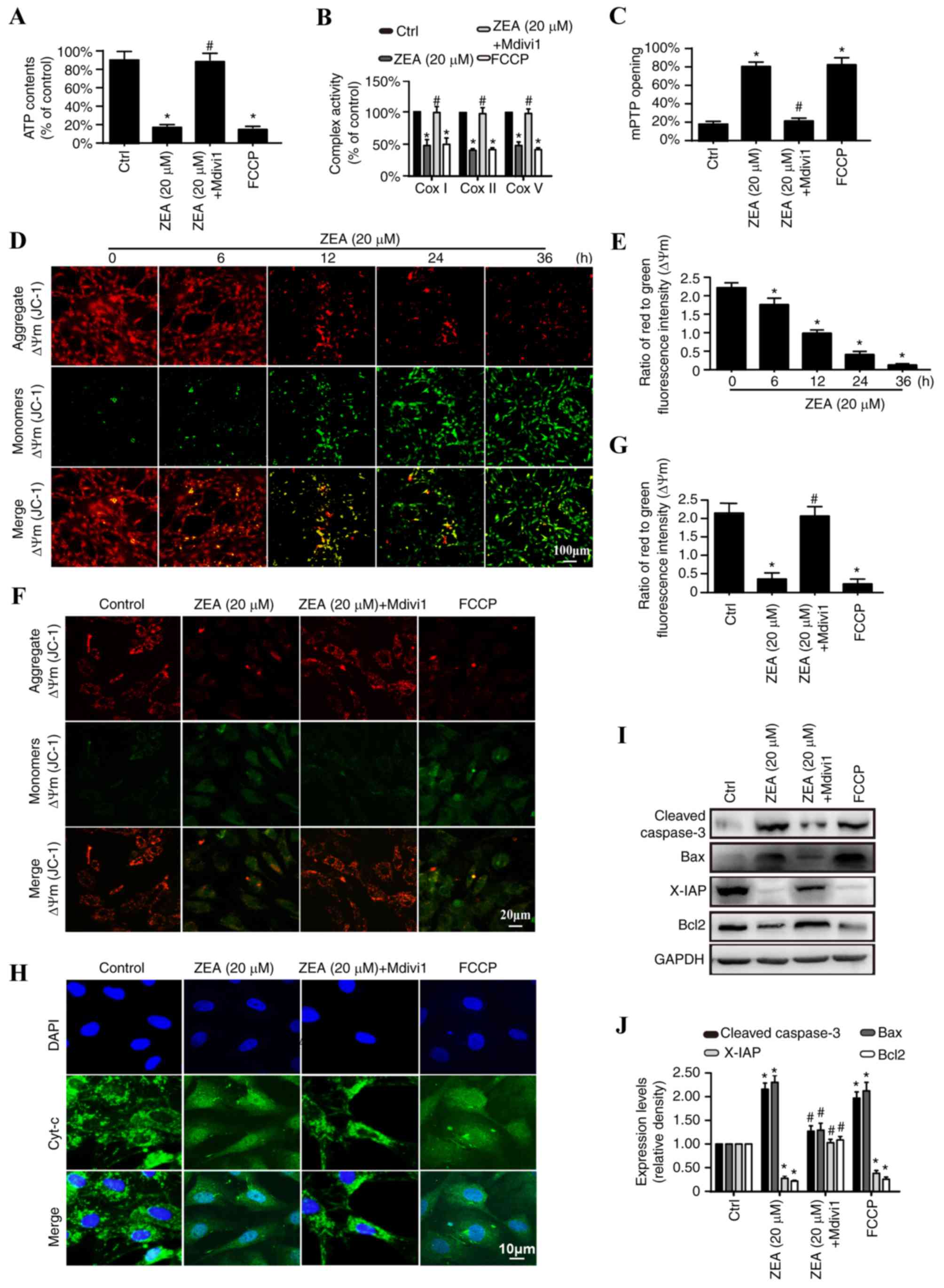 | Figure 3.Excessive mitochondrial fission
results in mitochondrial dysfunction and structural damage. Changes
in (A) ATP production, (B) ETCx I, II, and V activity and (C)
mitochondrial permeability mPTP opening were detected in ESCs
treated with 20 µM ZEA, 20 µM ZEA + Mdivi1 or FCCP. (D and E) The
effect of ZEA (20 µM) on ΔΨm at different time points as detected
by a JC-1 assay. Scale bar, 100 µm. (F) The effect of 20 µM ZEA, 20
µM ZEA + Mdivi1 or FCCP on ΔΨm as detected by a JC-1 assay. Scale
bar, 20 µm. (G) The ratio of red to green fluorescence in ESCs
treated with 20 µM ZEA, 20 µM ZEA + Mdivi1 or FCCP. (H)
Immunocytochemical analysis of cyt-c leakage from the
mitochondria into the cytoplasm. Scale bar, 10 µm. (I) Western blot
analysis of the protein levels of cleaved caspase-3, Bax, Bcl-2 and
X-IAP in ESCs treated with 20 µM ZEA, 20 µM ZEA + Mdivi1 or FCCP.
GADPH was used as an internal control. (J) Relative quantification
of the western blot analysis results. *P<0.05 vs. Ctrl;
#P<0.05 vs. ZEA group. ZEA, zearalenone; Mdivi1,
mitochondrial fission inhibitor; FCCP, carbonyl
cyanide-p-trifluoromethoxyphenylhydrazone; ATP, adenosine
triphosphate; ΔΨm, mitochondrial membrane potential; cyt-c,
cytochrome c; mPTP, mitochondrial permeability transition
pore; ESC, endometrial stromal cell; ETCx, electron transport chain
complex; Bcl-2, B-cell lymphoma 2; Bax, Bcl-2 associated X protein;
X-IAP, X-linked inhibitor of apoptosis; Ctrl, control. |
Western blot analysis
Cyt-c release triggers the formation of the
apoptosome, a complex comprising of apoptotic peptidase activating
factor 1, cyt-c, dATP (deoxy-adenosine triphosphate) and
pro-caspase-9 (45). The
apoptosome triggers caspase-9 activation, which subsequently
cleaves caspase-3 to its active form. Caspase-3 may then activate
the mitochondrial cell death effectors (46). Western blot analysis was used to
examine the downstream changes induced by cyt-c leakage. ZEA
treatment significantly increased levels of the pro-apoptotic
proteins, cleaved caspase-3 and Bax, and decreased the levels of
anti-apoptotic proteins, X-IAP and Bcl-2, compared with the levels
in the control group. Mdivi1 addition prevented the changes
observed in the ZEA-treated cells (Fig. 3I and J). These results identified
the potential pathological mechanism of ZEA on ESC apoptosis.
ZEA-induced mitochondrial fission may have promoted the destruction
of mitochondrial function and structure, which subsequently
activated the mitochondrial pathway of apoptosis.
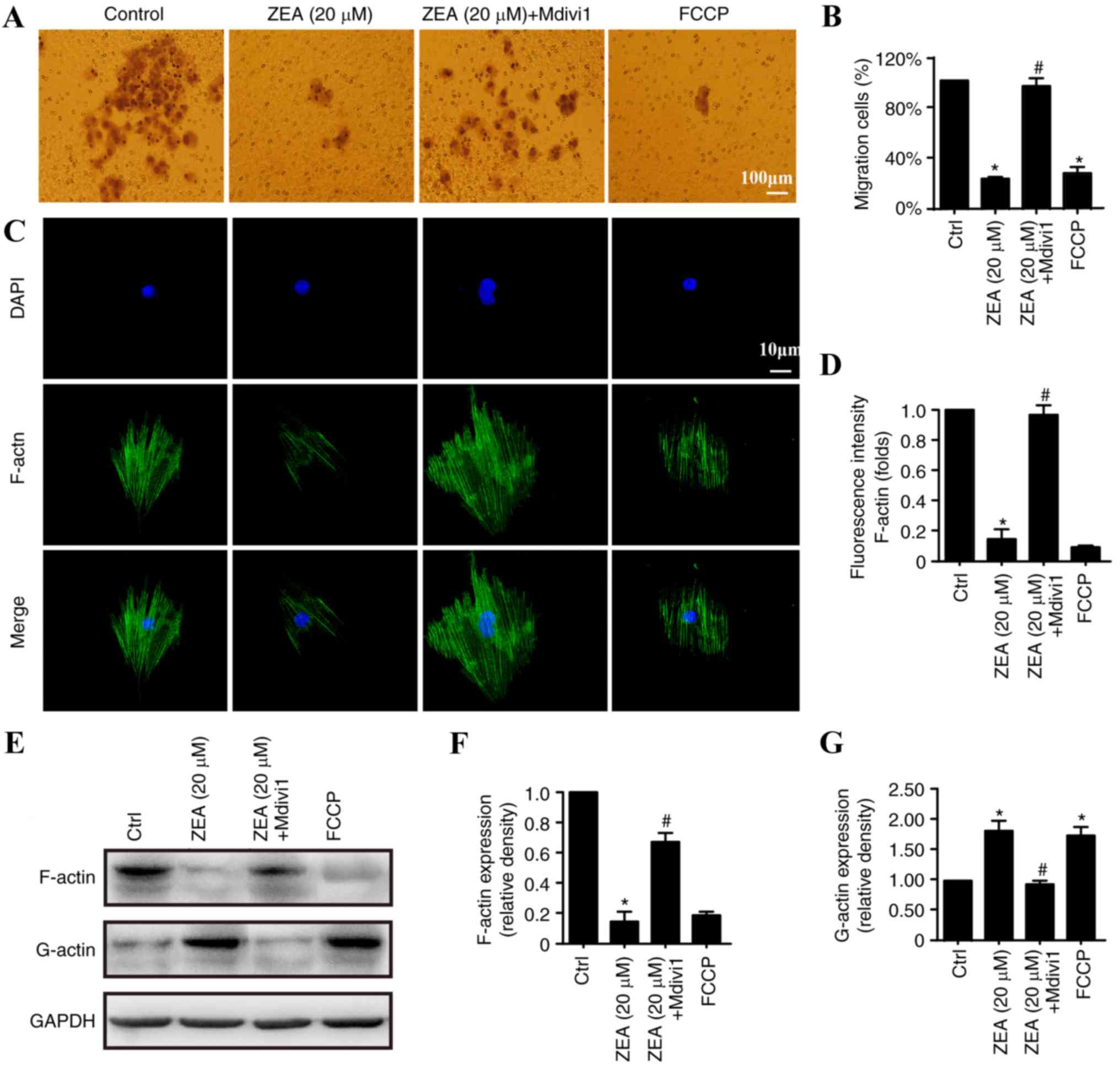
ZEA impairs ESC migration via the
activation of mitochondrial fission
A Transwell migration assay was performed to
investigate the association between ESC migration and endometriosis
progression. As ZEA reduced the number of ESCs through apoptosis
induction, ESCs were first treated with ZEA for 24 h. Equal numbers
of ESCs (1×105) were collected and placed in the upper
chamber of the Transwell plate. After 12 h, the number of ESCs that
had migrated to the underside of the insert membranes was counted.
As demonstrated in Fig. 4, ZEA
treatment for 24 h significantly decreased the migratory ability of
ESCs compared with the level observed in the control group.
Mitochondrial fission inhibition increased ESC migration,
suggesting that mitochondrial fission is involved in the migratory
process. As F-actin is the primary stress fiber that is essential
for cell mobility (47), it was
hypothesized that the decreased migration may be a result of
mitochondrial fission-induced F-actin depolymerization. ZEA
treatment significantly reduced F-actin fluorescence compared with
that observed in the control group, indicating a decrease in
F-actin activity (Fig. 4C and D).
Fission inhibition by Mdivi1 preserved the filamentary structure of
F-actin. Additionally, the decrease in F-actin occurred in parallel
with an accumulation of G-actin (Fig.
4E and G), an end-product of F-actin depolymerization (48), suggesting that mitochondrial
fission stimulated the dissociation and disrupted the synthesis of
F-actin. Mdivi1 reduced the conversion of F-actin to G-actin. These
results demonstrate that the ZEA treatment interfered with ESC
migration by increasing mitochondrial fission, which influenced the
balance of F-actin/G-actin.
ZEA regulates mitochondrial fission by
activation of the JNK/Drp1 pathway
JNK and Drp1 levels were analyzed to further
elucidate the mechanism of mitochondrial fission regulation by ZEA.
Drp1 is a large GTPase that controls mitochondrial fission in
mammalian cells, and its active phosphorylation site is a serine at
position 616 (Ser616) (40). Drp1
is activated by Ser616 phosphorylation, which may be performed by
JNK (39). Therefore, it was
speculated that the regulatory effects of ZEA on mitochondrial
fission may be due to JNK/Drp1 pathway activation. As demonstrated
in Fig. 5, the results revealed
that ZEA significantly increased p-JNK levels compared with the
levels in the control group, and this increase was inhibited by the
JNK pathway inhibitor SP600125 (Fig.
5A and B). JNK inhibition also significantly reduced Drp1
phosphorylation at Ser616 compared with the level observed in the
ZEA group, which resulted in a decrease in mitochondrial Drp1 and
an increase in cytoplasmic Drp1 compared with that observed in the
ZEA group (Fig. 5A, C and E).
These data indicate that mitochondrial fission is modulated by the
JNK/Drp1 pathway, which in turn is activated by ZEA.
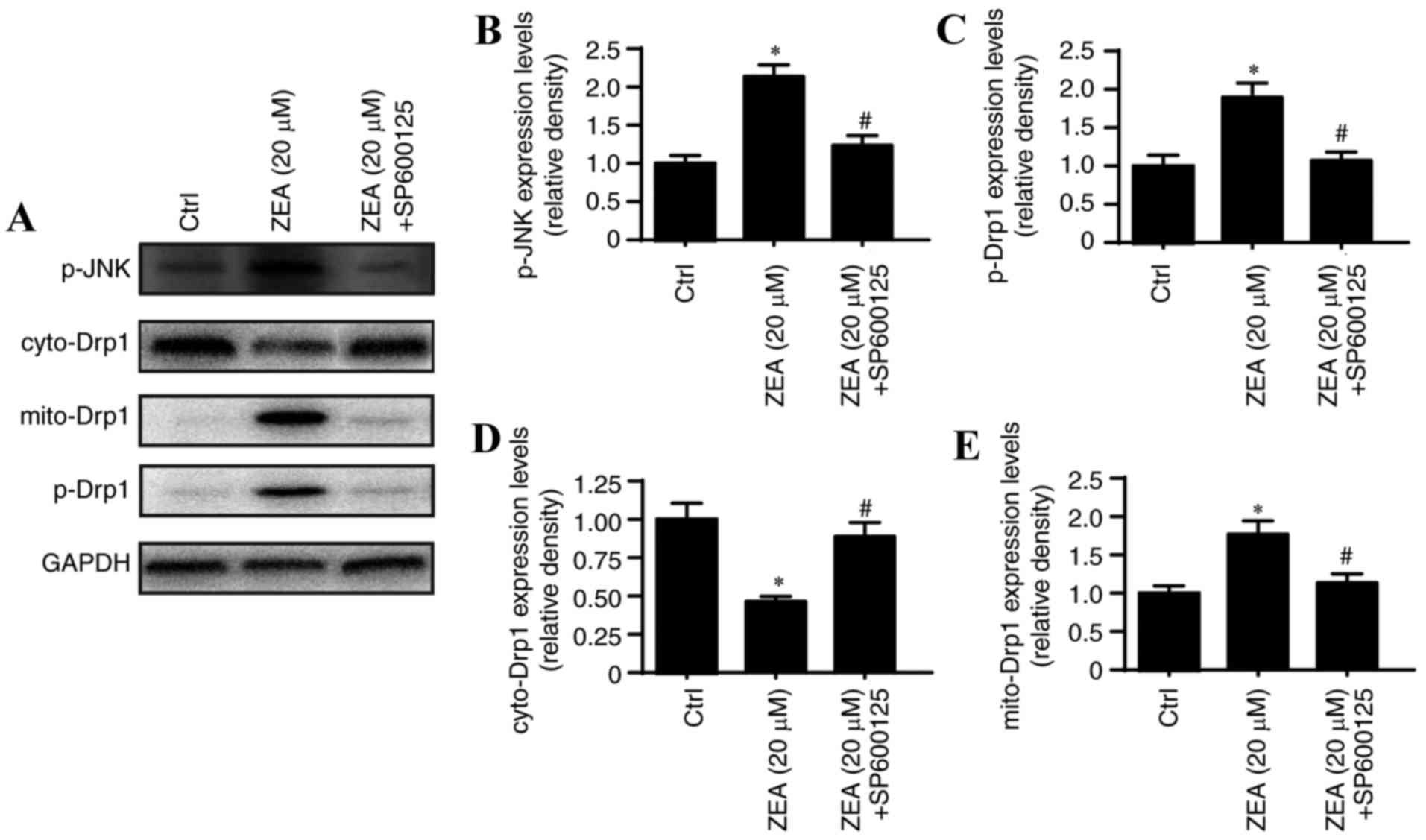 | Figure 5.ZEA regulates mitochondrial fission
by activating the JNK/Drp1 pathway. (A) Western blot analysis of
Drp1, p-Drp1 and p-JNK levels. The relative quantification of (B)
p-JNK, (C) p-Drp1, (D) cyto-Drp1 and (E) mito-Drp1 was calculated
from the results of the western blot analysis. *P<0.05 vs. Ctrl;
#P<0.05 vs. ZEA group. ZEA, zearalenone; p-,
phosphorylated; cyto-, cytoplasmic; mito-, mitochondrial; Drp1,
dynamin-related protein 1; JNK, c-Jun N-terminal kinase; SP600125,
JNK pathway inhibitor. |
Discussion
Endometriosis is a gynecological disease defined by
ectopic endometrial tissue implantation that forms functional
endometriotic lesions that are frequently located in the ovaries
and peritoneum (49).
Endometriosis affects ~10% of women of a reproductive age and may
cause severe pelvic pain and infertility (50). It has also been associated with an
elevated risk of ovarian cancer (51). The pathogenesis of endometriosis is
poorly defined and there are a limited number of effective
treatments to cure the disease or slow its progression (52). The relatively high incidence and
lack of effective therapeutic options presents a requirement for a
more in-depth understanding of the underlying mechanisms that
influence the development and severity of endometriosis (53). It has been demonstrated that the
increased migration and reduced apoptosis of ESCs contributes to
the progression of endometriosis (38). Therefore, methods to reduce the
mobility and enhance the apoptosis of ESCs are vital to improve the
clinical outcomes of patients with endometriosis.
ZEA, also known as the F-2 toxin, is a non-steroidal
mycotoxin produced by several species of Fusarium (54). It is a common fungal contaminant of
cereal crops worldwide and is typically found in feed and grains,
including maize, wheat and rye (55). ZEA is structurally similar to
estrogen and competes with estradiol for binding to estrogen
receptors, and stimulates estrogenic activity, which may cause
several physiological alterations in the reproductive tract
(56). In vitro study has
indicated that ZEA may regulate metabolic processes, including cell
proliferation, differentiation and apoptosis (57). The regulatory role of ZEA in
endometriosis has also been demonstrated (58). In the present study, ZEA was
revealed to induce the apoptosis and impair the migration of ESCs
through regulation of the JNK/Drp1 pathway and mitochondrial
fission.
The mitochondrion is present in all human body
cells, excluding erythrocytes. It is the primary organelle for cell
metabolism, signal transmission, cellular survival and apoptosis
(59). In addition to the vital
function of ATP production, it has also been identified that
mitochondrial fission is a prerequisite for intrinsic apoptosis in
some forms of cancer (60). An
increase in mitochondrial length has been demonstrated to decrease
mitochondrial fission and increase mitochondrial fusion, leading to
the inhibition of apoptotic initiation and the downstream catabolic
process of autophagy (6). In the
present study, mitochondrial fission was revealed to be responsible
for ESC apoptosis via the induction of structural and functional
mitochondrial damage. ZEA-induced mitochondrial fission caused
mitochondrial depolarization, which resulted in the leakage of
cyt-c into the cytoplasm. An increase in pro-apoptotic and a
decrease in anti-apoptotic proteins was detected, indicating that
the mitochondrial pathway of apoptosis was activated as a
consequence of cyt-c release.
Mitochondrial fission has been revealed to result in
VDAC1 oligomerization and HK2 separation from the outer
mitochondrial membrane, leading to mPTP opening that is responsible
for a reduction in ΔΨm (11).
Additionally, an increase of mROS in response to mitochondrial
fission has been demonstrated to induce cardiolipin peroxidation,
which mediates cyt-c release and the activation of the
mitochondrial pathway of apoptosis (61). Similarly, the present study
identified that mitochondrial fission was responsible for the
apoptosis observed, via the induction of structural and functional
mitochondrial damage. ZEA-induced mitochondrial fission was also
demonstrated to be involved in ESC migration, and mitochondrial
fission inhibition reduced ESC migration. These effects were
independent of cellular apoptosis.
As F-actin is the primary stress fiber that directly
modulates cellular migration, it was hypothesized that decreased
migration was the result of a mitochondrial fission-mediated
F-actin imbalance (62). The
results confirmed that mitochondrial fission resulted in F-actin
depolymerization to G-actin, thereby impeding cell migration.
Furthermore, successful mitochondrial fission has been revealed to
be dependent on Drp1, the Drp1 receptor and stress fibers (63). Previous study has suggested that
the brief accumulation of intracellular F-actin on the surface of
mitochondria is a prerequisite for subsequent mitochondrial
division (7). Under physiological
conditions, F-actin is regularly distributed to certain parts of
the cytoplasm that control cell migration through directional cues.
The initiation of mitochondrial fission transforms F-actin into
G-actin at the outer mitochondrial membrane, through the formation
of a contractile ring, which involves Drp1 and its receptor
(64). Considering the
indispensable nature of F-actin in fission, excessive fission would
likely consume large amounts of cytoplasmic F-actin and cause
uneven F-actin distribution, leading to the dysregulation of
F-actin homeostasis and impaired migration. However, further
evidence is required to support this hypothesis.
The JNK/Drp1 pathway was demonstrated to be involved
in the regulatory effects of ZEA on mitochondrial fission in the
present study. Previous study has identified that activated JNK
contributes to Drp1 phosphorylation (65). Drp1 is a large GTPase that
translocates into puncta on mitochondria, where it couples
guanosine 5′-triphosphate hydrolysis with mitochondrial membrane
constriction and fission (66).
Drp1 activity is determined by its phosphorylation state. Drp1
phosphorylation at Ser616 results in Drp1 activation (67), and activated Drp1 translocates from
the cytoplasm to the surface of the mitochondria to mediate
mitochondrial fission. The present study demonstrated that ZEA
increased p-JNK levels and p-Drp1. Notably, inhibition of JNK
prevents this change. ZEA-induced JNK pathway activation was also
responsible for an increase in mitochondrially-located Drp1 and a
decrease in cytoplasmic Drp1, suggesting that JNK regulated Drp1
activity and subsequent mitochondrial fission.
Collectively, the results of the present study
highlight the important role of mitochondrial fission in ESC
apoptosis and migration via the JNK/Drp1 pathway. Thus, the
regulation of mitochondrial fission may be an effective target for
endometriosis therapy. These data also provide evidence for the use
of ZEA as an effective method to increase mitochondrial fission.
However, further understanding of the underlying mechanisms is
required prior to potential clinical research and application.
Acknowledgements
Not applicable.
Funding
The present study was supported by the National
Natural Science Foundation of China (grant no. 81030002).
Availability of data and materials
All data generated or analyzed during the present
study are included in this published article.
Authors' contributions
HW and XZ conceived the research; CN, YD and YG
performed the experiments; all authors participated in discussing
and revising the manuscript.
Ethics approval and consent to
participate
All experimental protocols were approved by the
Ethics Committee of the Department of Gynecology and Obstetrics,
Beijing Tongren Hospital (Beijing, China). The ethics reference no.
2015TS1303525.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Ricci AG, Olivares CN, Bilotas MA,
Meresman GF and Baranao RI: Effect of vascular endothelial growth
factor inhibition on endometrial implant development in a murine
model of endometriosis. Reprod Sci. 18:614–622. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Crosignani P, Olive D, Bergqvist A and
Luciano A: Advances in the management of endometriosis: An update
for clinicians. Hum Reprod Update. 12:179–189. 2006. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Blitek A, Morawska E, Kiewisz J and Ziecik
AJ: Effect of conceptus secretions on HOXA10 and PTGS2 gene
expression and PGE2 release in co-cultured luminal epithelial and
stromal cells of the porcine endometrium at the time of early
implantation. Theriogenology. 76:954–966. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Daiber A, Oelze M, Steven S, Kroller-Schon
S and Munzel T: Taking up the cudgels for the traditional reactive
oxygen and nitrogen species detection assays and their use in the
cardiovascular system. Redox Biol. 12:35–49. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Chen HH, Chen YT, Yang CC, Chen KH, Sung
PH, Chiang HJ, Chen CH, Chua S, Chung SY, Chen YL, et al: Melatonin
pretreatment enhances the therapeutic effects of exogenous
mitochondria against hepatic ischemia-reperfusion injury in rats
through suppression of mitochondrial permeability transition. J
Pineal Res. 61:52–68. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Jin Q, Li R, Hu N, Xin T, Zhu P, Hu S, Ma
S, Zhu H, Ren J and Zhou H: DUSP1 alleviates cardiac
ischemia/reperfusion injury by suppressing the Mff-required
mitochondrial fission and Bnip3-related mitophagy via the JNK
pathways. Redox Biol. 14:576–587. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Shi C, Cai Y, Li Y, Li Y, Hu N, Ma S, Hu
S, Zhu P, Wang W and Zhou H: Yap promotes hepatocellular carcinoma
metastasis and mobilization via governing
cofilin/F-actin/lamellipodium axis by regulation of
JNK/Bnip3/SERCA/CaMKII pathways. Redox Biol. 14:59–71. 2018.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Zhou H, Zhu P, Guo J, Hu N, Wang S, Li D,
Hu S, Ren J, Cao F and Chen Y: Ripk3 induces mitochondrial
apoptosis via inhibition of FUNDC1 mitophagy in cardiac IR injury.
Redox Biol. 13:498–507. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Zhu H, Jin Q, Li Y, Ma Q, Wang J, Li D,
Zhou H and Chen Y: Melatonin protected cardiac microvascular
endothelial cells against oxidative stress injury via suppression
of IP3R-[Ca2+]c/VDAC-[Ca2+]m axis by
activation of MAPK/ERK signaling pathway. Cell Stress Chaperones.
23:101–113. 2015. View Article : Google Scholar
|
|
10
|
Zhou H, Li D, Shi C, Xin T, Yang J, Zhou
Y, Hu S, Tian F, Wang J and Chen Y: Effects of Exendin-4 on bone
marrow mesenchymal stem cell proliferation, migration and apoptosis
in vitro. Sci Rep. 5:128982015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Zhou H, Hu S, Jin Q, Shi C, Zhang Y, Zhu
P, Ma Q, Tian F and Chen Y: Mff-dependent mitochondrial fission
contributes to the pathogenesis of cardiac microvasculature
ischemia/reperfusion injury via induction of mROS-mediated
cardiolipin oxidation and HK2/VDAC1 Disassociation-involved mPTP
opening. J Am Heart Assoc. 6:e0053282017. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Chua S, Lee FY, Chiang HJ, Chen KH, Lu HI,
Chen YT, Yang CC, Lin KC, Chen YL, Kao GS, et al: The
cardioprotective effect of melatonin and exendin-4 treatment in a
rat model of cardiorenal syndrome. J Pineal Res. 61:438–456. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Du K, Ramachandran A and Jaeschke H:
Oxidative stress during acetaminophen hepatotoxicity: Sources,
pathophysiological role and therapeutic potential. Redox Biol.
10:148–156. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhou H, Wang S, Zhu P, Hu S, Chen Y and
Ren J: Empagliflozin rescues diabetic myocardial microvascular
injury via AMPK-mediated inhibition of mitochondrial fission. Redox
Biol. 15:335–346. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Zhou H, Wang J, Zhu P, Hu S and Ren J:
Ripk3 regulates cardiac microvascular reperfusion injury: The role
of IP3R-dependent calcium overload, XO-mediated oxidative stress
and F-action/filopodia-based cellular migration. Cell Signal.
45:12–22. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Kasahara A and Scorrano L: Mitochondria:
From cell death executioners to regulators of cell differentiation.
Trends Cell Biol. 24:761–770. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Paterniani E: Selection for reproductive
isolation between two populations of maize, zea mays l. Evolution.
23:534–547. 1969. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Banjerdpongchai R, Kongtawelert P,
Khantamat O, Srisomsap C, Chokchaichamnankit D, Subhasitanont P and
Svasti J: Mitochondrial and endoplasmic reticulum stress pathways
cooperate in zearalenone-induced apoptosis of human leukemic cells.
J Hematol Oncol. 3:502010. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Kowalska K, Habrowska-Gorczynska DE,
Dominska K and Piastowska-Ciesielska AW: The dose-dependent effect
of zearalenone on mitochondrial metabolism, plasma membrane
permeabilization and cell cycle in human prostate cancer cell
lines. Chemosphere. 180:455–466. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Bouaziz C, El Dein Sharaf O, El Golli E,
Abid-Essefi S, Brenner C, Lemaire C and Bacha H: Different
apoptotic pathways induced by zearalenone, T-2 toxin and ochratoxin
A in human hepatoma cells. Toxicology. 254:19–28. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
He B, Zhao Y, Xu L, Gao L, Su Y, Lin N and
Pu J: The nuclear melatonin receptor RORalpha is a novel endogenous
defender against myocardial ischemia/reperfusion injury. J Pineal
Res. 60:313–326. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Agorastos A and Linthorst AC: Potential
pleiotropic beneficial effects of adjuvant melatonergic treatment
in posttraumatic stress disorder. J Pineal Res. 61:3–26. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Kang JW, Hong JM and Lee SM: Melatonin
enhances mitophagy and mitochondrial biogenesis in rats with carbon
tetrachloride-induced liver fibrosis. J Pineal Res. 60:383–393.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Lee HY and Back K: Melatonin is required
for H2 O2-and NO-mediated defense signaling through
MAPKKK3 and OXI1 in Arabidopsis thaliana. J Pineal Res.
62:2017. View Article : Google Scholar
|
|
25
|
Carrasco-Pozo C, Tan KN, Reyes-Farias M,
De La Jara N, Ngo ST, Garcia-Diaz DF, Llanos P, Cires MJ and Borges
K: The deleterious effect of cholesterol and protection by
quercetin on mitochondrial bioenergetics of pancreatic β-cells,
glycemic control and inflammation: In vitro and in vivo studies.
Redox Biol. 9:229–243. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Lam SM, Wang Z, Li J, Huang X and Shui G:
Sequestration of polyunsaturated fatty acids in membrane
phospholipids of Caenorhabditis elegans dauer larva attenuates
eicosanoid biosynthesis for prolonged survival. Redox Biol.
12:967–977. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Gao Y, Xiao X, Zhang C, Yu W, Guo W, Zhang
Z, Li Z, Feng X, Hao J, Zhang K, et al: Melatonin synergizes the
chemotherapeutic effect of 5-fluorouracil in colon cancer by
suppressing PI3K/AKT and NF-kappaB/iNOS signaling pathways. J
Pineal Res. 62:2017. View Article : Google Scholar
|
|
28
|
Chen W, Zou P, Zhao Z, Chen X, Fan X,
Vinothkumar R, Cui R, Wu F, Zhang Q, Liang G and Ji J: Synergistic
antitumor activity of rapamycin and EF24 via increasing ROS for the
treatment of gastric cancer. Redox Biol. 10:78–89. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Zhou H, Yang J, Xin T, Zhang T, Hu S, Zhou
S, Chen G and Chen Y: Exendin-4 enhances the migration of
adipose-derived stem cells to neonatal rat ventricular
cardiomyocyte-derived conditioned medium via the phosphoinositide
3-kinase/Akt-stromal cell-derived factor-1α/CXC chemokine receptor
4 pathway. Mol Med Rep. 11:4063–4072. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Kim YD, Hwang SL, Lee EJ, Kim HM, Chung
MJ, Elfadl AK, Lee SE, Nedumaran B, Harris RA and Jeong KS:
Melatonin ameliorates alcohol-induced bile acid synthesis by
enhancing miR-497 expression. J Pineal Res. 62:2017.doi:
10.1111/jpi.12386. View Article : Google Scholar
|
|
31
|
Cai SY, Zhang Y, Xu YP, Qi ZY, Li MQ,
Ahammed GJ, Xia XJ, Shi K, Zhou YH, Reiter RJ, et al: HsfA1a
upregulates melatonin biosynthesis to confer cadmium tolerance in
tomato plants. J Pineal Res. 62:2017.doi: 10.1111/jpi.12387.
View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Mailloux RJ and Treberg JR: Protein
S-glutathionlyation links energy metabolism to redox signaling in
mitochondria. Redox Biol. 8:110–118. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Zhou H, Yang J, Xin T, Li D, Guo J, Hu S,
Zhou S, Zhang T, Zhang Y, Han T and Chen Y: Exendin-4 protects
adipose-derived mesenchymal stem cells from apoptosis induced by
hydrogen peroxide through the PI3K/Akt-Sfrp2 pathways. Free Radic
Biol Med. 77:363–375. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Zhang Y, Zhou H, Wu W, Shi C, Hu S, Yin T,
Ma Q, Han T, Zhang Y, Tian F and Chen Y: Liraglutide protects
cardiac microvascular endothelial cells against
hypoxia/reoxygenation injury through the suppression of the
SR-Ca(2+)-XO-ROS axis via activation of the
GLP-1R/PI3K/Akt/survivin pathways. Free Radic Biol Med. 95:278–292.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Feng D, Wang B, Wang L, Abraham N, Tao K,
Huang L, Shi W, Dong Y and Qu Y: Pre-ischemia melatonin treatment
alleviated acute neuronal injury after ischemic stroke by
inhibiting endoplasmic reticulum stress-dependent autophagy via
PERK and IRE1 signalings. J Pineal Res. 62:2017.doi:
10.1111/jpi.12395. View Article : Google Scholar
|
|
36
|
Areti A, Komirishetty P, Akuthota M, Malik
RA and Kumar A: Melatonin prevents mitochondrial dysfunction and
promotes neuroprotection by inducing autophagy during
oxaliplatin-evoked peripheral neuropathy. J Pineal Res.
62:2017.doi: 10.1111/jpi.12393. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Fuhrmann DC and Brune B: Mitochondrial
composition and function under the control of hypoxia. Redox Biol.
12:208–215. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Doskey CM, Buranasudja V, Wagner BA,
Wilkes JG, Du J, Cullen JJ and Buettner GR: Tumor cells have
decreased ability to metabolize H2O2:
Implications for pharmacological ascorbate in cancer therapy. Redox
Biol. 10:274–284. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Zhou H, Du W, Li Y, Shi C, Hu N, Ma S,
Wang W and Ren J: Effects of melatonin on fatty liver disease: The
role of NR4A1/DNA-PKcs/p53 pathway, mitochondrial fission and
mitophagy. J Pineal Res. 64:2018. View Article : Google Scholar
|
|
40
|
Zhou H, Zhang Y, Hu S, Shi C, Zhu P, Ma Q,
Jin Q, Cao F, Tian F and Chen Y: Melatonin protects cardiac
microvasculature against ischemia/reperfusion injury via
suppression of mitochondrial fission-VDAC1-HK2-mPTP-mitophagy axis.
J Pineal Res. 63:2017.doi: 10.1111/jpi.12413. View Article : Google Scholar :
|
|
41
|
Brasacchio D, Alsop AE, Noori T, Lufti M,
Iyer S, Simpson KJ, Bird PI, Kluck RM, Johnstone RW and Trapani JA:
Epigenetic control of mitochondrial cell death through
PACS1-mediated regulation of BAX/BAK oligomerization. Cell Death
Differ. 24:961–970. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Zhang R and Sun Y, Liu Z, Jin W and Sun Y:
Effects of melatonin on seedling growth, mineral nutrition and
nitrogen metabolism in cucumber under nitrate stress. J Pineal Res.
62:2017.doi: 10.1111/jpi.12403. View Article : Google Scholar
|
|
43
|
Banerjee K, Keasey MP, Razskazovskiy V,
Visavadiya NP, Jia C and Hagg T: Reduced FAK-STAT3 signaling
contributes to ER stress-induced mitochondrial dysfunction and
death in endothelial cells. Cell Signal. 36:154–162. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Dufour F, Rattier T, Shirley S, Picarda G,
Constantinescu AA, Morlé A, Zakaria AB, Marcion G, Causse S,
Szegezdi E, et al: N-glycosylation of mouse TRAIL-R and human
TRAIL-R1 enhances TRAIL-induced death. Cell Death Differ.
24:500–510. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Murphy PS, Wang J, Bhagwat SP, Munger JC,
Janssen WJ, Wright TW and Elliott MR: CD73 regulates
anti-inflammatory signaling between apoptotic cells and
endotoxin-conditioned tissue macrophages. Cell Death Differ.
24:559–570. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Park J, Tran Q, Mun K, Masuda K, Kwon SH,
Kim SH, Kim DH, Thomas G and Park J: Involvement of S6K1 in
mitochondria function and structure in HeLa cells. Cell Signal.
28:1904–1915. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Van Nostrand JL, Bowen ME, Vogel H, Barna
M and Attardi LD: The p53 family members have distinct roles during
mammalian embryonic development. Cell Death Differ. 24:575–579.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Perdiz D, Lorin S, Leroy-Gori I and Pous
C: Stress-induced hyperacetylation of microtubule enhances
mitochondrial fission and modulates the phosphorylation of Drp1 at
616Ser. Cell Signal. 39:32–43. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Xu S, Pi H, Zhang L, Zhang N, Li Y, Zhang
H, Tang J, Li H, Feng M, Deng P, et al: Melatonin prevents abnormal
mitochondrial dynamics resulting from the neurotoxicity of cadmium
by blocking calcium-dependent translocation of Drp1 to the
mitochondria. J Pineal Res. 60:291–302. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Laschke MW and Menger MD: Anti-angiogenic
treatment strategies for the therapy of endometriosis. Hum Reprod
Update. 18:682–702. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Lagana AS, Vitale SG, Salmeri FM, Triolo
O, Ban Frangež H, Vrtačnik-Bokal E, Stojanovska L, Apostolopoulos
V, Granese R and Sofo V: Unus pro omnibus, omnes pro uno: A novel,
evidence-based, unifying theory for the pathogenesis of
endometriosis. Med Hypotheses. 103:10–20. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
52
|
Kowalska K, Habrowska-Gorczynska DE and
Piastowska-Ciesielska AW: Zearalenone as an endocrine disruptor in
humans. Environ Toxicol Pharmacol. 48:141–149. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
53
|
Smith MC, Madec S, Coton E and Hymery N:
Natural co-occurrence of mycotoxins in foods and feeds and their in
vitro combined toxicological effects. Toxins. 8:942016. View Article : Google Scholar : PubMed/NCBI
|
|
54
|
Danicke S and Winkler J: Invited review:
Diagnosis of zearalenone (ZEN) exposure of farm animals and
transfer of its residues into edible tissues (carry over). Food
Chem Toxicol. 84:225–249. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
55
|
Mally A, Solfrizzo M and Degen GH:
Biomonitoring of the mycotoxin Zearalenone: Current state-of-the
art and application to human exposure assessment. Arch Toxicol.
90:1281–1292. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
56
|
Hu J, Xu M, Dai Y, Ding X, Xiao C, Ji H
and Xu Y: Exploration of Bcl-2 family and caspases-dependent
apoptotic signaling pathway in Zearalenone-treated mouse
endometrial stromal cells. Biochem Biophys Res Commun. 476:553–559.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
57
|
Stopa E, Babinska I, Zielonka L, Gajecki M
and Gajecka M: Immunohistochemical evaluation of apoptosis and
proliferation in the mucous membrane of selected uterine regions in
pre-pubertal bitches exposed to low doses of zearalenone. Pol J Vet
Sci. 19:175–186. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
58
|
Ellenrieder L, Rampelt H and Becker T:
Connection of protein transport and organelle contact sites in
mitochondria. J Mol Biol. 429:2148–2160. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
59
|
Senft D and Ronai ZA: Regulators of
mitochondrial dynamics in cancer. Curr Opin Cell Biol. 39:43–52.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
60
|
Ribas V, Garcia-Ruiz C and Fernandez-Checa
JC: Mitochondria, cholesterol and cancer cell metabolism. Clin
Transl Med. 5:222016. View Article : Google Scholar : PubMed/NCBI
|
|
61
|
Nomura K, Imai H, Koumura T, Kobayashi T
and Nakagawa Y: Mitochondrial phospholipid hydroperoxide
glutathione peroxidase inhibits the release of cytochrome c from
mitochondria by suppressing the peroxidation of cardiolipin in
hypoglycaemia-induced apoptosis. Biochem J. 351:183–193. 2000.
View Article : Google Scholar : PubMed/NCBI
|
|
62
|
Prieto-Dominguez N, Ordonez R, Fernandez
A, Méndez-Blanco C, Baulies A, Garcia-Ruiz C, Fernández-Checa JC,
Mauriz JL and González-Gallego J: Melatonin-induced increase in
sensitivity of human hepatocellular carcinoma cells to sorafenib is
associated with reactive oxygen species production and mitophagy. J
Pineal Res. 61:396–407. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
63
|
Zhou H, Ma Q, Zhu P, Ren J, Reiter RJ and
Chen Y: Protective role of melatonin in cardiac
ischemia-reperfusion injury: From pathogenesis to targeted therapy.
J Pineal Res. 2018.doi: 10.1111/jpi.12471. View Article : Google Scholar
|
|
64
|
Estaquier J and Arnoult D: Inhibiting
Drp1-mediated mitochondrial fission selectively prevents the
release of cytochrome c during apoptosis. Cell Death Differ.
14:1086–1094. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
65
|
Bereiter-Hahn J, Voth M, Mai S and
Jendrach M: Structural implications of mitochondrial dynamics.
Biotechnol J. 3:765–780. 2008. View Article : Google Scholar : PubMed/NCBI
|
|
66
|
Lin C, Chao H, Li Z, Xu X, Liu Y, Hou L,
Liu N and Ji J: Melatonin attenuates traumatic brain injury-induced
inflammation: A possible role for mitophagy. J Pineal Res.
61:177–186. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
67
|
Yang X, Wang H, Ni HM, Xiong A, Wang Z,
Sesaki H, Ding WX and Yang L: Inhibition of Drp1 protects against
senecionine-induced mitochondria-mediated apoptosis in primary
hepatocytes and in mice. Redox Biol. 12:264–273. 2017. View Article : Google Scholar : PubMed/NCBI
|