Introduction
Severe acute pancreatitis (SAP) is an inflammatory
disease of the pancreas characterized by elevated pancreatic
enzymes in the blood which can lead to multiple organ dysfunction
and is associated with high mortality rates (1). SAP involves a complex cascade of
events. The progression of SAP leads to acinar cell injury, and as
a result of inflammation and tissue damage, inflammatory cells
including macrophages, neutrophils and dendritic cells are actively
recruited to the diseased site and induce the rapid production and
release of inflammatory cytokines such as tumor necrosis factor
(TNF)-α and interleukin (IL)-6, which play the dominant role in
local pancreatic inflammation and systemic complications. Our
previous research showed that the phosphatidylinositol 3-kinase
(PI3K)/protein kinase B (Akt) inhibitor wortmannin could decrease
the production of inflammatory cytokines, alleviate the severity of
inflammation in the pancreas and improve the survival rate in SAP
rats, indicating that the PI3K/Akt signaling pathway is involved in
the development of SAP and the severity of inflammation through the
regulation of inflammatory cytokines (2). However, the mechanism of PI3K/Akt in
SAP is not fully understood.
One Study confirmed that Toll-like receptors (TLRs)
and PI3K interact to modulate inflammation (3) TLRs are the first-line of host defense
guards that sense invading microbes to induce innate and adaptive
immune responses. Since the activation of downstream signaling
pathways culminates in the production of proinflammatory mediators,
TLRs are closely involved in the developmental stages of a variety
of inflammatory diseases, include SAP (4–7).
TLR4 activation by lipopolysaccharide (LPS) triggers the
association of myeloid differentiation primary response gene 88
(MyD88) with the cytoplasmic domain of TLR4, which recruits IL-1
receptor-associated kinase 4 (IRAK4) and sequentially
phosphorylates IRAK-1, activating a series of signaling molecules,
kinases, transcription factors and inflammatory genes (8,9).
NF-κB and p38MAPK are two major downstream signaling pathways in
the TLR4 signaling pathway (9,10).
It is well known that NF-κB and p38MAPK play critical roles in
regulating the expression of proinflammatory genes such as TNF-α,
IL-β and IL-6 (11,12). Our previous findings indicated that
these two signaling pathways were activated in SAP rats (2,13)
and pancreatitis-associated ascitic fluid stimulated THP-1 cells
(14), which was suppressed by
wortmannin (2). In the present
study, we examined the expression of TLR4, PI3K, Akt, p38MAPK,
NF-κBp65 and MyD88 in vivo in SAP rats and in
vitro in RAW264.7 murine macrophages (stimulated by LPS)
following treatment with the PI3K agonist insulin-like growth
factor (IGF)-1, and further explored the possible mechanisms by
evaluating the correlation between PI3K/Akt and TLR4 in
inflammatory reactions associated with SAP. Our results demonstrate
for the first time to our knowledge evidence that PI3K/Akt may
regulate the TLR4 signaling pathway and take part in the
progression of the SAP inflammation reaction.
Materials and methods
Animals and treatments
Thirty healthy male Sprague-Dawley rats weighing
230–270 g were purchased from the Animal Center of Shanghai Jiao
Tong University (Shanghai, China). Animals were housed in cages
under a controlled temperature of 22±1°C in 12-h light-dark cycles,
were fed standard laboratory chow and provided water ad libitum and
allowed to acclimatize for at least 1 week. The experiment was
designed in accordance with the guidelines for the care and use of
laboratory animals in research and was approved by the Ethical and
Research Committee of Shanghai Jiao Tong University. Sprague-Dawley
rats were randomly divided into five groups: Normal group (Normal),
sham operation group (SO), SAP model group (SAP), high-dose IGF-1
treatment group (High-dose IGF-1) and low-dose IGF-1 treatment
group (Low-dose IGF-1), n=6 per group. Surgical anesthesia was
accomplished by intraperitoneal injection of 2% pentobarbital
sodium (40 mg/kg bw). SAP was induced by retrograde infusion of
5.0% sodium taurocholate (1 ml/kg body weight) into the pancreatic
and biliary duct. Half an hour before inducing SAP, rats were
injected intraperitoneally with 20 or 200 µg/kg of IGF-1
(Sigma-Aldrich, USA) in the low- and high-dose IGF-1 groups,
respectively. Rats in the sham group underwent a sham operation
with nothing infused. All animals were anaesthetized by 2%
pentobarbital sodium (50 mg/kg bw) prior to sacrifice and
sacrificed by cervical dislocation. Blood, ascitic fluid and
pancreatic tissue samples were obtained 6 h after ductal infusion
of sodium taurocholate. Arterial blood samples were obtained from
the abdominal aorta. Blood samples were centrifuged at 3,000 g for
10 min at 5°C and the supernatants were separated with sterile
pipettes and frozen at −80°C for the following assays. Before the
rats were sacrificed, the sutured abdominal incisions were opened.
Changing the rat's position allowed the ascites to accumulate,
which was then collected by 10 ml syringe. The maximum volume of
ascites observed in the study was 5.06% (i.e., 12 ml in a rat of
235 g. These 12 ml of ascites weighted 11.89 g). Ascites fluid was
stored at −80°C until assay. A portion of the pancreatic tissue
samples was immediately frozen and maintained at −80°C for
real-time polymerase chain reaction (PCR), and the remaining tissue
was fixed in 10% formaldehyde for histopathological examination.
Pathological examination was as follows: The pancreatic tissue from
each rat was collected and fixed in 10% neutral-buffered
formaldehyde, embedded in paraffin and stained with hematoxylin and
eosin. Edema, inflammation, as well as hemorrhage and necrosis of
the pancreas were each graded from 0 to 4 as described below: Edema
(0, no edema; 1, focally increased between lobules; 2, diffusely
increased between lobules; 3, tense acini, widely separated
lobules; and 4, gross lobular separation); inflammation (0, no
leukocytes; 1, 2–10 leukocytes/high-power magnification [HP]; 2,
11–20 leukocytes/HP; 3, 21–30 leukocytes/HP; and 4, >30
leukocytes/HP); hemorrhage (0, no hemorrhage; 1, blood in 25% of
parenchyma; 2, blood in 25–50% of parenchyma; 3, blood in 50–75% of
parenchyma; and 4, blood in 100% of lobules); necrosis (0, no
necrosis; 1, periductal parenchymal destruction; 2, focal
parenchymal necrosis; 3, diffuse loss of lobules; and 4, severe
loss of lobules). The pathological scores in the study were
presented as the sum of these different scores.
Cell culture and treatment
RAW264.7 murine macrophages from the Chinese Academy
of Science Cell Bank were grown in RPMI 1640 supplemented with 10%
fetal bovine serum at 37°C in a 5% CO2/air environment.
The cells were resuspended at a concentration of 5×105
cells/ml in culture medium. A 1-ml cell suspension was transferred
to a six-well plate and incubated with a final concentration of 1
µg/ml LPS (Sigma-Aldrich; Merck KGaA, Darmstadt, Germany). Cells
were divided into five groups: Blank group, LPS group, IGF-1 group
[cells were incubated with IGF-1 (100 ng/ml) 1 h before treatment
with LPS], wortmannin group [cells were incubated with wortmannin
(100 nM; Sigma-Aldrich; Merck KGaA) 30 min before treatment with
LPS] and IGF-1+wortmannin group (cells were incubated with both
IGF-1 (100 ng/ml) and wortmannin (100 nM) before treatment with
LPS). The Blank group was incubated in culture medium without
drugs. The reaction was stopped after a 6-h incubation. After
centrifugation (1,000 × g, 6 min), the supernatant and cell lysates
were collected and stored at −80°C for subsequent analysis.
Cell viability assay
We used a Cell Counting Kit-8 (CCK-8) assay (Dojindo
Molecular Technologies, Inc., Kumamoto, Japan) to evaluate cell
viability. Cells were collected in the logarithmic growth phase and
plated at a density of 1×103-5×103 cells per
well in 96-well plates in triplicate, incubated at 37°C for 24 h,
then treated with increasing concentrations of LPS (0, 0.1, 0.3, 1,
3 and 10 µg/ml), IGF-1 (0, 1, 10, 30, 100 and 300 ng/ml) and
wortmannin (0, 3, 10, 20, 100 and 300 nM), while the Blank group
was incubated with culture medium without drugs. After 24 h
incubation at 37°C, every well was incubated with CCK-8 reagent in
complete medium at a ratio of 1:10 for 1 h. The absorbance value of
each well was measured at 450 nm using a microplate reader
(Molecular Devices, LLC, Sunnyvale, CA, USA).
Histopathological examination
At 6 h after inducing SAP, pancreatic tissue was
removed and fixed in 10% neutral-buffered formalin. Samples were
embedded in paraffin, sliced into 4-µm sections and stained with
hematoxylin-eosin in preparation for blinded histologic assessment.
Histologic changes were evaluated in random, nonconsecutive fields
(Olympus BX51/Olympus DP71; Olympus Corporation, Tokyo, Japan).
Edema, inflammation, as well as hemorrhage and necrosis of the
pancreas were each graded from 0 to 4 by the standard of Schmidt
et al (15), and the total
score was obtained by addition of the four aspect scores.
Ascite amylase activity and ascite
volume measurement
The amylase activity in samples of ascites fluid was
measured with an automated biochemistry analyzer. Ascite volume was
also measured.
Expression of TNF-α and IL-6
assay
IL-6 levels in sera and TNF-α and IL-6 levels in
cell supernatants were assayed with ELISA kits (Nanjing Jiancheng
Bioengineering Institute, Nanjing, China). ELISA was performed
according to the manufacturer's instructions.
Quantitative PCR (qPCR)
qPCR was used to investigate the expression of TLR4,
IGF-1 receptor (IGF-1R), p38MAPK and NF-κBp65 mRNA in pancreatic
tissue, and expression of PI3K, Akt, TLR4, MyD88, p38MAPK and
NF-κBp65 mRNA in RAW264.7 murine macrophages. Total RNA was
prepared from tissue and cells using RNAiso (Takara Bio, Inc.,
Otsu, Japan) according to the manufacturer's protocol. Reverse
transcription of total RNA was performed for synthesis of cDNA
using a Takara RNA PCR kit. qPCR was performed using the Light
Cycler Nano instrument (Roche Diagnostics, Indianapolis, IN, USA).
Calculations were performed using GAPDH as an internal
reference gene. The gene-specific primers used in pancreatic tissue
were as follows. TLR4: Sense 5′-ATGCTAAGGTTGGCACTCTC-3′,
antisense 5′-CAGGCAGGAAAGGAACAATG-3′; NF-κBp65: Sense
5′-AGACCTGGAGCAAGCCATTAG-3′, antisense 5′-CGGACCGCATTCAAGTCATAG-3′;
p38MAPK: Sense 5′-TTCCCAGCAGTCCTATCC-3′, antisense
5′-CAGATGGCAAGGGTTCAG-3′; IGF-1R: Sense
5′-CGCAGGATGGCTATCTGTTC-3′, antisense 5′-ATCACCACCGCACACTTC-3;
GAPDH: Sense 5′-GTCGGTGTGAACGGATTTG-3′, antisense
5′-TCCCATTCTCAGCCTTGAC-3′. The PCR amplification cycling conditions
were as follows: 95°C for 10 min, followed by 40 cycles of 95°C for
15 sec, 60°C for 1 min and 72°C for 1 min.
The gene-specific primers used in RAW364.7 murine
macrophages were as follows. TLR4: Sense
5′-CTATGAACAAAGGGTCTATCAG-3′, antisense 5′-AAGAACAGCAACCACTAAAG-3′;
MyD88: Sense 5′-CACTCGCAGTTTGTTGGATG-3′, antisense
5′-TGTAAAGGCTTCTCGGACTC-3′; PI3K: Sense
5′-ATGCCAGAAAGGAGAATG-3′, antisense 5′-TGTTGGACTCAGCAATAC-3′;
Akt: Sense 5′-GGGCACATCAAGATAACG-3′, antisense
5′-TGGTCCTGGTTGTAGAAG-3′; NF-κBp65: Sense
5′-CCCGAAACTCAACTTCTG-3′; antisense 5′-ATCTGCCCTGATGGTAAC-3′;
p38MAPK: Sense 5′-GTGTTCACACCCGCAAGGTC-3′, antisense
5′-CGGTCAGCTTCTGGCACTTC-3′; GAPDH: Sense
5′-ATCACTGCCACCCAGAAG-3′, antisense 5′-TCCACGACGGACACATTG-3′. The
PCR amplification cycling conditions were as follows: 95°C for 10
min, followed by 40 cycles of 95°C for 15 sec and 60°C for 45 sec.
All reactions were performed in triplicate. Relative gene
expression data were determined by the 2−ΔΔCq
method.
Western blotting
Western blot analysis was used to investigate Akt
activation in pancreatic tissue and RAW264.7 cells. Protein samples
were prepared using PRO-PREP protein extraction solution for total
fractions according to the manufacturer's instructions. Total
protein was separated with sodium dodecyl sulfate-polyacrylamide
gel electrophoresis and transferred to polyvinylidene fluoride
membranes. The membranes were blocked with 5% nonfat milk for 1 h,
incubated overnight at 4°C with Phospho-Akt antibody (1:1,000; Cell
Signaling Technology, Inc., Danvers, MA, USA) or Akt antibody
(1:1,000; Cell Signaling Technology, Inc.) and then incubated with
the appropriate secondary antibodies for 1 h. GAPDH (1:10,000;
Abcam, Cambridge, MA, USA) was used as an internal control. The ECL
Plus kit from Amersham; (GE Healthcare Life Sciences, Chalfont, UK)
was used for chemiluminescent detection. Densitometric analyses
were conducted using Optimus software (Optimus Corp, Fort Collins,
CO, USA) for quantitation.
Statistical analysis
Results are presented as means ± standard deviation.
All data were analyzed by one-way ANOVA tests with LSD and SNK post
hoc tests using SPSS 13.0 (IBM Corp., Armonk, NY, USA). P<0.05
was considered to indicate a statistically significant
difference.
Results
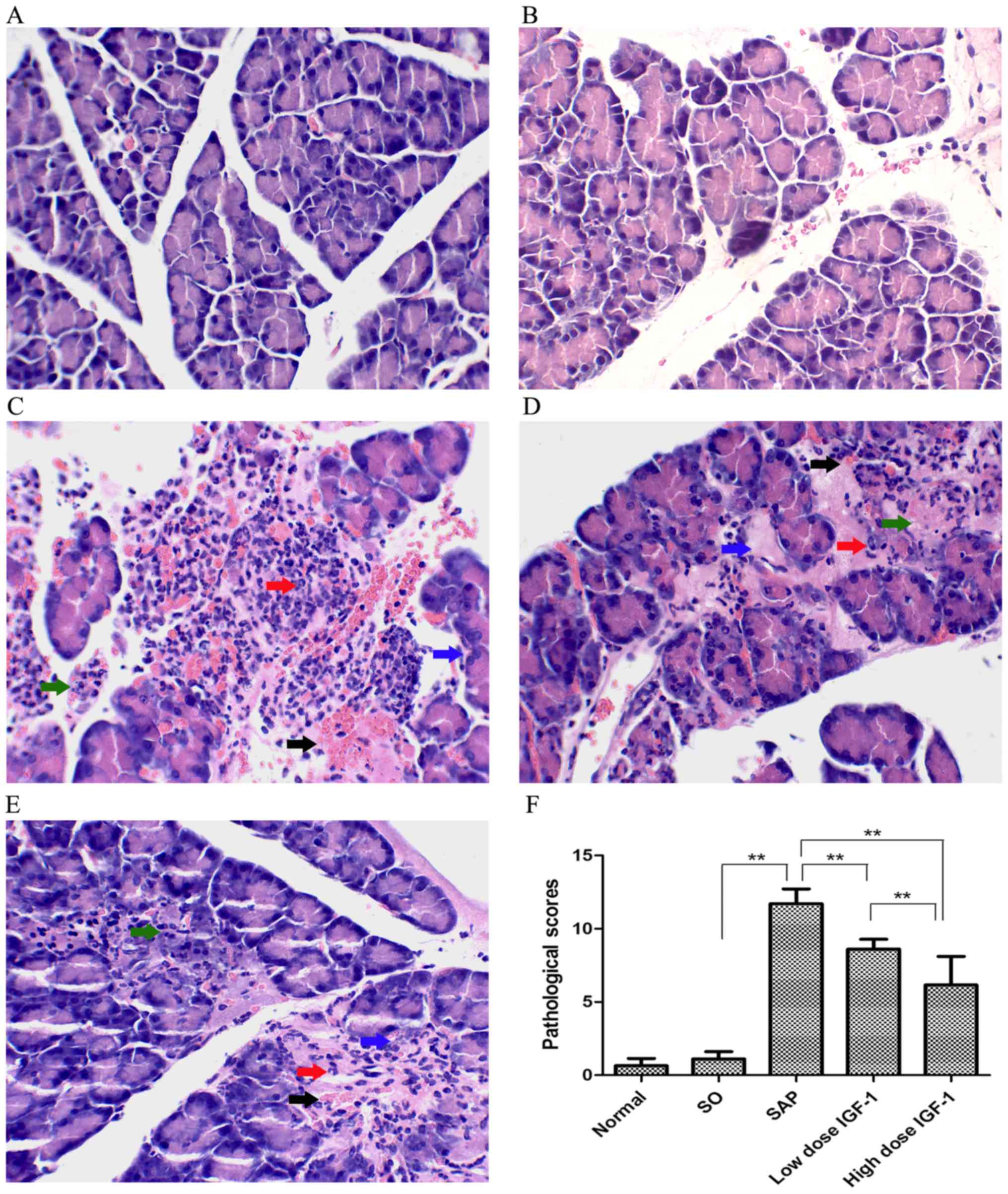
Histological examination of pancreatic
tissue
Histological examination found no difference between
the normal and SO groups. Compared with the SO group, pancreatic
injury in SAP rats was characterized by tissue edema, leukocyte
infiltration and acinar cell necrosis, and the hemorrhage
pathological scores for pancreatic tissues were markedly increased
(Fig. 1). Treatment with IGF-1
obviously ameliorated the severity of the inflammatory response in
SAP animals, alleviating tissue edema, leukocyte infiltration and
acinar cell necrosis, decreasing the hemorrhage pathological scores
for pancreatic injury. As the IGF-1 dosage increased, pancreatic
injury was lessened and the pathological scores significantly
declined, as shown in Fig. 1.
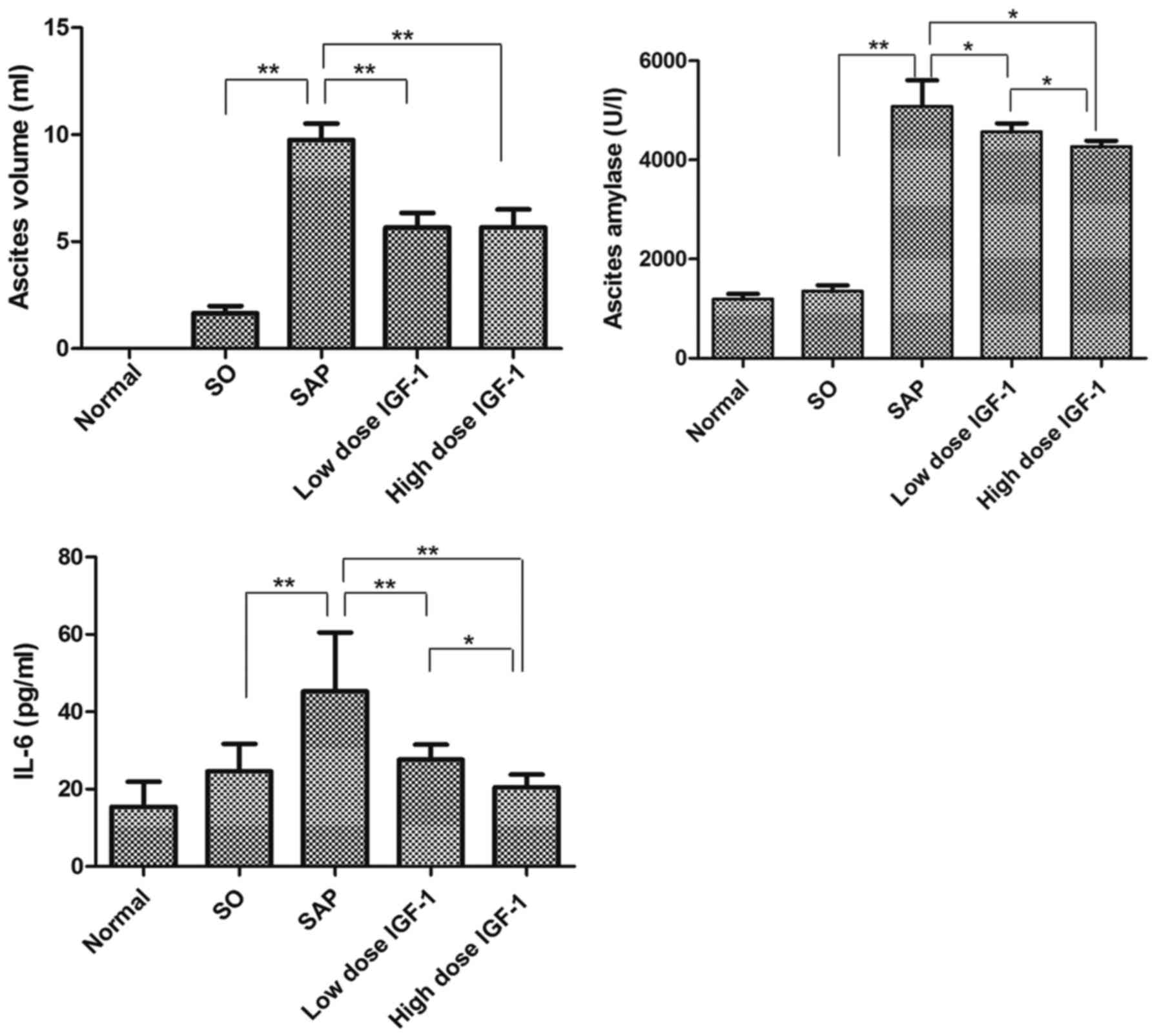
Ascites volume and ascites amylase
activity
Ascites volume gradually increased after the
exposure to LPS in the SAP group. The volume of ascites was
significantly reduced by treatment with IGF-1, but not in a
dose-dependent manner. Ascites amylase activity from the normal
rats is shown in Fig. 2. In
agreement with the histological changes, amylase activity in the
SAP rats significantly increased compared with the SO group. The
IGF-1 treatment groups had markedly decreased levels of ascites
amylase compared with the SAP rats, and as the IGF-1 dosage
increased the levels of amylase activity declined substantially
(Fig. 2).
Inflammatory cytokine levels in
serum
The measurement of serum inflammatory cytokine
levels showed that SAP rats had increased expression of the
proinflammatory cytokine IL-6 compared with the SO group. In
contrast, the level of IL-6 was reduced by treatment with IGF-1 in
a dose-dependent manner (Fig. 2).
The results indicated that IGF-1 could effectively attenuate
pancreatic injury.
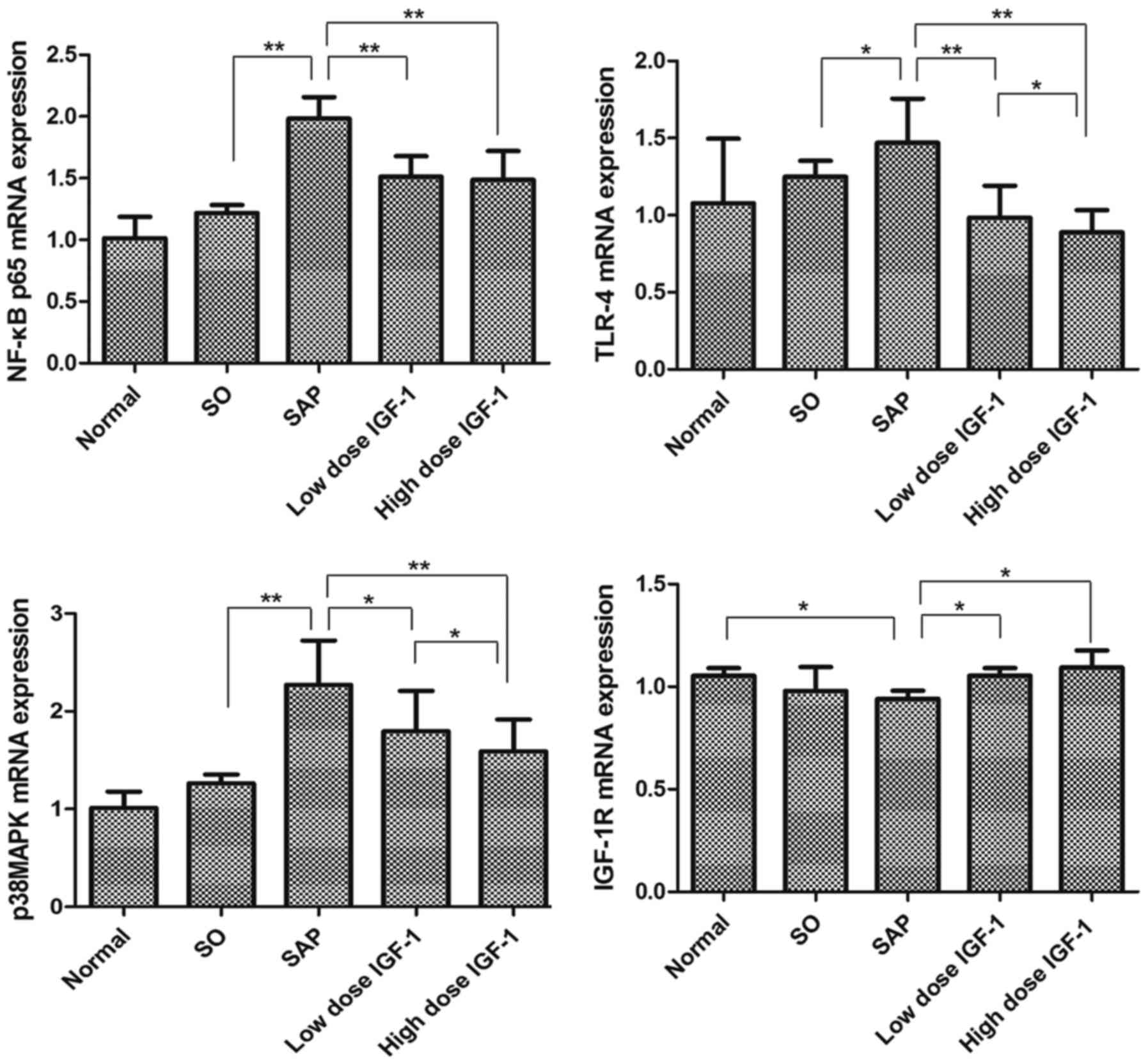
Effect of IGF-1 on TLR4, p38MAPK,
NF-κBp65 and IGF-1R mRNA levels in pancreatic tissue
The levels of TLR4, p38MAPK, NF-κBp65 and IGF-1R
mRNA in the five groups were assessed (Fig. 3). There was no significant
difference between the normal and SO group. Compared with the SO
group, TLR4, p38MAPK and NF-κBp65 mRNA markedly increased in
pancreatic tissues of SAP rats (P<0.05). High and low dose IGF-1
treatment decreased the expression of TLR4, p38MAPK and NF-κBp65
mRNA in the pancreas observably. Because TLR4, NF-κBp65 and p38MAPK
are important signaling molecules in the inflammatory reaction
associated with SAP, these results suggested that IGF-1 attenuated
the sodium taurocholate-induced inflammation in the pancreas.
IGF-1R, a member of the family of transmembrane
tyrosine kinases, binds IGF-1 with high affinity and initiates the
physiological response to this ligand (16). Compared with the normal and SO
groups, the level of IGF-1R mRNA expression fell in pancreatic
tissues of SAP rats, and was significantly decreased compared with
the normal group (P<0.05). In contrast, high and low dose IGF-1
treatment produced similar significant increases in expression of
IGF-1R mRNA in the pancreas (P<0.05). These results suggested
that exogenous IGF-1 increased the expression of IGF-1R in the same
manner as endogenous IGF-1.
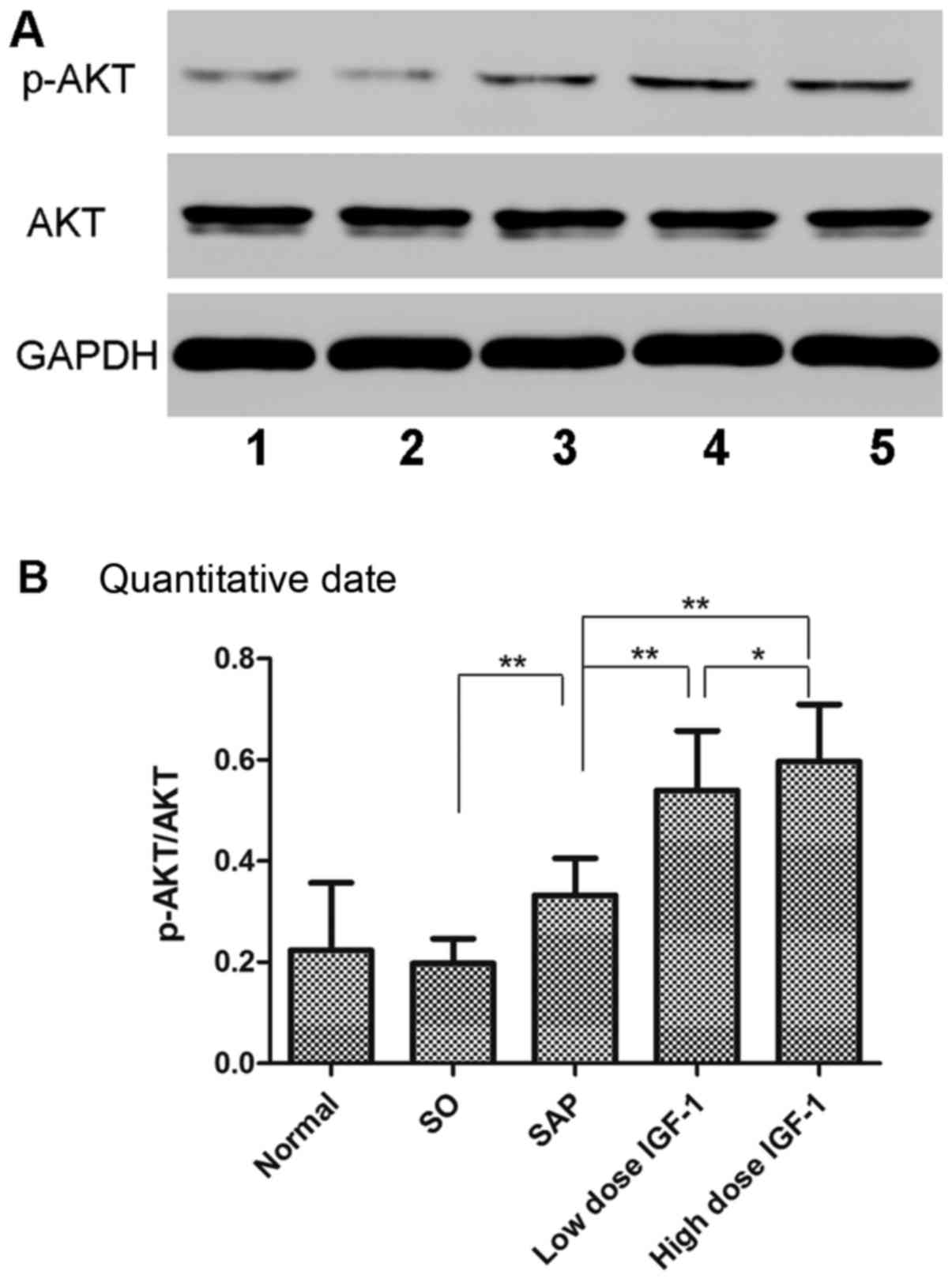
Effect of IGF-1 on phosphorylation of
Akt in pancreatic tissue
Akt is the downstream signaling molecule of PI3K in
inflammation. The phosphorylation of Akt in the SAP group was
markedly upregulated compared to the SO group. Moreover, IGF-1
treatment resulted in increased protein expression of p-Akt/Akt in
the pancreas (Fig. 4). These
results suggested that IGF-1 could increase the phosphorylation of
Akt, consistent with the results of another study (17).
Assay for cellular viability
Increasing concentrations of LPS, wortmannin and
IGF-1 had no significant effect on the proliferative activity or
viability of RAW264.7 cells compared with Blank cells after 6 h of
treatment. The final concentration of LPS, IGF-1 and wortmannin was
therefore set at 1 g/ml, 100 ng/ml and 100 nM, respectively, for
the next set of experiments.
Effect of IGF-1 and wortmannin on
TNF-α and IL-6 levels in RAW264.7 cells
Since macrophages play prominent roles in various
inflammatory conditions, especially in SAP (18), we analyzed whether IGF-1 and
wortmannin influenced the production of proinflammatory cytokines
in response to the TLR4 ligand LPS in RAW264.7 macrophages
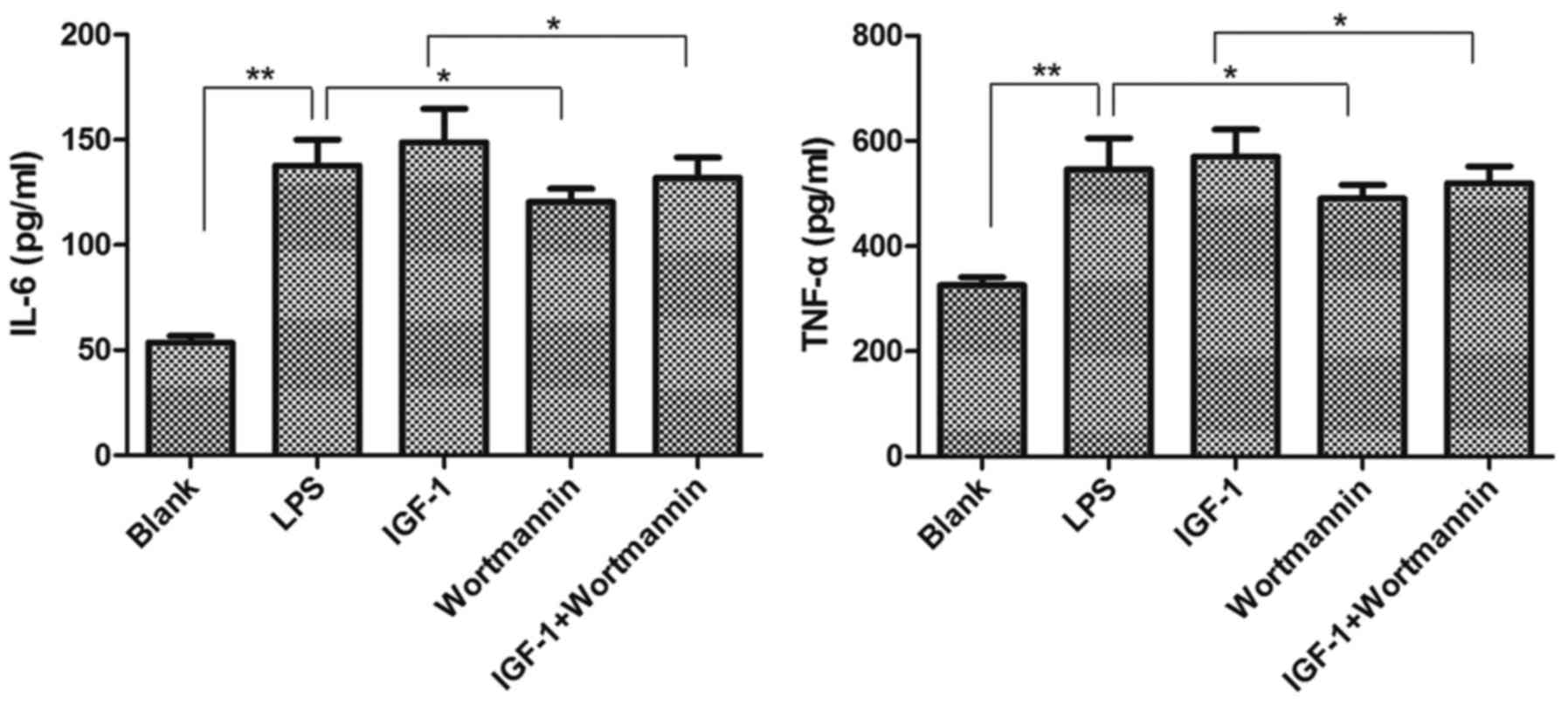
(Fig. 5). The protein levels of
TNF-α and IL-6 were higher in the LPS group than in the Blank group
(P<0.01). Compared with the LPS group, the levels of these
cytokines were increased in the IGF-1 group, but not different from
each other in statistics (P>0.05). We added IGF-1+wortmannin
group and compared it with IGF-1 group, we found that the
expression of IL-6 and TNF-α were different from each other in
statistics (P<0.05; Fig. 5).
Meanwhile, the level of TNF-α and IL-6 were significantly decreased
in the wortmannin group (P<0.05). These data demonstrated that
agonists and inhibitors of PI3K/Akt were able to affect LPS-induced
production of proinflammatory cytokines in RAW264.7 cells.
Effect of IGF-1 and wortmannin on
PI3K, Akt, TLR4, MyD88, p38MAPK and NF-κBp65 mRNA in RAW264.7
cells
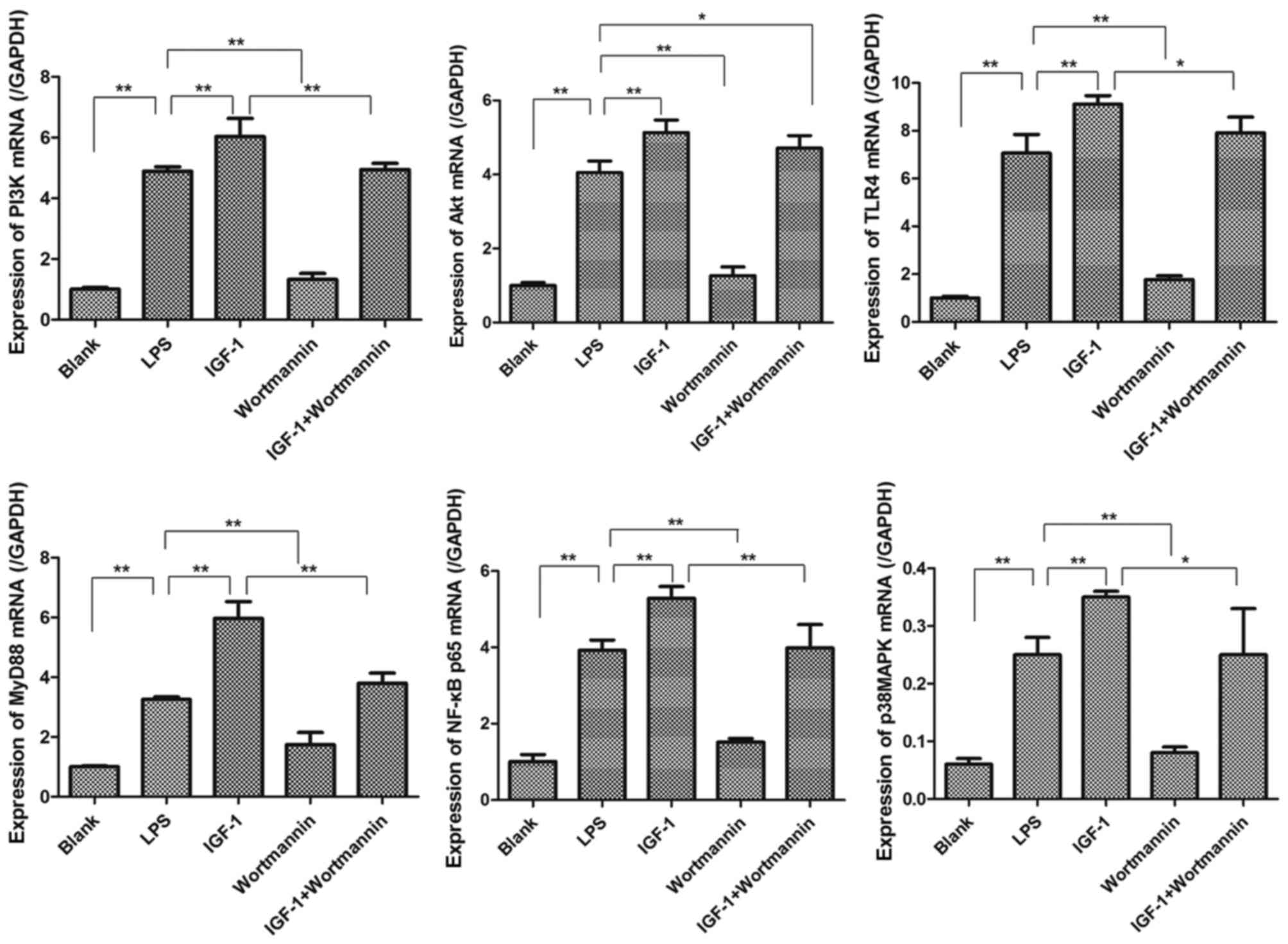
In response to LPS, overexpression of TLR4, MyD88,
PI3K, Akt, p38MAPK and NF-κBp65 mRNA was observed in RAW264.7 cells
compared with Blank cells. As shown in Fig. 6, the level of each of these factors
was higher in the IGF-1 group compared with the LPS group, whereas
their expression in the wortmannin group was lower than in the LPS
group. Moreover, the mRNA level of TLR4, MyD88, PI3K, p38MAPK and
NF-κBp65 in the IGF-1+wortmannin group was lower than in the IGF-1
group (P<0.05). While the mRNA level of Akt in the
IGF-1+wortmannin group was lower than in the IGF-1 group, the
difference was not statistically significant (Fig. 6). These data demonstrated that
agonists and inhibitors of PI3K/Akt were able to change the level
of PI3K and Akt and affect gene expression of TLR4, MyD88, p38MAPK
and NF-κBp65 in RAW264.7 cells.
Effect of IGF-1 on phosphorylation of
Akt in RAW264.7 cells
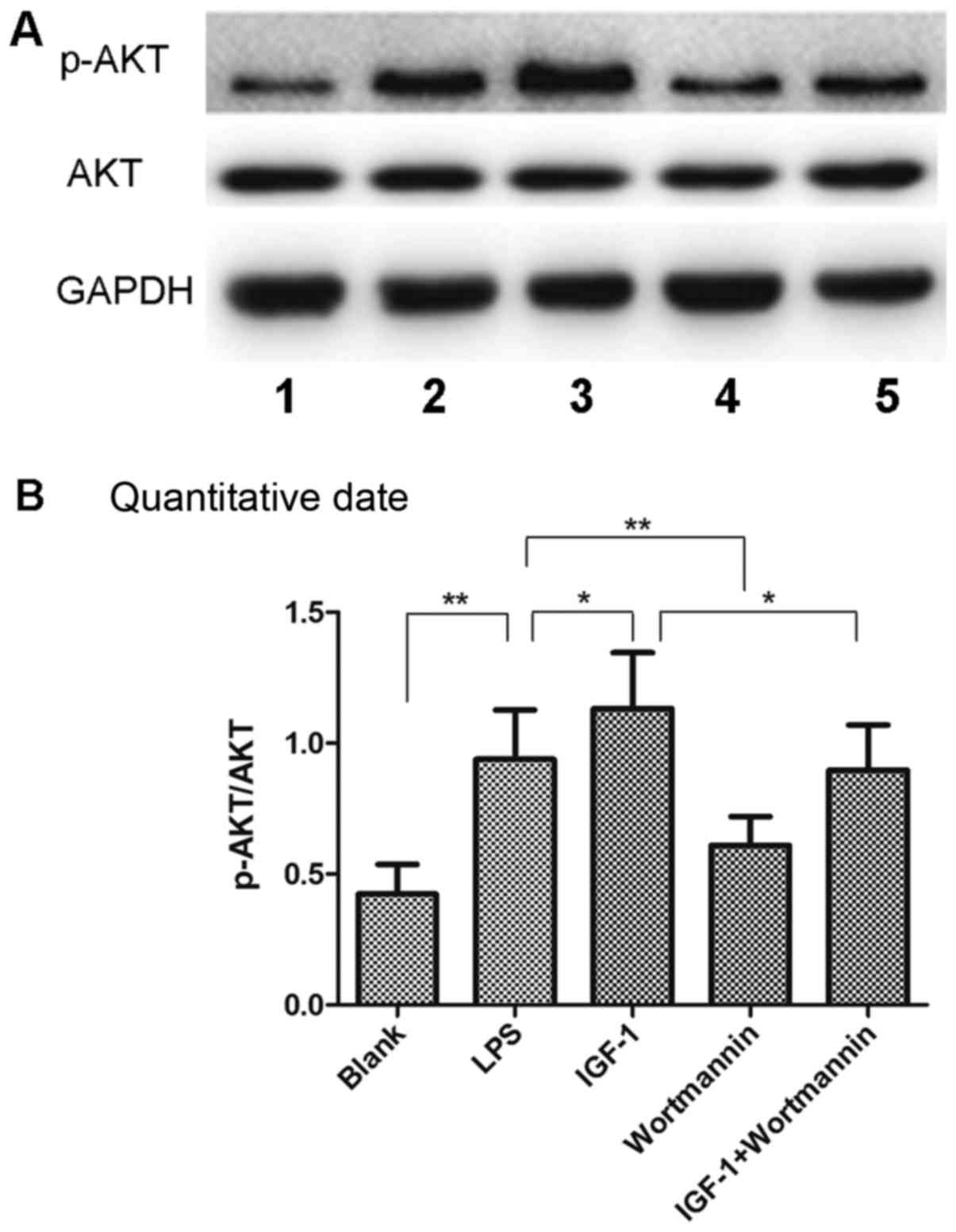
The phosphorylation of Akt in the LPS group was
markedly upregulated compared to the Blank cells (Fig. 7). Moreover, IGF-1 treatment
resulted in increased protein expression of p-Akt/Akt in the cells.
The phosphorylation of Akt in the wortmannin group was lower than
in the LPS group, and was also lower in the IGF-1+wortmannin group
compared to the IGF-1 group (P<0.05; Fig. 7). These results suggested that
IGF-1 could increase the phosphorylation of Akt in RAW264.7
cells.
Discussion
The PI3K/Akt pathway has been shown to control a
variety of cellular processes, including cell survival and
proliferation. Several lines of evidence have recently shown that
PI3K/Akt are involved in several inflammatory diseases (19,20).
We have shown previously that the PI3K/Akt inhibitor wortmannin
decreased inflammatory cytokines, alleviated the severity of
inflammation in the pancreas and improved the survival rate in SAP
rats, indicating that PI3K/Akt take part in the development of SAP
(2). In the present study, we used
IGF-1, an agonist of PI3K, to test the hypothesis that PI3K/Akt
regulate proinflammatory cytokines and inflammation through TLR4
and its downstream signaling pathway, thereby controlling SAP
inflammation.
We examined the severity of inflammation in SAP rats
by histological examination of the pancreas, ascites volume,
activity of ascites amylase and TNF-α level following IGF-1
treatment. The results showed that IGF-1 not only decreased the
severity of pancreatic damage but also greatly attenuated the
expression of the proinflammatory cytokine IL-6 in a dose-dependent
manner, which was consistent with the histological changes we
observed in the pancreas, and suggested that IGF-1 exerts an
anti-inflammatory effect in SAP rats by reducing the expression of
proinflammatory cytokines. To determine if the modulating effect of
IGF-1 on inflammation was associated with TLR4 gene expression,
TLR4 mRNA levels were determined by real-time PCR. The results
showed that IGF-1 decreased TLR4 expression in a dose-dependent
manner. It is well known that TLR4-mediated signaling activates
NF-κB and p38MAPK, which play critical roles in regulating
expression of proinflammatory genes. Thus, we examined the
expression of NF-κBp65 and p38MAPK and found that they changed in
accordance with expression of TLR4. All of these results indicate
that IGF-1 may exert a modulating effect on TLR4 and its downstream
signaling pathway in SAP rats. However, there are some caveats
regarding this conclusion. First, IGF-1 as an agonist of PI3K
showed the same anti-inflammatory effect as the PI3K inhibitor
wortmannin. Second, inhibitors were not used to inhibit the effect
of IGF-1 and TLR4 in our SAP rats. Thus, it was difficult to prove
that the anti-inflammatory effect of IGF-1 was mediated by TLR4 and
its downstream signaling pathway. Additional study will be required
to unambiguously demonstrate this effect.
To determine the relationship between PI3K/Akt and
TLR4, RAW264.7 cells were treated with LPS in the presence or
absence of a PI3K inhibitor (wortmannin) and agonist (IGF-1).
Stimulation of RAW264.7 cells with LPS induces the production of
various proinflammatory cytokines. Our data showed that IL-6 and
TNF-α secretion were increased by co-stimulation with LPS and IGF-1
compared to LPS stimulation alone. This modulation was suppressed
by using wortmannin, indicating that IGF-1 promotes the
inflammatory effect in RAW264.7 cells through PI3K/Akt.
Furthermore, the expression of TLR4 and MyD88 mRNA, which was
increased by IGF-1 overexpression following LPS treatment, was
downregulated by wortmannin. Moreover, the expression of Akt,
NF-κBp65 and p38MAPK were in line with the expression of TLR4 and
proinflammatory cytokines. It has been shown that TLR4 activation
by LPS triggers the activity of MyD88, changes a series of adapter
molecules and kinases and then stimulates NF-κB and p38MAPK
translocation and expression of target genes (9). These findings suggest that PI3K/Akt
have a relationship with TLR4 in which upstream and downstream TLR4
signaling can be affected by inhibitors and agonists of PI3K in
RAW264.7 cells.
There are many TLRs. The expressions of TLR2, TLR4
and TLR9 were found to be increased in experimental AP in rodents,
and there is strong data indicating that TLR4 plays a significant
pro-inflammatory role in the progression of AP apart from TLR2 and
TLR9 (5–7). Activation of TLR4 signaling is
involved in the injury of the pancreas, intestine, lung, liver and
kidney in SAP; meanwhile, expression of TLR4 was increased in the
early phase of SAP in those organs (5–7,21,22).
TLR4 (−/−) mice showed significantly less SAP and reduced lung
injury compared to wild-type mice after stimulation in the SAP
animal model (5). Furthermore,
another study found that TLR4 but not TLR2 regulates inflammation
and tissue damage in AP (23).
TLR4 may trigger inflammatory responses through a series of signal
transduction cascades, including MYD88. Knockdown of MYD88
attenuates LPS-induced inflammatory responses in pancreatic ductal
cells (24). Meanwhile, acute
pancreatitis patients have high level of TLR4 and decreasing the
expression of TLR4 reduces the risk of systemic complications and
mortality (25). NF-κB and p38MAPK
are the two main signaling pathways activated by TLR4-mediated
signaling, and both play an important role in the regulation of
several inflammatory cytokines and mediators. Previous work from
our group has shown that the activation of NF-κB and p38MAPK is a
prominent response during pancreatic injury and that activation of
these molecules plays a key role not only in the release of
inflammatory factors, but also in increasing neutrophil
infiltration of the pancreas (14,26).
All the above research thus suggests that TLR4 plays an important
role in the synthesis and release of proinflammatory cytokines, and
indicates that upregulation of the TLR4 gene may be related to the
development of inflammation in SAP. Our present study is consistent
with those previous results.
Both in vitro and in vivo data in our
present study suggested that inhibitors and agonists of PI3K could
regulate the expression of TLR4 and its downstream molecules
(MyD88, NF-κB, p38MAPK, IL-6 and TNF-α), indicating a relationship
between PI3K and TLR4 in the progression of SAP. In fact, several
reports have shown just such a relationship in other cells
(27–32), as well as hypoxic stress, not only
in acute pancreatitis. One study demonstrated that treatment with a
PI3K inhibitor could significantly decrease LPS-induced TLR4
protein and mRNA expression in vascular smooth muscle cells
(27). Other studies have shown
that activation of the PI3K pathway could increase TLR4-induced
TNF-α and IL-6 production in mast cells (28). It has also been demonstrated that
PI3K/Akt contribute to increased expression of TLR4 in macrophages
exposed to hypoxic stress (29).
Meanwhile, several investigators have determined that PI3K/Akt are
important mediators involved in regulating NF-κB and
p38MAPK-dependent gene expression in various cell types (30,31).
In addition, PI3K has been shown to positively regulate cytokine
expression through the formation of a complex between the p85
regulatory subunit, TLR4 and MyD88, which limits LPS-induced
inflammation via inhibition of TLR4/MyD88/PI3K complex formation
that in turn inhibits activation of downstream signaling pathways
(32). Our previous data showed
that an inhibitor of PI3K (wortmannin) alleviated the severity of
SAP by suppressing phosphorylation of Akt, which inhibited the
activation of NF-κB and p38MAPK (2). In the present study, the
downregulation of TLR4 and amelioration of inflammation were
accompanied by a decline in the level of Akt in SAP rats.
Meanwhile, our in vitro experiments produced similar
results, in which inhibition of PI3K attenuated the expression of
Akt, TLR4, MyD88, NF-κBp65 and p38MAPK and downregulated the level
of proinflammatory cytokines; in contrast, activation of PI3K with
IGF-1 produced the opposite result, and wortmannin restrained the
proinflammatory effect of IGF-1. In agreement with previous reports
from other laboratories, we have uncovered the relationship between
PI3K/Akt and TLR4, and our results provide evidence for the first
time to our knowledge that PI3K/Akt may control inflammation
through regulation of TLR4 and its downstream signaling in the
progression of SAP.
However, there was a very mixed reaction to the
application of IGF-1 in vivo and vitro. IGF-1, a peptide
hormone, is synthesized in the liver and kidney and has pleiotropic
effects on cell growth and metabolism (33). IGF-1R binds IGF-1 with high
affinity and initiates the physiological response to this ligand,
such as induce strong activation of the PI3K pathway (28). Some reports have shown that IGF-1
has an anti-inflammatory effect, and one report determined that
IGF-1 alleviates ox-LDL-induced inflammation (34). In our study we employed IGF-1 as an
agonist of PI3K in our study and testd the level of IGF-1R. We
found that IGF-1 increased the phosphorylation of Akt, consistent
with the results of other studies (17). However, IGF-1 activated RAW264.7
cells in vitro, but failed to do so in vivo. Those
results demonstrated that IGF-1 can protect SAP rats in other ways
than by IGF-1R in pancreatic tissue. Study reported that
administration of IGF-1 attenuates pancreatic damage in part due to
the increase in IL-10 production, the reduction in liberation of
IL-1β and the improvement in pancreatic blood flow (35). Moreover, insulin protects
pancreatic acinar cells from cytosolic calcium overload via
inhibition of the plasma membrane calcium pump (36). Thus, the expression of IGF-1R mRNA
only reflect the effect of exogenous IGF-1. Further in-depth
studies will be necessary to interpret the discrepancy between
IGF-1 in vivo and in vitro effects.
Taken together, our results suggest that PI3K/Akt
may regulate TLR4 and its downstream adapter molecules, kinases,
transcription factors and expression of inflammatory cytokinse,
thereby controlling SAP inflammation. Akt and TLR4 knockdown
experiment would be done to futher confirm this conculsion in the
future. We also found that IGF-1 could inhibit inflammation in SAP
rats. Based on our previous and present studies, we suggest that
pharmacologic inhibition of the PI3K/Akt pathway may represent a
new therapeutic approach to limit the progress and development of
inflammation in SAP. It is also important to note that because
signaling pathways in inflammation are linked together in an
interrelated network, further studies will be required to
accurately investigate the mechanisms of PI3K/Akt and TLR4 in the
progression of SAP.
Acknowledgements
Not applicable.
Funding
This study was supported by Medical leading
professional project of Songjiang district, Shanghai Health Bureau
(grant no. 201358) and Shanghai Health Bureau Youth Scientific
Research Project. (grant no. 20144Y0161).
Availability of data and materials
The datasets used and analyzed during the current
study are available from the corresponding author on reasonable
request.
Authors' contributions
JW conceived and performed the experiments and was a
major contributor in writing the manuscript. PX designed the
experiments and analyzed the cell data. CZ and ZWY analyzed and
interpreted the data regarding the animal studies and performed
polymerase chain reaction and western blot analysis of the
pancreases and cells. CZW and YXL performed the histological
examination of the pancreases. All authors read and approved the
final manuscript.
Ethics approval and consent to
participate
The experiment was designed in accordance with the
guidelines for the care and use of laboratory animals in research
and was approved by the Ethical and Research Committee of Shanghai
Jiao Tong University.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
Glossary
Abbreviations
Abbreviations:
|
PI3K/Akt
|
phosphoinositide 3-kinase/protein
kinase B
|
|
TLR4
|
Toll-like receptor 4
|
|
SAP
|
severe acute pancreatitis
|
|
TNF-α
|
tumor necrosis factor-α
|
|
IL-6
|
interleukin-6
|
References
|
1
|
Chiang DT, Anozie A, Fleming WR and Kiroff
GK: Comparative study on acute pancreatitis management. ANZ J Surg.
74:218–221. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Xu P, Wang J, Yang ZW, Lou XL and Chen C:
Regulatory roles of the PI3K/Akt signaling pathway in rats with
severe acute pancreatitis. PLoS One. 8:e817672013. View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Troutman TD, Hu W, Fulenchek S, Yamazaki
T, Kurosaki T, Bazan JF and Pasare C: Role for B-cell adapter for
PI3K (BCAP) as a signaling adapter linking Toll-like receptors
(TLRs) to serine/threonine kinases PI3K/Akt. Proc Natl Acad Sci
USA. 109:273–278. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Akira S, Uematsu S and Takeuchi O:
Pathogen recognition and innate immunity. Cell. 124:783–801. 2006.
View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Sharif R, Dawra R, Wasiluk K, Phillips P,
Dudeja V, Kurt-Jones E, Finberg R and Saluja A: Impact of toll-like
receptor 4 on the severity of acute pancreatitis and
pancreatitis-associated lung injury in mice. Gut. 58:813–819. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Ding JL, Zhou ZG, Zhou XY, Zhou B, Wang L,
Wang R, Zhan L, Sun XF and Li Y: Attenuation of acute pancreatitis
by peroxisome proliferator-activated receptor-α in rats: The effect
on toll-like receptor signaling pathways. Pancreas. 42:114–122.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hoque R, Sohail M, Malik A, Sarwar S, Luo
Y, Shah A, Barrat F, Flavell R, Gorelick F, Husain S and Mehal W:
TLR9 and the NLRP3 inflammasome link acinar cell death with
inflammation in acute pancreatitis. Gastroenterology. 141:358–369.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Mogensen TH: Pathogen recognition and
inflammatory signaling in innate immune defenses. Clin Microbiol
Rev. 22:240–273. 2009. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Takeda K and Akira S: TLR signaling
pathways. Semin Immunol. 16:3–9. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Gay NJ and Gangloff M: Structure and
function of Toll receptors and their ligands. Annu Rev Biochem.
76:141–165. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Schmid RM and Adler G:
NF-kappaB/rel/IkappaB: Implications in gastrointestinal diseases.
Gastroenterology. 118:1208–1228. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Zarubin T and Han J: Activation and
signaling of the p38 MAP kinase pathway. Cell Res. 15:11–18. 2005.
View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Xu P, Zhou XJ, Chen LQ, Chen J, Xie Y, Lv
LH and Hou XH: Pioglitazone attenuates the severity of sodium
taurocholate-induced severe acute pancreatitis. World J
Gastroenterol. 13:1983–1988. 2007. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Wang J, Xu P, Hou YQ, Xu K, Li QH and
Huang L: Pancreatitis-associated ascitic fluid induces
proinflammatory cytokine expression in THP-1 cells by inhibiting
anti-inflammatory signaling. Pancreas. 42:855–860. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Schmidt J, Rattner DW, Lewandrowski K,
Compton CC, Mandavilli U, Knoefel WT and Warshaw AL: A better model
of acute pancreatitis for evaluating therapy. Ann Surg. 215:44–56.
1992. View Article : Google Scholar : PubMed/NCBI
|
|
16
|
LeRoith D, Werner H, Beitner-Johnson D and
Roberts CT Jr: Molecular and cellular aspects of the insulin-like
growth factor I receptor. Endocr Rev. 16:143–163. 1995. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Zhang L, Yue Y, Ouyang M, Liu H and Li Z:
The effects of IGF-1 on TNF-α-treated DRG neurons by modulating
ATF3 and GAP-43 expression via PI3K/Akt/S6K signaling pathway.
Neurochem Res. 42:1403–1421. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Metz M and Maurer M: Mast cells-key
effector cells in immune responses. Trends Immunol. 28:234–241.
2007. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Konrad S, Ali SR, Wiege K, Syed SN,
Engling L, Piekorz RP, Hirsch E, Nürnberg B, Schmidt RE and Gessner
JE: Phosphoinositide 3-kinases gamma and delta, linkers of
coordinate C5a receptor-Fcgamma receptor activation and immune
complex-induced inflammation. J Biol Chem. 283:33296–33303. 2008.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
González-García A, Sánchez-Ruiz J, Flores
JM and Carrera AC: Phosphatidylinositol 3-kinase gamma inhibition
ameliorates inflammation and tumor growth in a model of
colitis-associated cancer. Gastroenterology. 138:1374–1383. 2010.
View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Sawa H, Ueda T, Takeyama Y, Yasuda T,
Shinzeki M, Matsumura N, Nakajima T and Kuroda Y: Expression of
toll-like receptor 2 and 4 in intestinal mucosa in experimental
severe acute pancreatitis. Hepatogastroenterology. 55:2247–2251.
2008.PubMed/NCBI
|
|
22
|
Zhang X, Zhu C, Wu D and Jiang X: Possible
role of toll-like receptor 4 in acute pancreatitis. Pancreas.
39:819–824. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Awla D, Abdulla A, Regnér S and Thorlacius
H: TLR4 but not TLR2 regulates inflammation and tissue damage in
acute pancreatitis induced by retrograde infusion of taurocholate.
Inflamm Res. 60:1093–1098. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Y and Li Y, Chen KL, Zhou B, Lv ZY,
Zhou ZG and Li Y: Knockdown of myeloid differentiation factor 88
attenuates lipopolysaccharide-induced inflammatory response in
pancreatic ductal cells. Pancreas. 45:755–760. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Gorsky VA, Agapov MA, Khoreva MV and
Leonenko IV: The effect of lornoxicam on TLR2 and TLR4 messenger
RNA expression and tumor necrosis factor-α, interleukin-6, and
interleukin-8 secretion in patients with systemic complications of
acute pancreatitis. Pancreas. 44:824–830. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Xu P, Xu K, Wang J, Jiang JP and Chen LQ:
Pioglitazone: A promising therapeutic tool in sodium
taurocholate-induced severe acute pancreatitis. Dig Dis Sci.
56:1082–1089. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Jiang D, Li D, Cao L, Wang L, Zhu S, Xu T,
Wang C and Pan D: Positive feedback regulation of proliferation in
vascular smooth muscle cells stimulated by lipopolysaccharide is
mediated through the TLR 4/Rac1/Akt pathway. PLoS One.
9:e923982014. View Article : Google Scholar : PubMed/NCBI
|
|
28
|
Hochdörfer T, Kuhny M, Zorn CN, Hendriks
RW, Vanhaesebroeck B, Bohnacker T, Krystal G and Huber M:
Activation of the PI3K pathway increases TLR-induced TNF-α and IL-6
but reduces IL-1β production in mast cells. Cell Signal.
23:866–875. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Kim SY, Jeong E, Joung SM and Lee JY:
PI3K/Akt contributes to increased expression of Toll-like receptor
4 in macrophages exposed to hypoxic stress. Biochem Biophys Res
Commun. 419:466–471. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Steelman LS, Pohnert SC, Shelton JG,
Franklin RA, Bertrand FE and McCubrey JA: JAK/STAT, Raf/MEK/ERK,
PI3K/Akt and BCR-ABL in cell cycle progression and leukemogenesis.
Leukemia. 18:189–218. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Kang X, Wang LZ, Wang YG, Liu L, Fan ZW,
Bai LZ and Lu XG: Expression and significance of
phosphatidylinositol 3-kinase/protein kinase B signal transduction
pathway in severe acute pancreatitis-associated lung injury.
Zhonghua Yi Xue Za Zhi. 90:732–737. 2010.(In Chinese). PubMed/NCBI
|
|
32
|
Endale M, Park SC, Kim S, Kim SH, Yang Y,
Cho JY and Rhee MH: Quercetin disrupts tyrosine-phosphorylated
phosphatidylinositol 3-kinase and myeloid differentiation factor-88
association, and inhibits MAPK/AP-1 and IKK/NF-κB-induced
inflammatory mediators production in RAW 264.7 cells.
Immunobiology. 218:1452–1467. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Payne JF, Tangpricha V, Cleveland J, Lynn
MJ, Ray R and Srivastava SK: Serum insulin-like growth factor-I in
diabetic retinopathy. Mol Vis. 17:2318–2324. 2011.PubMed/NCBI
|
|
34
|
Yu X, Xing C, Pan Y, Ma H, Zhang J and Li
W: IGF-1 alleviates ox-LDL-induced inflammation via reducing HMGB1
release in HAECs. Acta Biochim Biophys Sin (Shanghai). 44:746–751.
2012. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Warzecha Z, Dembinski A, Ceranowicz P,
Konturek SJ, Tomaszewska R, Stachura J and Konturek PC: IGF-1
stimulates production of interleukin-10 and inhibits development of
caerulein-induced pancreatitis. J Physiol Pharmacol. 54:575–590.
2003.PubMed/NCBI
|
|
36
|
Mankad P1, James A, Siriwardena AK,
Elliott AC and Bruce JI: Insulin protects pancreatic acinar cells
from cytosolic calcium overload and inhibition of plasma membrane
calcium pump. J Biol Chem. 287:1823–1836. 2012. View Article : Google Scholar : PubMed/NCBI
|