Introduction
Osteoarthritis (OA) is the most common type of
arthritis and a leading cause of pain and disability, which places
a great burden on the economy of health and reduces quality of life
(1–3). OA involves the degeneration of
numerous tissues, including subchondral bone, ligaments, muscle,
tendons, and the meniscus and synovium (1). Numerous factors may affect OA
progression, including age, gender, obesity, genetics and joint
injury (4); however, how these
factors affect the development of OA requires further investigation
and no effective method has been developed for the relief of pain.
In addition, molecular biology studies have identified numerous
biomarkers and biological processes that contribute to OA,
including the erosion of the extracellular matrix (5), the expression of chemokines
(chemokine C-C ligands 9 and 5, and interleukin-8) (6) and the upregulation of inflammatory
genes (7). Further investigation
into the molecular events associated with cartilage degeneration is
required.
Over the past decades, the development of
high-throughput technologies has resulted in the large amount of
accumulation of omics data for various complex diseases. For OA,
gene expression profiling via microarray or high-throughput
sequencing has become a promising method for the analysis of the
mechanisms underlying its initiation and progression (8). For example, Rasheed et al
(9) performed an integrated study
of microRNA (miRNA) expression profiles in OA chondrocytes and
OA-associated genes, and identified numerous miRNAs associated with
the development of OA. Sun et al (10) reported several potential biomarkers
for OA via differential expression and network analysis based on
gene microarray datasets. Microarray analysis in the study of
Loeser et al (11)
indicated the link between age-associated differences in gene
expression and the development of OA. In addition, epigenetic
modifications serve important roles in gene expression regulation,
and DNA methylation is one of the most common types of epigenetic
modification. Recently, an increasing number of studies have
focused on the associations between methylation status and the
progression of OA (12,13). In contrast to cancer, in which CpG
sites are frequently hypermethylated, the majority of studies
investigating OA reported a higher frequency of hypomethylation
(14,15). DNA methylation may also affect the
allelic imbalance of specific small nucleotide polymorphisms, and
thus the development of OA (16).
Combined analysis of gene expression and DNA methylation profiles
may contribute to the screening of potential biomarkers of OA,
early diagnosis and treatment; to the best of our knowledge, an
investigation into this is yet to be performed.
In the present study, combined analysis of publicly
accessible gene expression and DNA methylation microarray datasets
of OA was conducted. Functional enrichment and network analysis was
performed for the identification of potential biomarkers. Numerous
known and novel targets were obtained and their involvement in OA
was further confirmed via reverse transcription-quantitative
polymerase chain reaction (RT-qPCR) analysis.
Materials and methods
Microarray datasets
The publicly accessible data were all obtained from
the Gene Expression Omnibus (GEO, www.ncbi.nlm.nih.gov/geo). Gene expression profiles
deposited by Klinger et al (17), accession no. GSE43923, containing
six samples (three osteophytic cartilage and three corresponding
articular cartilage samples from the knee joints of patients with
OA) were employed in the present study. The genome-wide expression
profiles were quantified using the commercial gene microarray
GPL570 [HG-U133_Plus_2] Affymetrix Human Genome U133 Plus 2.0 Array
(Affymetrix; Thermo Fisher Scientific, Inc., Waltham, MA, USA). The
DNA methylation profiles (GSE73626) (18) of five hip OA, six knee OA and seven
hip healthy cartilage samples were detected via Illumina
HumanMethylation450 BeadChip assay (Illumina, Inc., San Diego, CA,
USA), which contains >480,000 methylation sites, covering 99% of
RefSeq (https://www.ncbi.nlm.nih.gov/refseq/) genes and 96% of
University of California, Santa Cruz (http://genome.ucsc.edu/)-defined CpG sites with an
average of 17 CpG sites/gene across different genomic regions,
including the promoter, 5′ untranslated region (UTR), first exon,
gene body, intergenic and 3′UTR.
Microarray data analysis
The present study conducted differential expression
analysis for osteophytic and articular cartilage samples from
patients with OA. The raw CEL data were imported into R version
3.2.2 (http://www.R-project.org/) and
normalized via the affy package (19); subsequently, the limma package
(20) was used for the screening
of differentially expressed genes (DEGs) with the criteria of fold
change >1.5 and false discovery rate (FDR)<0.05. For the
methylation dataset, site-level analysis was performed based on the
Illumina Methylation Analyzer package (Illumina, Inc.) (21) to obtain the differentially
methylated CpG sites (DMSs) between hip/knee OA cartilage and
healthy cartilage samples, with thresholds of db value >0.2 and
FDR<0.05. DMSs were mapped to the corresponding genes (DMGs) and
genomic regions based on the full annotation file of the microarray
and following this, cross analysis was performed via the
‘intersect’ function of R version 3.2.2 (http://www.R-project.org/) using DEGs and DMGs to
reveal overlapping genes. In addition, differences between
distributions of DMSs relative to CpG islands and genes were
compared using the χ2 test.
Functional clustering analysis
Investigation into the functions of enriched DEGs
and DMGs may improve understanding of their involvement in OA. In
the present study, functional clustering analysis of DEGs and DMGs
based on the Database for Annotation, Visualization and Integrated
Discovery (DAVID; david.abcc.ncifcrf.gov) (22) was conducted. Clusters with an
enrichment score >1, and Gene Ontology (GO) terms and Kyoto
Encyclopedia of Genes and Genomes (KEGG; www.genome.jp/kegg) pathways with P<0.05 were
retained in the present study.
Protein-protein interaction network
analysis
Genes are likely to function together rather than
alone in complex diseases; hub nodes in the network may represent
key biomarkers. In the present study, protein-protein interaction
(PPI) network analysis was performed to investigate the overlaps
between DEGs and DMGs based on the Human Protein Reference Database
(HPRD; www.hprd.org) (23). The network was visualized using
Cytoscape 3.6.0 software (http://www.cytoscape.org/), and the topological
property of every gene [degree (number of direct interactions in
the network)] was additionally analyzed for the assessment of their
importance.
RT-qPCR
Normal and OA tissues were obtained from the
articular cartilage of 26 females and 20 males with a median age of
56.35 years (43.58–69.12 years) between April 2012 and April 2015
in Zibo Central Hospital (Zibo, China). Patients exhibiting
temporomandibular joint pain that were not suffering from any form
of rheumatic disease or cancer were included in the present study.
The study was approved by the ethics committee of Zibo Central
Hospital. Written informed consent was obtained from all
patients.
Total RNA was extracted from OA and normal tissues
(50–100 mg) using an RNeasy Mini kit (Qiagen GmbH, Hilden, Germany)
and quantified with a NanoDrop system (Thermo Fisher Scientific,
Inc.), and subsequently subjected to RT-qPCR using EasyScript
Reverse Transcriptase kit (Promega Corporation, Madison, WI, USA).
The temperature protocol used for RT was as follows: 95°C for 10
min, 55°C for 1 min and 68°C for 10 min.
The 7500 Real-Time PCR system (Applied Biosystems;
Thermo Fisher Scientific, Inc.) was used for qPCR. Reactions were
conducted in triplicate in each reaction tube using AceQ qPCR SYBR
Green Master Mix (Vazyme Biotech Co., Ltd., Nanjing, China). The
temperature protocol used for qPCR was as follows: 94°C for 5 min;
followed by 40 cycles of 95°C for 10 sec and 60°C for 30 sec;
followed by 95°C for 15 sec, 60°C for 60 sec and 95°C for 15 sec.
Data were analyzed via the 2−ΔΔCq method using GAPDH as
internal control (24). Primer
sequences were: G protein subunit α1 (GNAI1) forward,
5′-TTAGGGCTATGGGGAGGTTGA-3′, and reverse,
5′-GGTACTCTCGGGATCTGTTGAAA-3′; runt related transcription factor 2
(RUNX2) forward, 5′-TGGTTACTGTCATGGCGGGTA-3′ and reverse,
5′-TCTCAGATCGTTGAACCTTGCTA-3′; integrin subunit β2 (ITGB2) forward,
5′-TGCGTCCTCTCTCAGGAGTG-3′ and reverse, 5′-GGTCCATGATGTCGTCAGCC-3′;
and GAPDH forward, 5′-ACAACTTTGGTATCGTGGAAGG-3′ and reverse,
5′-GCCATCACGCCACAGTTTC-3′.
Statistical analysis
R version 3.2.2 (http://www.R-project.org/) was used for all of the
statistical analysis. The relative mRNA expression levels in the
RT-qPCR analysis were presented as the mean ± standard deviation of
the three replicates. RT-qPCR data were analyzed using a Student's
t-test. P<0.05 was considered to indicate a statistically
significant difference.
Results
Gene expression profile analysis
The raw microarray data was normalized and used for
the following differential expression analysis. As a result, a
total of 466 genes were detected to be DEGs in osteophytic
cartilage samples compared with articular samples, which contained
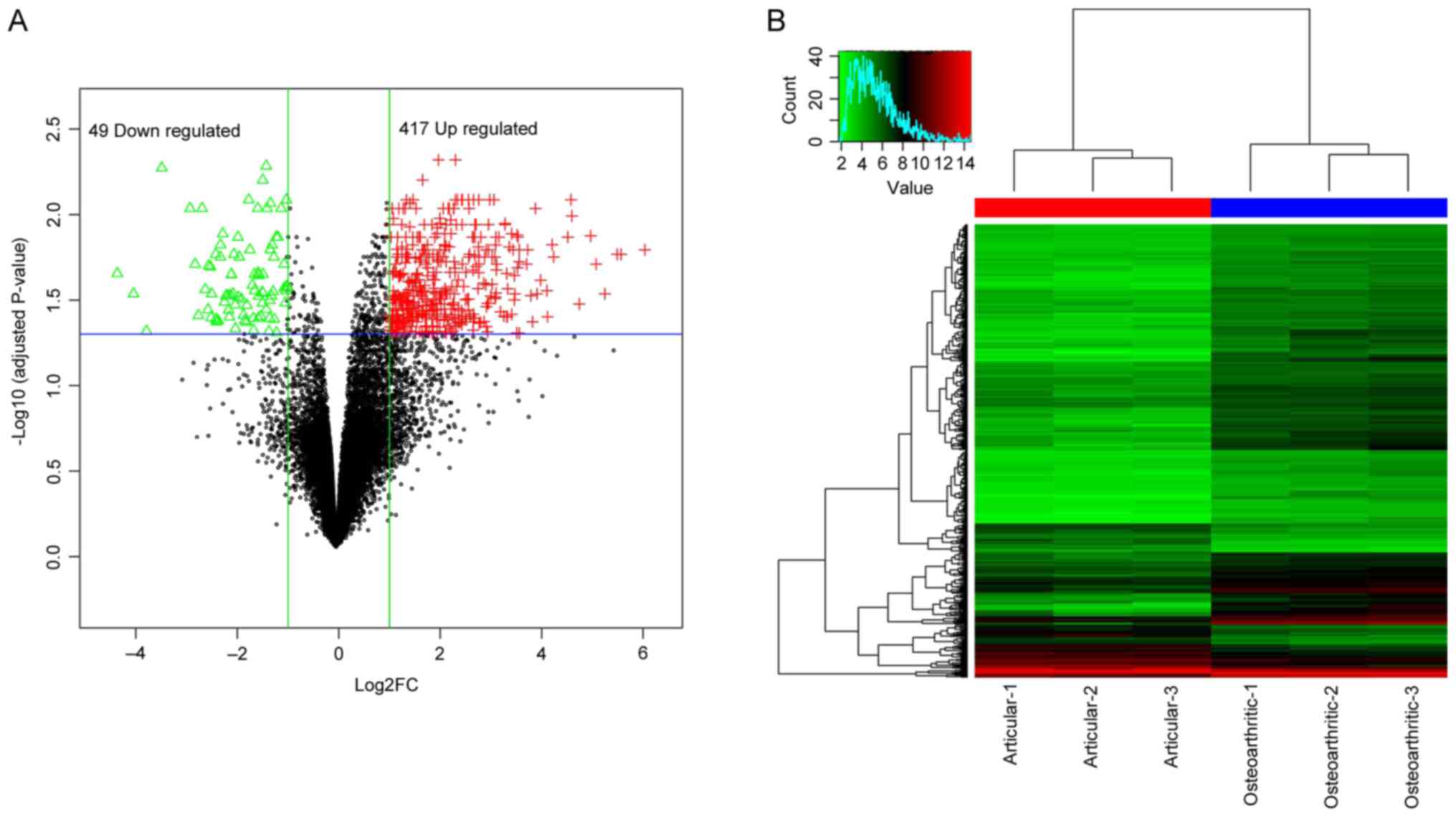
49 downregulated and 417 upregulated genes (Fig. 1A). The two-way supervised
clustering indicated notable differences between osteophytic and
articular cartilage samples from patients with OA, with blue and
red coloring indicating low and high expression levels,
respectively (Fig. 1B). The full
list of DEGs is provided in Table
I.
 | Table I.Previously identified biomarkers of
osteoarthritis with the PMID of each corresponding reference. |
Table I.
Previously identified biomarkers of
osteoarthritis with the PMID of each corresponding reference.
| Author, date | Gene | (Refs.) |
|---|
| Mabey T et
al, 2014 | Angiopoietin 2 | (43,44) |
| Gao W et al,
2013 |
|
|
| Valverde-Franco G,
2016 | Ephrin B2 | (39) |
| Leijten JCH,
2013 | Gremlin 1 DAN
family BMP antagonist | (37,45) |
| Yi J, 2016 |
|
|
| Fan Y, 2017 | Integrin subunit
β2 | (46,47) |
| Hopwood B,
2007 |
|
|
| Xiao JL, 2015 | Runt related
transcription factor 2 | (35,48) |
| Liao L, 2017 |
|
|
DMSs
Comparisons were performed between DMSs in hip/knee
cartilage samples and healthy cartilage samples. As presented in
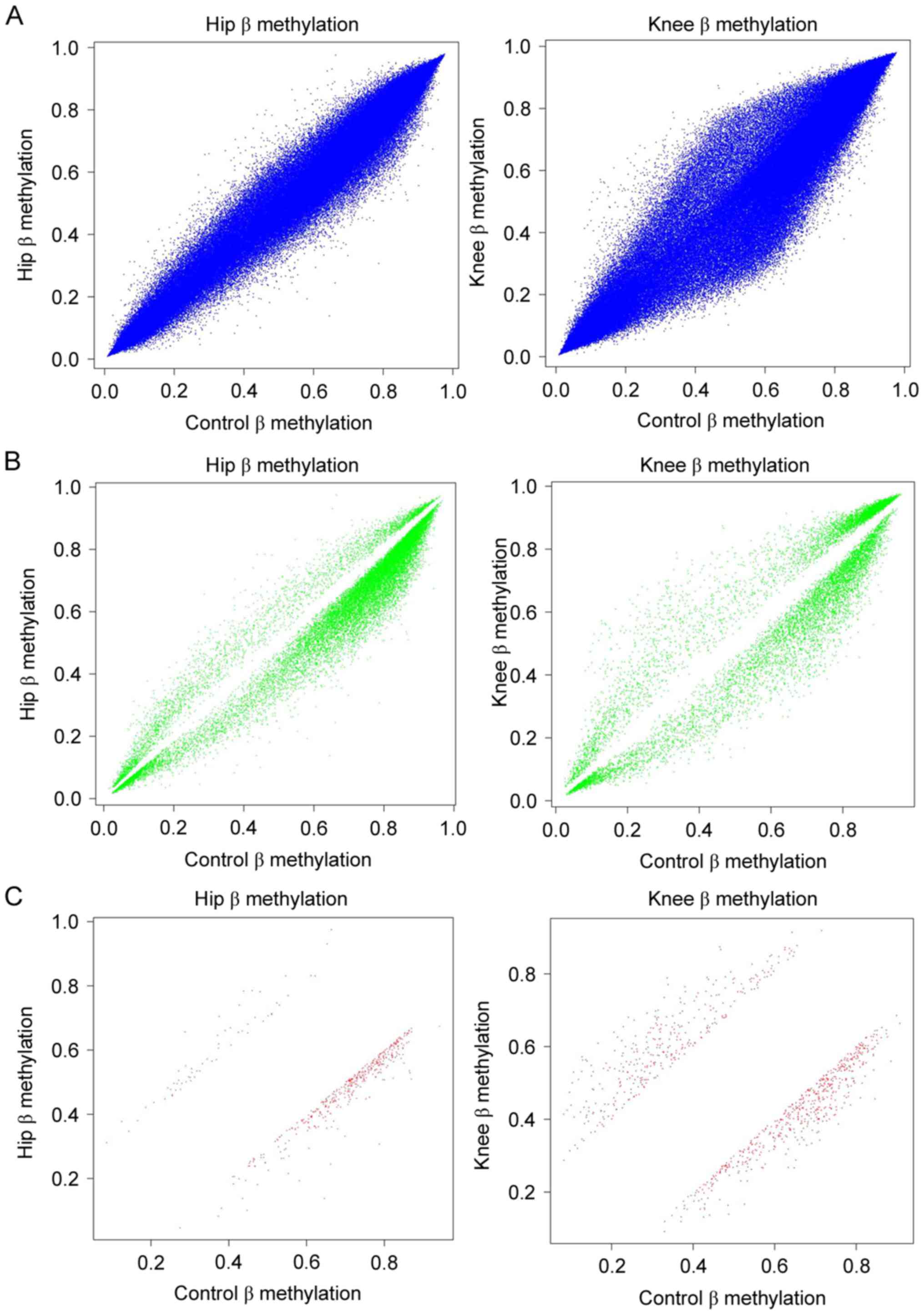
Fig. 2A, the β values of OA hip
compared with healthy hip tissue, and OA knee compared with healthy
knee tissue, of all CpG sites in the microarray were obtained. The
β values for OA hip compared with healthy hip tissue, and OA knee
compared with healthy knee tissue, of CpG sites satisfied the
criterion of P<0.05; the frequencies of hypomethylated sites
were increased compared with hypermethylated sites in OA hip and
knee samples (Fig. 2B).
Additionally, the number of hypomethylated sites with P<0.05 and
db>0.2 were increased compared with hypermethylated sites
(Fig. 2C). This was consistent
with the results of the DEG analysis (the number of upregulated
genes was increased compared with downregulated genes), as
hypermethylation of the promoter may result in the downregulation
of the corresponding gene.
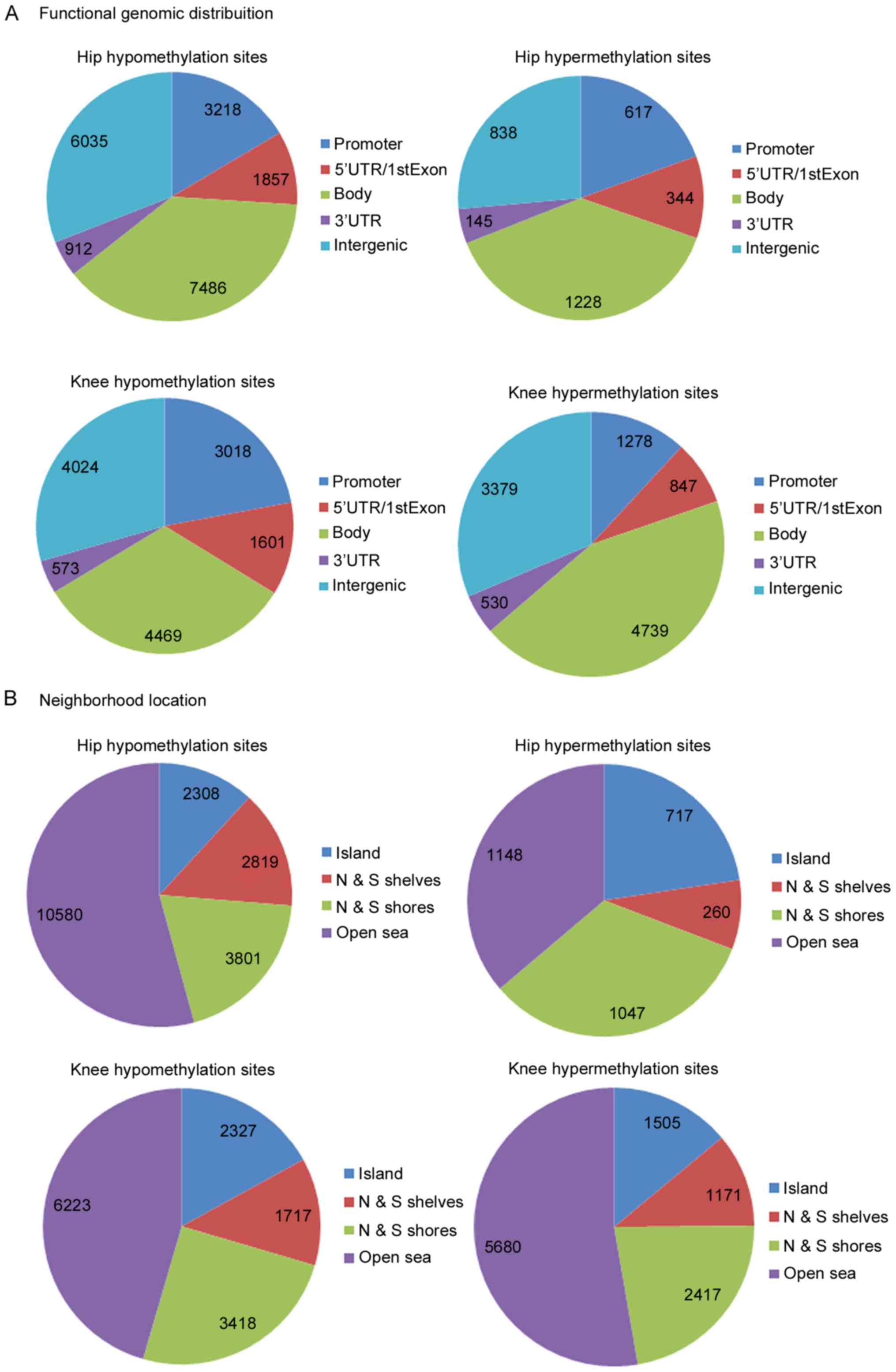
To improve understanding of the functional
significance of DMSs, the functional locations of DMSs were
investigated. As presented in Fig.
3A, the majority of the DMSs were reported to be in intergenic,
gene body and promoter regions. Additionally, four neighborhood
locations were defined in the Illumina HumanMethylation450 BeadChip
assay: 31% CpG islands, 23% shores (0–2 kb from canonical CpG
islands), 10% shelves (2–4 kb from canonical) and open sea (rest of
the sequence). Consistent with the BeadChip assay, the majority of
the hypo- and hypermethylated sites in hip and knee cartilage
samples were detected in open sea, and following the north and
south shores (upstream and downstream shores). To investigate the
associations between functional and neighborhood locations with
differential methylation status, a χ2 test for data
presented in Fig. 3. The results
indicated P<5×10−7 in all of the cases, demonstrating
that functional and neighborhood locations are associated with
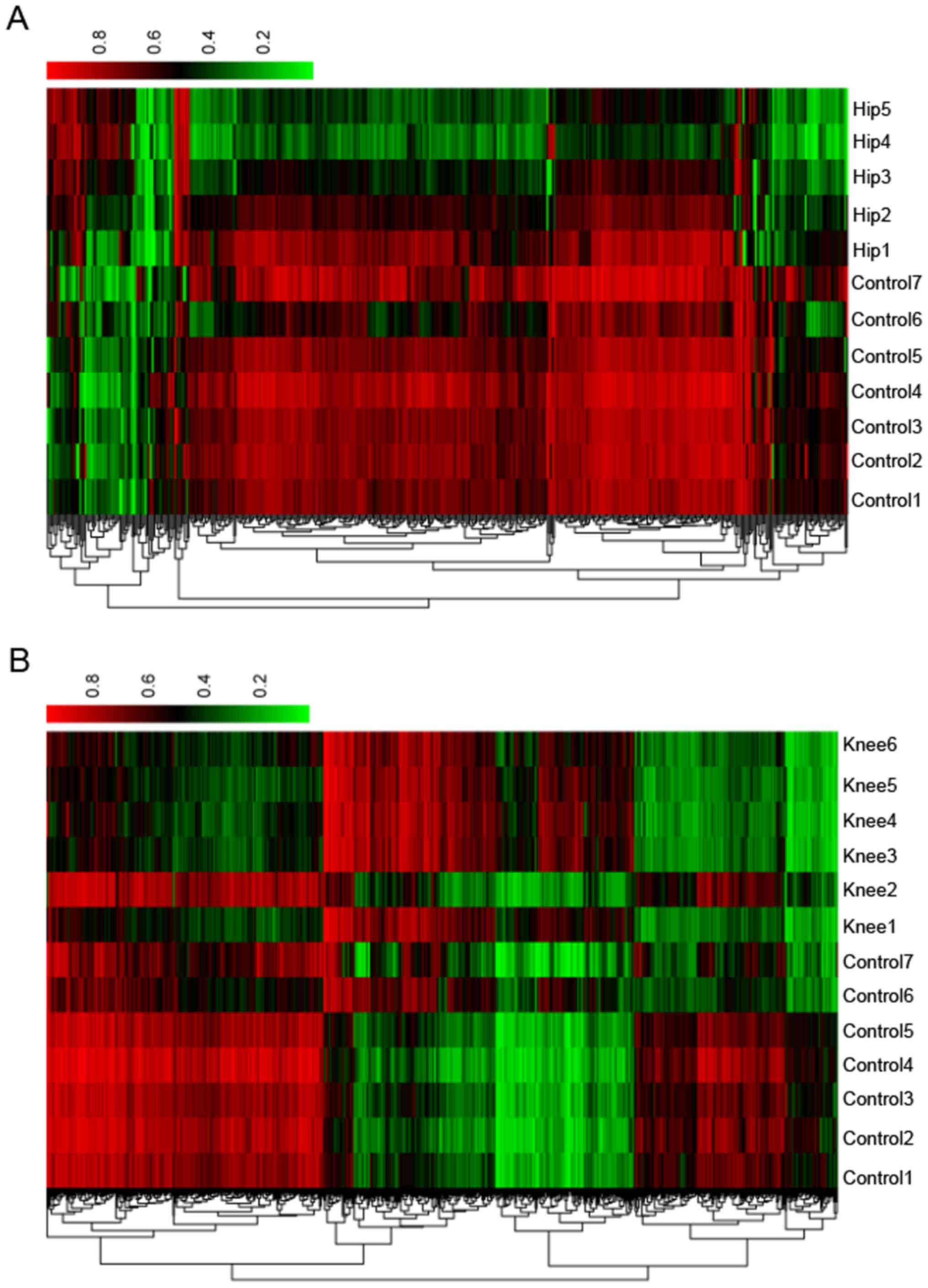
differential methylation status. One-way clustering of DMSs of hip
and knee cartilage samples is presented in Fig. 4.
Functional clusters
Functional clustering analysis in DAVID resulted in
three functional clusters for DEGs and DMGs. DEGs were primarily
involved in the GO terms and KEGG pathways that were associated
with ‘cell structure’, ‘inflammatory and immune response’,
‘substance synthetic’ and ‘metabolic’. ‘Guanosine 5′-triphosphaate
(GTP)ase activity’, ‘gene expression regulation’ and ‘inflammatory
and immune response’ were reported to be significantly enriched in
DMGs (data not shown).
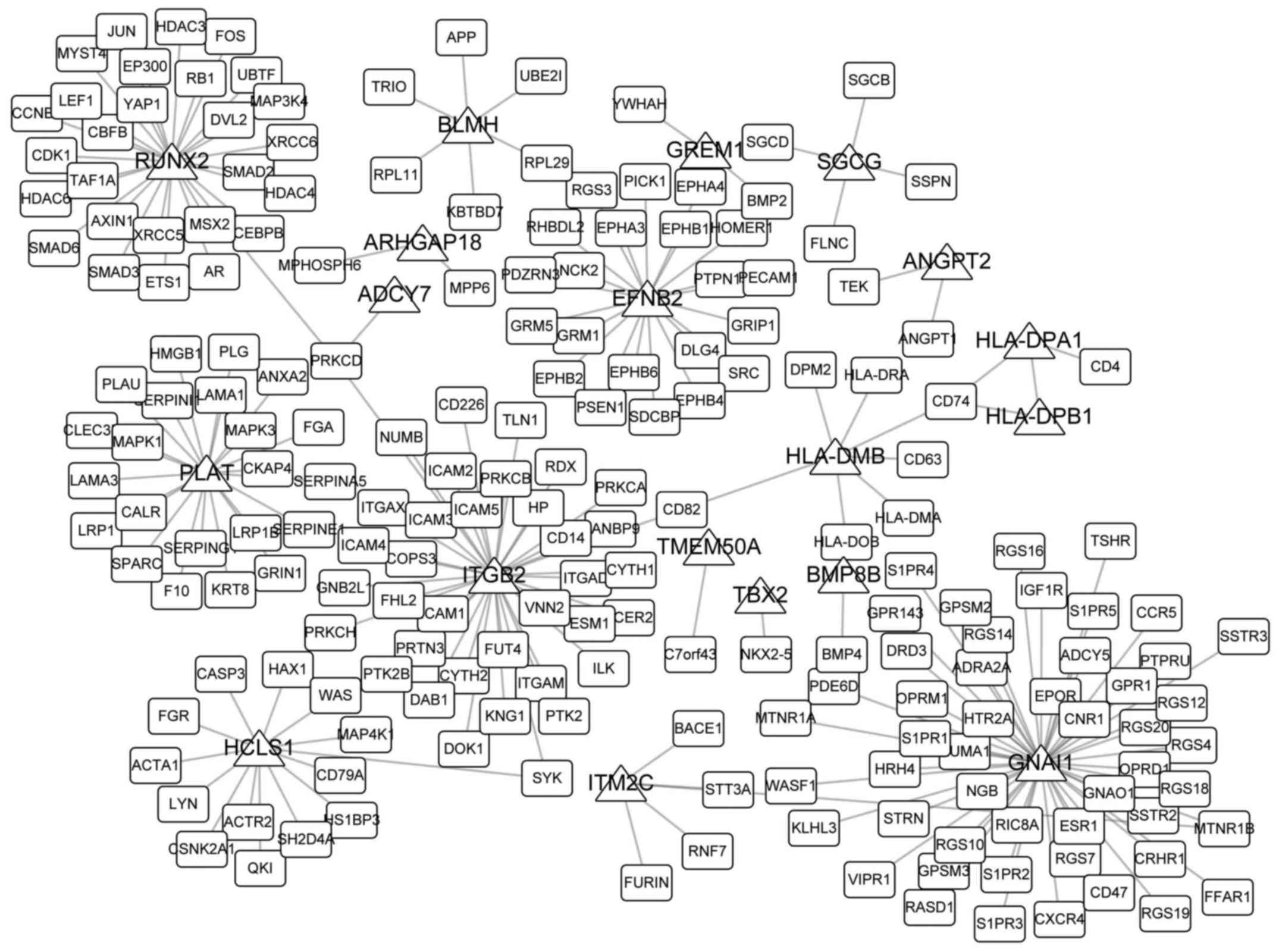
Network analysis
Network topological properties are important
representations of their roles in specific biological processes and
diseases. In the present study, 30 overlaps were obtained among
DMGs of hip and knee cartilage samples and DEGs; 20 of these
overlaps were reported to interact with other genes from the HPRD.
PPI networks are presented in Fig.
5. GNAI1 directly interacted with 50 genes, which was markedly
higher compared with the degree of the other 19 overlaps in the
network, potentially indicating its important roles in the
development of OA. Table I
includes the five previously identified biomarkers of OA and their
corresponding PMID nos.
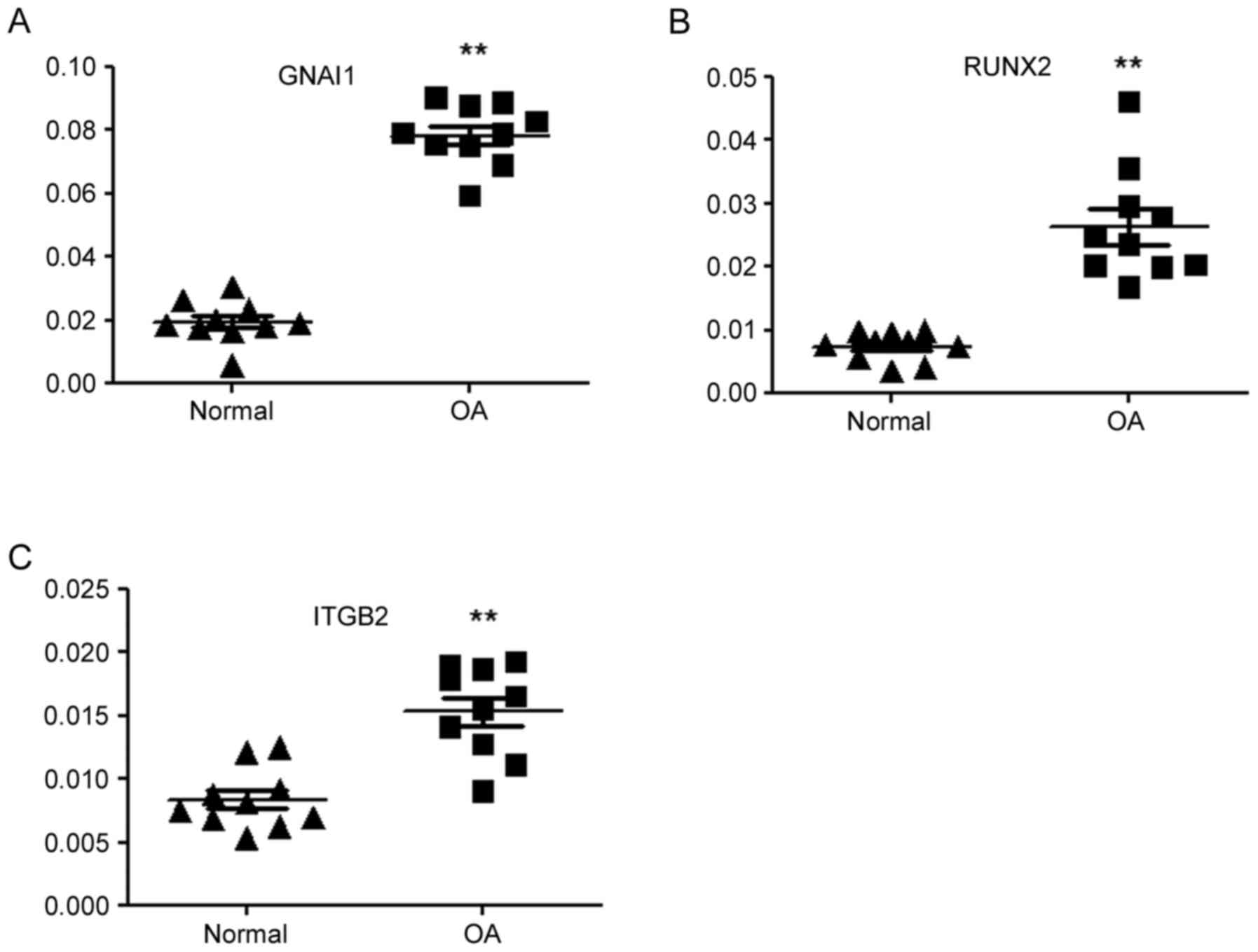
RT-qPCR analysis
A total of three hub genes, GNAI1, RUNX2 and
integrin subunit β 2 (ITGB2), were subjected to RT-qPCR analysis
for the quantification of their relative abundance in OA and
control samples. The results of the RT-qPCR analysis were
consistent with the results of the gene microarray analysis; the
relative mRNA expression levels of GNAI1, RUNX2 and ITGB2 in OA
samples were significantly increased compared with the control
samples (Fig. 6).
Discussion
OA is the most common degenerative disease of the
synovial membrane, comprising the destruction and loss of articular
tissues (25). The initiation and
development of OA have been reported to be associated with numerous
factors, including age, joint injury, obesity and chronic
inflammation, in addition to genetic factors, including epigenetic
modification and altered gene or miRNA expression (26). In-depth understanding of gene
regulation in OA may contribute to diagnosis and treatment. For
this purpose, DNA methylation and gene expression analysis of hip
and knee cartilage of OA patients was performed, and potential
targets for OA were identified and verified in the present
study.
DNA methylation has been revealed to repress gene
expression by blocking the sites at the promoter where
transcription factors bind; hypermethylation of the promoter is
associated with no or low transcription (27). In the present study, the majority
of the DEGs were observed to be upregulated and DMSs were
hypomethylated, consistent with the roles of DNA methylation.
Unlike cancer, which is characterized by the hypermethylation of
tumor suppressor genes, genome-wide hypomethylation in OA has been
widely observed previously (28–30)
and in the present study. It is known that OA is influenced by
inflammatory chemokines (31),
which was also demonstrated by the functional enrichment analysis
of the present study. The hypomethylation and increased expression
of a number of inflammation-associated genes have been observed to
be associated with OA, including IL-8 (32), nuclear factor-κB (33) and pleckstrin (34), which may serve as therapeutic
targets for OA. In the present study, numerous genes that were
differentially expressed and methylated simultaneously were
identified, including ITGB2, GNAI1 and RUNX2. To the best of our
knowledge, the roles of the aforementioned genes in the development
of OA have not been investigated; the RT-qPCR analysis conducted in
the present study revealed their relative abundance in OA and
adjacent tissues, which may indicate their association with the
progression of OA. Furthermore, numerous genes were observed to
directly interact with other genes in the PPI network, including
RUNX2, bleomycin hydrolase and gremlin 1 DAN family BMP antagonist
(GREM1). The majority of these genes have been demonstrated to be
closely associated with the progression of OA; for example, RUNX2
polymorphisms may affect temporomandibular joint OA in females
(35) and may influence the
expression of other genes in OA (36). The mRNA expression levels of GREM1
are correlated with OA and may be regulated by OA-associated
factors (37); in addition, GREM1
is a key regulator of synoviocyte hyperplasia and invasiveness
(38). Valverde-Franco et
al (39) reported that
ephrin-B2 may be essential for normal bone growth, and its absence
may lead to knee and hip OA. In addition, a number of genes with a
high degree in the PPI network have not been proven to be
associated with the development of OA. For example, GNAI1, also
known as Gi, is a protein that can hydrolyze GTP and interact with
other proteins, and T cell differentiation may alter its structure
(40). Additionally, the altered
expression of GNAI1 was observed to be associated with the
progression of inflammatory and immune diseases (41,42),
and may be considered to be a novel biomarker for OA.
Certain limitations of the present study were noted.
Only three genes were analyzed via RT-qPCR and future studies are
required to investigate more genes. Furthermore, osteophytes may be
considered to contribute to regeneration in OA joints; however,
further studies investigating the differences regarding whole
genome expression between OA and healthy tissues should be employed
for further analysis in the present study.
In conclusion, the present study provided a pipeline
for the combined analysis of gene expression and DNA methylation
datasets. In addition, numerous known and potential novel markers
were proposed in the present study, which may contribute to
diagnosis and treatment targets for OA; however, further
investigation is required for confirmation of the functions
exhibited by these markers.
Acknowledgements
Not applicable.
Funding
No funding was received.
Availability of data and materials
The datasets analyzed in the present study are
available in the GEO repository, www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE43923
and www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE73626.
Authors' contributions
DS analyzed and interpreted the microarray datasets
and produced the manuscript. WQ and ML modified the syntax; CY and
KT designed experiments of the present study. FZ made substantial
contributions to the conception and design of the study, and
submitted the manuscript. All authors read and approved the final
manuscript.
Ethics approval and consent to
participate
The present study was approved by the ethics
committee of Zibo Central Hospital, Zibo, China. Written informed
consent was obtained from all patients.
Consent for publication
Not applicable.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
Pan Q, O'Connor MI, Coutts RD, Hyzy SL,
Olivares-Navarrete R, Schwartz Z and Boyan BD: Characterization of
osteoarthritic human knees indicates potential sex differences.
Biol Sex Differ. 7:272016. View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Young IC, Chuang ST, Hsu CH, Sun YJ, Liu
HC, Chen YS and Lin FH: Protective effects of aucubin on
osteoarthritic chondrocyte model induced by hydrogen peroxide and
mechanical stimulus. BMC Complement Altern Med. 17:912017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Rhee J, Park SH, Kim SK, Kim JH, Ha CW,
Chun CH and Chun JS: Inhibition of BATF/JUN transcriptional
activity protects against osteoarthritic cartilage destruction. Ann
Rheum Dis. 76:427–434. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
4
|
Zhang M, Egan B and Wang J: Epigenetic
mechanisms underlying the aberrant catabolic and anabolic
activities of osteoarthritic chondrocytes. Int J Biochem Cell Biol.
67:101–109. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Schwager J, Hoeller U, Wolfram S and
Richard N: Rose hip and its constituent galactolipids confer
cartilage protection by modulating cytokine, and chemokine
expression. BMC Complement Altern Med. 11:1052011. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Nair A, Gan J, Bush-Joseph C, Verma N,
Tetreault MW, Saha K, Margulis A, Fogg L and Scanzello CR: Synovial
chemokine expression and relationship with knee symptoms in
patients with meniscal tears. Osteoarthritis Cartilage.
23:1158–1164. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Hashimoto S, Rai MF, Gill CS, Zhang Z,
Sandell LJ and Clohisy JC: Molecular characterization of articular
cartilage from young adults with femoroacetabular impingement. J
Bone Joint Surg Am. 95:1457–1464. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Park R and Ji JD: Unique gene expression
profile in osteoarthritis synovium compared with cartilage:
Analysis of publicly accessible microarray datasets. Rheumatol Int.
36:819–827. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Rasheed Z, Al-Shobaili HA, Rasheed N, Al
Salloom AA, Al-Shaya O, Mahmood A, Alajez NM, Alghamdi AS and
Mehana el-SE: Integrated study of globally expressed micrornas in
il-1beta-stimulated human osteoarthritis chondrocytes and
osteoarthritis relevant genes: A microarray and bioinformatics
analysis. Nucleosides Nucleotides Nucleic Acids. 35:335–355. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Sun J, Yan B, Yin W and Zhang X:
Identification of genes associated with osteoarthritis by
microarray analysis. Mol Med Rep. 12:5211–5216. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Loeser RF, Olex AL, McNulty MA, Carlson
CS, Callahan MF, Ferguson CM, Chou J, Leng X and Fetrow JS:
Microarray analysis reveals age-related differences in gene
expression during the development of osteoarthritis in mice.
Arthritis Rheum. 64:705–717. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
12
|
Housman G, Havill LM, Quillen EE, Comuzzie
AG and Stone AC: Assessment of DNA methylation patterns in the bone
and cartilage of a nonhuman primate model of osteoarthritis.
Cartilage. 1:19476035187591732018.
|
|
13
|
Peffers M, Balaskas P and Smagul A:
Osteoarthritis year in review 2017: Genetics and epigenetics.
Osteoarthritis Cartilage. 26:304–311. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
14
|
Zhang Y, Fukui N, Yahata M, Katsuragawa Y,
Tashiro T, Ikegawa S and Lee MT: Genome-wide DNA methylation
profile implicates potential cartilage regeneration at the late
stage of knee osteoarthritis. Osteoarthritis Cartilage. 24:835–843.
2016. View Article : Google Scholar : PubMed/NCBI
|
|
15
|
Rushton MD, Young DA, Loughlin J and
Reynard LN: Differential DNA methylation and expression of
inflammatory and zinc transporter genes defines subgroups of
osteoarthritic hip patients. Ann Rheum Dis. 74:1778–1782. 2015.
View Article : Google Scholar : PubMed/NCBI
|
|
16
|
Reynard LN, Bui C, Syddall CM and Loughlin
J: CpG methylation regulates allelic expression of GDF5 by
modulating binding of SP1 and SP3 repressor proteins to the
osteoarthritis susceptibility SNP rs143383. Hum Genet.
133:1059–1073. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Klinger P, Beyer C, Ekici AB, Carl HD,
Schett G, Swoboda B, Hennig FF and Gelse K: The transient
chondrocyte phenotype in human osteophytic cartilage: A role of
pigment epithelium-derived factor? Cartilage. 4:249–255. 2013.
View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Aref-Eshghi E, Zhang Y, Liu M, Harper PE,
Martin G, Furey A, Green R, Sun G, Rahman P and Zhai G: Genome-wide
DNA methylation study of hip and knee cartilage reveals embryonic
organ and skeletal system morphogenesis as major pathways involved
in osteoarthritis. BMC Musculoskelet Disord. 16:2872015. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Gautier L, Cope L, Bolstad BM and Irizarry
RA: Affy-analysis of affymetrix genechip data at the probe level.
Bioinformatics. 20:307–315. 2004. View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Diboun I, Wernisch L, Orengo CA and
Koltzenburg M: Microarray analysis after RNA amplification can
detect pronounced differences in gene expression using limma. BMC
Genomics. 7:2522006. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Wang D, Yan L, Hu Q, Sucheston LE, Higgins
MJ, Ambrosone CB, Johnson CS, Smiraglia DJ and Liu S: IMA: An R
package for high-throughput analysis of Illumina's 450K Infinium
methylation data. Bioinformatics. 28:729–730. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Dennis G Jr, Sherman BT, Hosack DA, Yang
J, Gao W, Lane HC and Lempicki RA: DAVID: Database for annotation,
visualization, and integrated discovery. Genome Biol. 4:P32003.
View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Prasad Keshava TS, Goel R, Kandasamy K,
Keerthikumar S, Kumar S, Mathivanan S, Telikicherla D, Raju R,
Shafreen B, Venugopal A, et al: Human protein reference
database-2009 update. Nucleic Acids Res. 37:D767–D772. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2 (-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Yang F, Zhou S, Wang C, Huang Y, Li H,
Wang Y, Zhu Z, Tang J and Yan M: Epigenetic modifications of
interleukin-6 in synovial fibroblasts from osteoarthritis patients.
Sci Rep. 7:435922017. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Zhou X, Chen L, Grad S, Alini M, Pan H,
Yang D, Zhen W, Li Z, Huang S and Peng S: The roles and
perspectives of microRNAs as biomarkers for intervertebral disc
degeneration. J Tissue Eng Regen Med. 11:3481–3487. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
27
|
Suzuki MM and Bird A: DNA methylation
landscapes: Provocative insights from epigenomics. Nat Rev Genet.
9:465–476. 2008. View
Article : Google Scholar : PubMed/NCBI
|
|
28
|
Yang J and Wang N: Genome-wide expression
and methylation profiles reveal candidate genes and biological
processes underlying synovial inflammatory tissue of patients with
osteoarthritis. Int J Rheum Dis. 18:783–790. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
29
|
Johnson AA, Akman K, Calimport SR, Wuttke
D, Stolzing A and de Magalhaes JP: The role of DNA methylation in
aging, rejuvenation, and age-related disease. Rejuvenation Res.
15:483–494. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Ezura Y, Sekiya I, Koga H, Muneta T and
Noda M: Methylation status of CpG islands in the promoter regions
of signature genes during chondrogenesis of human synovium-derived
mesenchymal stem cells. Arthritis Rheum. 60:1416–1426. 2009.
View Article : Google Scholar : PubMed/NCBI
|
|
31
|
Rogers EL, Reynard LN and Loughlin J: The
role of inflammation-related genes in osteoarthritis.
Osteoarthritis Cartilage. 23:1933–1938. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Takahashi A, de Andres MC, Hashimoto K,
Itoi E and Oreffo RO: Epigenetic regulation of interleukin-8, an
inflammatory chemokine, in osteoarthritis. Osteoarthritis
Cartilage. 23:1946–1954. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
33
|
Imagawa K, de Andrés MC, Hashimoto K, Pitt
D, Itoi E, Goldring MB, Roach HI and Oreffo RO: The epigenetic
effect of glucosamine and a nuclear factor-kappa B (NF-κB)
inhibitor on primary human chondrocytes-implications for
osteoarthritis. Biochem Biophys Res Commun. 405:362–367. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Fernández-Tajes J, Soto-Hermida A,
Vázquez-Mosquera ME, Cortés-Pereira E, Mosquera A, Fernández-Moreno
M, Oreiro N, Fernández-López C, Fernández JL, Rego-Pérez I and
Blanco FJ: Genome-wide DNA methylation analysis of articular
chondrocytes reveals a cluster of osteoarthritic patients. Ann
Rheum Dis. 73:668–677. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Xiao JL, Meng JH, Gan YH, Zhou CY and Ma
XC: Association of GDF5, SMAD3 and RUNX2 polymorphisms with
temporomandibular joint osteoarthritis in female Han Chinese. J
Oral Rehabil. 42:529–536. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Ji Q, Xu X, Xu Y, Fan Z, Kang L, Li L,
Liang Y, Guo J, Hong T, Li Z, et al: miR-105/Runx2 axis mediates
FGF2-induced ADAMTS expression in osteoarthritis cartilage. J Mol
Med (Berl). 94:681–694. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Leijten JC, Bos SD, Landman EB, Georgi N,
Jahr H, Meulenbelt I, Post JN, van Blitterswijk CA and Karperien M:
GREM1, FRZB and DKK1 mRNA levels correlate with osteoarthritis and
are regulated by osteoarthritis-associated factors. Arthritis Res
Ther. 15:R1262013. View
Article : Google Scholar : PubMed/NCBI
|
|
38
|
Han EJ, Yoo SA, Kim GM, Hwang D, Cho CS,
You S and Kim WU: GREM1 is a key regulator of synoviocyte
hyperplasia and invasiveness. J Rheumatol. 43:474–485. 2016.
View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Valverde-Franco G, Lussier B, Hum D, Wu J,
Hamadjida A, Dancause N, Fahmi H, Kapoor M, Pelletier JP and
Martel-Pelletier J: Cartilage-specific deletion of ephrin-B2 in
mice results in early developmental defects and an
osteoarthritis-like phenotype during aging in vivo. Arthritis Res
Ther. 18:652016. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Kaya AI, Lokits AD, Gilbert JA, Iverson
TM, Meiler J and Hamm HE: A conserved hydrophobic core in Gαi1
regulates G protein activation and release from activated receptor.
J Biol Chem. 291:19674–19686. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Diehl SA, McElvany B, Noubade R, Seeberger
N, Harding B, Spach K and Teuscher C: G proteins galphai1/3 are
critical targets for bordetella pertussis toxin-induced vasoactive
amine sensitization. Infect Immun. 82:773–782. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Rivetti S, Lauriola M, Voltattorni M,
Bianchini M, Martini D, Ceccarelli C, Palmieri A, Mattei G, Franchi
M, Ugolini G, et al: Gene expression profile of human colon cancer
cells treated with cross-reacting material 197, a diphtheria toxin
non-toxic mutant. Int J Immunopathol Pharmacol. 24:639–649. 2011.
View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Mabey T, Honsawek S, Saetan N, Poovorawan
Y, Tanavalee A and Yuktanandana P: Angiogenic cytokine expression
profiles in plasma and synovial fluid of primary knee
osteoarthritis. Int Orthop. 38:1885–1892. 2014. View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Gao W, Sweeney C, Walsh C, Rooney P,
McCormick J, Veale DJ and Fearon U: Notch signalling pathways
mediate synovial angiogenesis in response to vascular endothelial
growth factor and angiopoietin 2. Ann Rheum Dis. 72:1080–1088.
2013. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Yi J, Jin Q, Zhang B, Wu X and Ge D:
Gremlin-1 concentrations are correlated with the severity of knee
osteoarthritis. Med Sci Monit. 22:4062–4065. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
Fan Y, Chen J, Yang Y, Lin J and Wu Z:
Genome-wide expression and methylation profiling reveal candidate
genes in osteoarthritis. Clin Exp Rheumatol. 35:983–990.
2017.PubMed/NCBI
|
|
47
|
Hopwood B, Tsykin A, Findlay DM and
Fazzalari NL: Microarray gene expression profiling of
osteoarthritic bone suggests altered bone remodelling, WNT and
transforming growth factor-β/bone morphogenic protein signalling.
Arthritis Res Ther. 9:R1002007. View
Article : Google Scholar : PubMed/NCBI
|
|
48
|
Liao L, Zhang S, Gu J, Takarada T, Yoneda
Y, Huang J, Zhao L, Oh CD, Li J, Wang B, et al: Deletion of Runx2
in articular chondrocytes decelerates the progression of
DMM-induced osteoarthritis in adult mice. Sci Rep. 7:23712017.
View Article : Google Scholar : PubMed/NCBI
|