Introduction
Colorectal cancer (CRC) is one of the most commonly
diagnosed cancers worldwide (1).
It is the second most prevalent cancer type among women, following
breast cancer, and the third most prevalent cancer type among men
(2,3). In recent decades, diagnoses and
treatment of CRC have significantly improved. However, the disease
remains a major public health problem worldwide. In 2016, the
American Cancer Society estimated 70,820 new male cases and 63,670
new female cases of CRC would be diagnosed, and that there would be
49,190 CRC associated deaths in the USA (4). The high rate of mortality of the
disease is in part due to limitations in the currently available
therapies for CRC treatment, which is the result of an incomplete
understanding of the biological mechanisms underlying the
disease.
It has been well established that genes have an
important role in tumor pathogenesis, and that the transcriptional
activation or inhibition of certain genes serves an important role
in the development and progression of the majority of human tumors
(5). The analysis of genome-wide
gene expression levels via DNA microarray experiments is a recently
developed systematic approach that is used to gain comprehensive
information regarding tumor associated gene transcription profiles
(6). This information can lead to
the identification of prognostic biomarkers (7,8),
enable the discrimination between various histological subtypes of
tumors (9,10).
Interactions between genes can affect the
pathogenesis of CRC and tumor progression. The aim of the present
study was to identify biologically relevant genes that may be
associated with the pathogenesis of CRC, via analysis of
genome-wide expression profiles in CRC. Following the
identification of differentially expressed genes (DEGs), the
present study further aimed to determine the hub genes involved in
the regulatory pathways of CRC progression, by Gene Ontology (GO)
analysis, and to subsequently derive a gene interaction network.
Identification of hub genes in CRC and the development of a gene
interaction network may reveal the biological characteristics of
CRC tumor development and enable the development of novel
molecular-targeted therapies for the treatment of CRC.
Materials and methods
Data collection
The gene expression profiling dataset from
previously published relevant study was retrieved from the NCBI GEO
database (accession number GSE32323; www.ncbi.nlm.nih.gov/geo) (11). Data regarding a total of 17 paired
samples of CRC tumors and adjacent tissues were available from the
GSE32323 dataset.
Data processing
Raw RNA expression profile datasets were
preprocessed using R 3.4.1 statistical software (https://www.r-project.org/) together with Affy package
(12). In accordance with the
R-package, the gcrma algorithm (13) was used to adjust for background
intensities in the Affymtrix array data by including optical noise
and non-specific binding. The background adjusted probe intensities
were then converted into expression measures using the
normalization and summarization methods included in the multiarray
average algorithm (12). The
k-Nearest Neighbor algorithm (14)
was subsequently used to generate the missing values.
Microarray and statistical
analysis
The R-package and limma (www.bioconductor.org.uk) (15) were used to investigate gene
expression microarray data. Limma uses linear models, as well as
empirical Bayesian methods, to investigate the differential
expression levels in the GSE32323 dataset. Fold-change (FC) values
were calculated using the ratio of the geometric means of the gene
expression levels between the two treatment groups (CRC tissue vs.
adjacent tissue). A Wilcoxon Rank-Sum Test was performed to
determine the statistical significance of differences between the
groups (P<0.05 was considered statistically significant). Genes
were selected for if the log2-FC values were >1 (equivalent to a
FC value of >2.0 or <0.5), and P-values determined by the
Wilcoxon Rank-Sum Test were P<0.0002. Hierarchical clustering of
the selected genes was performed with R software using the
Euclidean distance and complete linkage method. Regarding
clustering, the expression data were standardized via conversion to
z scores (mean=0, variance=1) for each probe. Following this,
protein-protein interactions among the selected genes were analyzed
using the reference data contained in the String database of known
protein-protein interactions, version 10.5 (string-db.org/). Furthermore, the weight of each edge
of the gene clusters was calculated using molecular complex
detection (MCODE) software (version 1.4.2) from Cytoscape, version
3.5.1 (apps.cytoscape.org/apps/mcode) (16). In addition, the Cytoscape
(www.cytoscape.org/) plug-in CytoHubba,
version 0.1 (http://apps.cytoscape.org/apps/cytohubba) was used to
identify hub genes among the selected genes. Finally, BiNGO
software version 3.0.3 (http://apps.cytoscape.org/apps/bingo) from Cytoscape
was used to determine which GO categories represented identified
genes, and Kyoto Encyclopedia of Genes and Genomes (KEGG) pathways
(http://www.genome.jp/kegg/) were
analyzed using DAVID functional annotation tools (version 6.8)
(17,18).
Collection of CRC samples
The present study also investigated 3 patients (2
males and 1 female; aged 48–56 years old) with CRC who had
previously undergone radical resection of CRC in the
Gastroenterology Department of Renji Hospital Affiliated to
Shanghai Jiaotong University School of Medicine (Shanghai, China)
between November 2017 and December 2017. The inclusion criteria for
patients were as follows: i) colorectal adenocarcinoma confirmed by
pathological examination; ii) the patients must have not received a
radical resection prior and must not have been treated with
preoperative chemoradiotherapy. Characteristics of patients
included in the present study are presented in Table I. Written informed consent was
obtained from all patients included in the present study. The
Ethics Committee of Renji Hospital Affiliated to Shanghai Jiaotong
University School of Medicine approved the present study and
protocols. CRC tissue samples (diameter of 0.5 cm) were obtained
via tumor dissection. Paracancerous tissue (distal tissue 3 cm away
from the tumor) was also obtained from patients via dissection.
Paracancerous tissues and cancerous tissues were collected as
normal tissues and tumor tissues, respectively. RNA was extracted
from these tissues by TRIzol (Invitrogen; Thermo Fisher Scientific,
Inc., Waltham, MA, USA) for future experiments.
 | Table I.Characteristics of patients with
colorectal cancer in the present study. |
Table I.
Characteristics of patients with
colorectal cancer in the present study.
| Patient | Age | Sex | Stage | Preoperative
chemoradiotherapy | Surgical
resection |
|---|
| 1 | 48 | Male | 2a | No | Radical
correction |
| 2 | 53 | Male | 3c | No | Radical
correction |
| 3 | 56 | Female | 2b | No | Radical
correction |
Reverse transcription-quantitative
polymerase chain reaction (RT-qPCR)
In order to further investigate the results of the
microarray analysis, the 20 hub genes identified to be associated
with CRC were subjected to RT-qPCR analysis. A total of 1 µg of RNA
from tumor or normal tissue samples was reverse transcribed to cDNA
using SuperScript™ II reverse transcriptase (Invitrogen; Thermo
Fisher Scientific, Inc.; heat mixture to 65°C for 5 min, incubate
at 42°C for 50 min, inactivate the reaction by heating at 70°C for
15 min. RT-qPCR was performed using cDNA, iQ™ SYBR Green Supermix
(Invitrogen; Thermo Fisher Scientific, Inc.) and primers (2.5 µM).
The reaction was performed using the Bio-Rad RT-qPCR detection
system (Bio-Rad Laboratories, Inc., Hercules, CA, USA) under the
following PCR conditions: Initial denaturation at 95°C for 5 min;
35 cycles at 95°C for 30 sec; 60°C for 30 sec; 72°C for 30 sec; and
final extension at 72°C for 5 min. Expression levels of these genes
were normalized to the house-keeping gene, β-actin via the
2−ΔΔCq method (19).
Primer sequences used were as follows: WD repeat-containing protein
3 (WDR3) forward, 5′-AATCCAGCGGGTGACTAA-3′ and reverse,
5′-ACAGGCTGAGGAGTAGGC-3′; WD repeat-containing protein 43 (WDR43)
forward, 5′-TGACTTATTGGCTCTTGG-3′ and reverse,
5′-GGCTGATACATAGGGAAC-3′; UTP14 small subunit processome component
(UTP14A) forward, 5′-GCTGTGGAGGCGAGTAAG-3′, and reverse,
5′-GCCAATGGTGGGTAAATG-3′; damage specific DNA binding protein 1 and
cullin4 associated factor 13 (DCAF13) forward,
5′-GGGATGAACAAAGAACTAA-3′ and reverse, 5′-AGATTTATCGAAACTAGCAG-3′;
KRR1 small subunit processome component (KRR1) forward,
5′-GGCAGCATGACTGTTTGT-3′ and reverse, 5′-TAAGCCGTTGTCTTCGTT-3′;
digestion organ expression factor (DIEXF) forward,
5′-GAGAAGCATCCGACTCTA-3′, and reverse, 5′-TTAAACCTGGCATCAATC-3′;
HEAT repeat containing 1 (HEATR1) forward,
5′-ATTCACTTGTCGCCTTAC-3′, and reverse, 5′-TCTTGTCTCGTGGTATGG-3′; WD
repeat-containing protein 75 (WDR75) forward,
5′-ATGCATTGCGAATATCCAAAAGAGC-3′, and reverse,
5′-GCTCTTTTGGATATTCGCAATGCAT-3′; UTP 18 small subunit processome
component (UTP18) forward, 5′-GTTTATGTTTGGGATGTGA-3′, and reverse,
5′-GGCTTTGGGTTTGTTTCT-3′; UTP 3 small subunit processome component
(UTP3) forward, 5′-AATGCCGATGATGATGGT-3′ and reverse,
5′-CAAGTATTGGCTTCCTTTT-3′; RNA terminal phosphate cyclase-like
protein (RCL1) forward, 5′-TGGAGGAAATCTACAGGG-3′ and reverse,
5′-CACTTGAGGGTCTTGCTAA-3′; small subunit processome complement 20
(UTP20) forward, 5′-AGACCGAGAACACCTACC-3′ and reverse,
5′-CAACCTCCTCCTCATAGC-3′; testis-expressed protein 10 (TEX10)
forward, 5′-ATGTCGAGAATGACTAAAAAAAGAA-3′ and reverse,
5′-TTCTTTTTTTAGTCATTCTCGACAT-3′; WD repeat domain 12 (WDR12)
forward, 5′-ACATACTGGTTGGGTGAC-3′ and reverse,
5′-ATGGGAAGTGGTAGGTGA-3′; exosome component 5 (EXOSC5) forward,
5′-GAGGAGACGCATACTGACGC-3′ and reverse, 5′-ACACCAGGCAGCCCAATC-3′;
neutral invertase (regulatory particle non-ATPase 12) binding
protein 1 (NOB1) forward, 5′-TGTGAGCCTGAGAACCTGG-3′ and reverse,
5′-CTGCTGGATCTGCTTGATGT-3′; SDA1 domain containing 1 (SDAD1)
forward, 5′-TGCCGCAGTTACAGAATC-3′, and reverse,
5′-GTGCCATAAACATCACCAG-3′; DEAD-box helicase 27 (DDX27) forward,
5′-GCCCGTGGACTTGACATT-3′ and reverse, 5′-GCCGCTTTGCTGTATTGA-3′;
ribosome production factor 2 (RPF2) forward,
5′-TCCGCCTGGCTGGATTAG-3′ and reverse, 5′-TTCCTGGTTTGTAGTTTGCTTA-3′;
GTP binding protein 4 (GTPBP4) forward, 5′-CTAAAGATTATGTGCGACTG-3′,
and reverse, 5′-ATGGTGTTCCTATCCTCC-3′; and β-actin forward,
5′-CTCCATCCTGGCCTCGCTGT-3′ and reverse,
5′-GCTGTCACCTTCACCGTTCC-3′.
Statistical analysis
SPSS software (version 22.0; IBM Corp., Armonk, NY,
USA) was used to perform statistical analysis. Expression levels of
genes in paracancerous tissues and cancerous tissues were compared
using two-tailed Student's t-tests. The bar plot was generated by
GraphPad Prism (version 6.0; GraphPad Software, Inc., La Jolla, CA,
USA). The data in the present study was presented as the mean ±
standard deviation. P<0.05 was considered to indicate a
statistically significant difference.
Results
Characterization of differentially
expressed genes
A total of 3,264 genes were identified by the
Affymetrix Human Genome Array analysis as being differentially
expressed in CRC tissues compared with adjacent tissue (P<0.05;
data not shown). This included 1,594 genes that were downregulated
(FC<0.5 in expression in CRC vs. adjacent tissue) and 1,670
genes that were upregulated (FC>2.0 in expression in CRC vs.
adjacent tissue) compared with the adjacent tissue. The former
group was defined as the ‘lower group’ and the latter group as the
‘higher group’. GO and KEGG enrichment analyses were then performed
to identify the biological characteristics of each of these gene
subgroups.
In the lower group, a total of 1,594 downregulated
genes were revealed to be significantly enriched in 75 KEGG
pathways, the top 20 of which are presented in Fig. 1A (P<0.05). The protein kinase,
guanosine monophosphate-protein kinase G signaling pathway
(hsa04022) was the most notably enriched pathway in the lower group
(Fig. 1A). In addition, numerous
well-established signaling pathways were also enriched, including
the mitogen-activated kinase-like protein signaling pathway
(hsa04010), cyclic adenosine monophosphate signaling pathway
(hsa04024) and the phosphoinositide 3 kinase-protein kinase B
signaling pathway (hsa04151). Furthermore, pathways associated with
melanoma (hsa05218) and bladder cancer (hsa05219) were also
enriched in CRC, with 10 and 7 genes identified as being associated
with these diseases, respectively. In addition, the results of GO
classification of the identified genes into the categories
‘biological process’, ‘molecular function’ and ‘cellular
component’, as performed by BiNGO from Cytoscape, revealed that in
the ‘biological process’ category, the genes were notably enriched
in the following GO terms: ‘Positive regulation of IκB
kinase/nuclear factor-κB signaling’ (GO: 0043123), ‘negative
regulation of cell growth’ (GO: 0030308) and ‘negative regulation
of growth’ (GO: 0045926; Fig. 1B).
GO terms associated with the identified enriched genes belonging to
the ‘cellular component’ and ‘molecular function’ categories are
presented in Fig. 1C and D,
respectively.
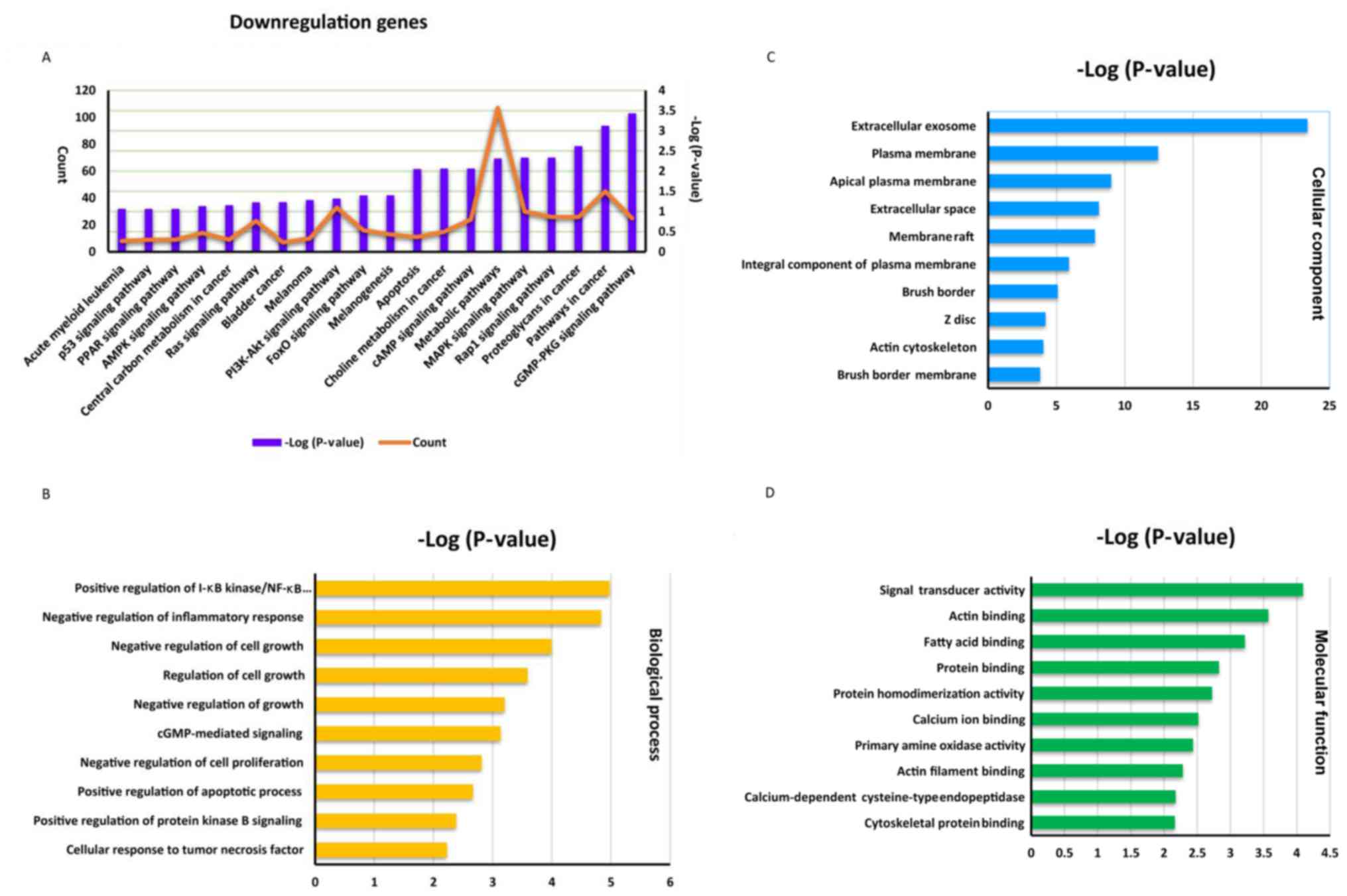 | Figure 1.Enrichment analysis of the 1,594
downregulated genes. (A) The top 20 enriched pathways (purple
columns) in the Kyoto Encyclopedia of Genes and Genomes database
and the gene count (orange line) enriched in each pathway are
presented. GO enrichment analysis revealing the top 10 GO terms
associated with (B) ‘biological process’, (C) ‘cellular component’
and (D) ‘molecular function’ are presented. GO, Gene Ontology; p53,
cellular tumor antigen p53; PPAR, peroxisome proliferator-activated
receptor; AMPK, AMP protein kinase; PI3K-Akt, phosphoinositide
3-kinase-protein kinase B; FoxO, forkhead box sub-group O; cAMP,
cyclic adenosine monophosphate; MAPK, mitogen-activated protein
kinase; Rap1, Rab5 activating protein 1; cGMP-PKG, guanosine
monophosphate-protein kinase G. |
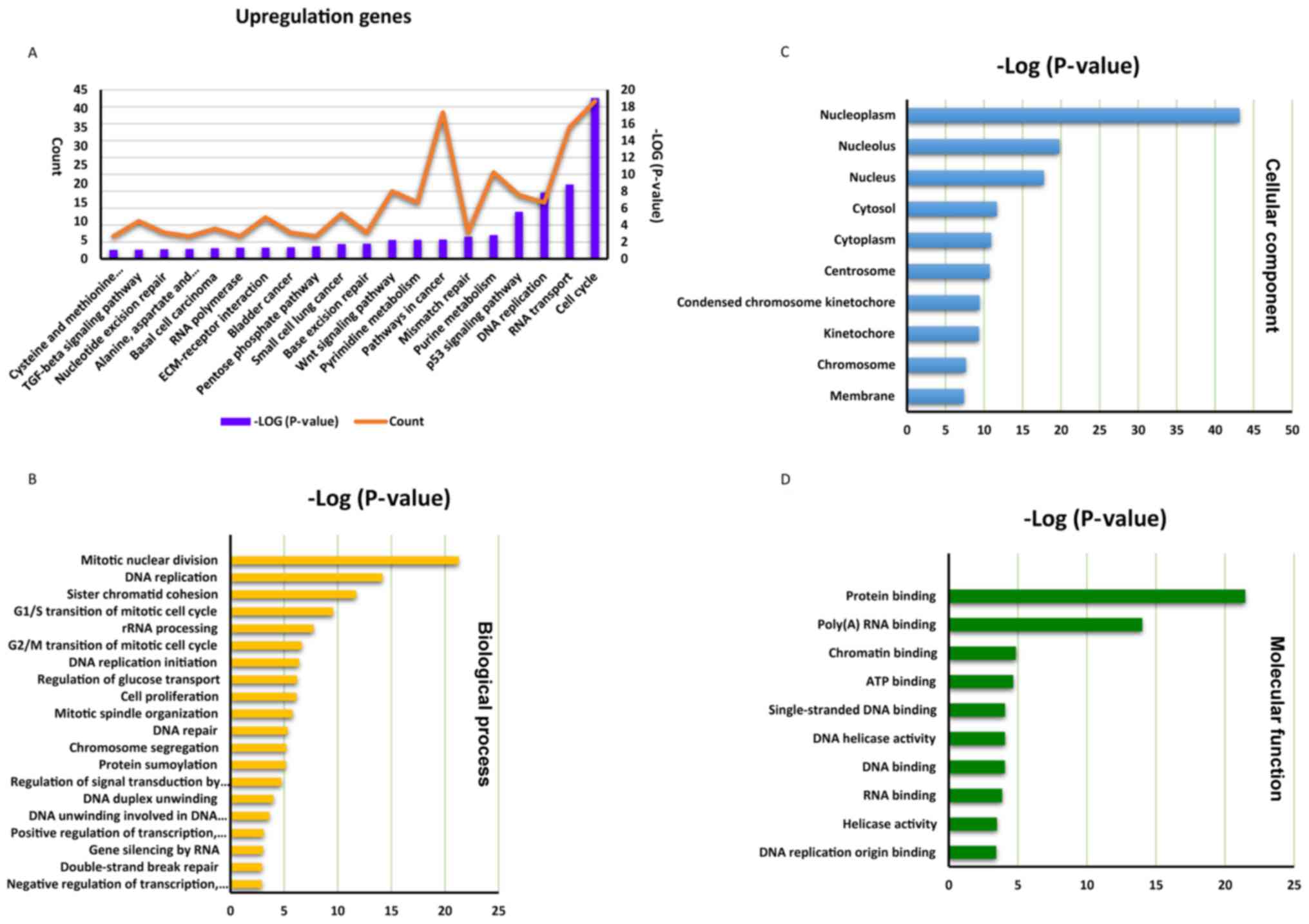
In the higher group, a total of 1,670 upregulated
genes were revealed to be significantly enriched in 28 KEGG
signaling pathways, the top 20 genes of which are presented in
Fig. 2A. The ‘cell cycle’
(hsa04110) pathway represents the most enriched pathway within this
group, followed by the ‘cellular tumor antigen p53 signaling
pathway’ (hsa04115) and the ‘Wnt protein signaling pathway’
(hsa04310). In the ‘biological process’ GO category, genes were
notably enriched in the following GO terms: ‘Mitotic nuclear
division’ (GO: 0007067), ‘DNA replication’ (GO: 0006260), ‘sister
chromatid cohesion’ (GO: 0007062) and ‘G1/S transition of mitotic
cell cycle’ (GO: 0000082; Fig.
2B). In the ‘cellular component’ category, genes were
significantly enriched in the GO term associated with the
‘nucleoplasm’ (GO: 0005654; Fig.
2C), and in the ‘molecular function’ category, the genes were
most significantly enriched in GO terms associated with ‘protein
binding’ (GO: 0005515) and ‘poly (A) RNA binding’ (GO: 0044822;
Fig. 2D).
Analysis of protein-protein
interactions of the differentially expressed genes
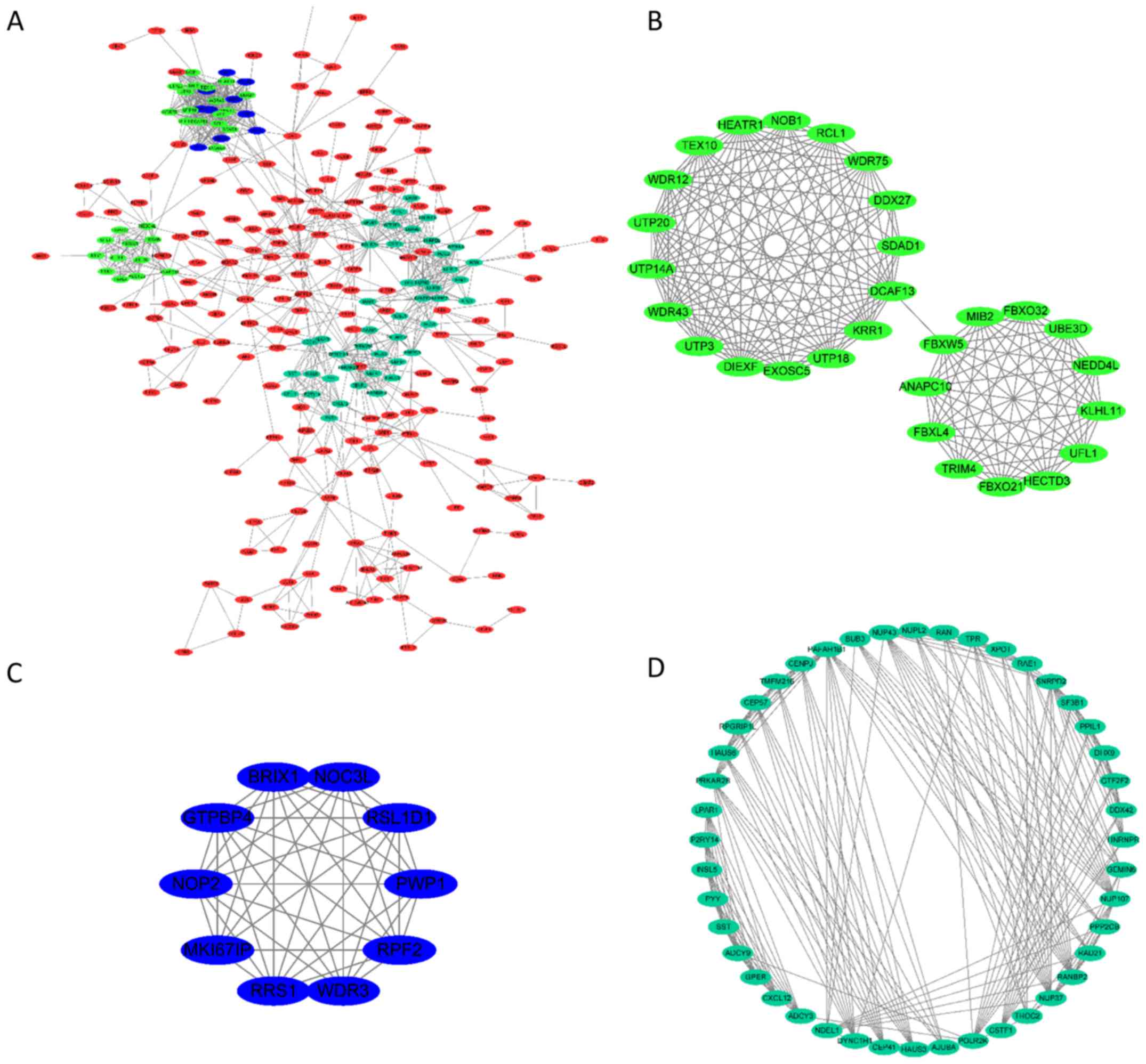
To determine the protein-protein interactions of the
selected genes, the String tool was used. The results revealed that
306 of the genes were clustered in a complex interaction network
(Fig. 3A), thus suggesting close
interactions between the proteins encoded by such genes.
Furthermore, calculation of the weighted index between each encoded
protein revealed the existence of three subgroups within the larger
network, which were closely associated with each other (subgroups
are represented in blue, green and light blue shading in Fig. 3A). Each subgroup was extracted and
their interactions are separately presented in Fig. 3B.
Identification of hub genes for
CRC
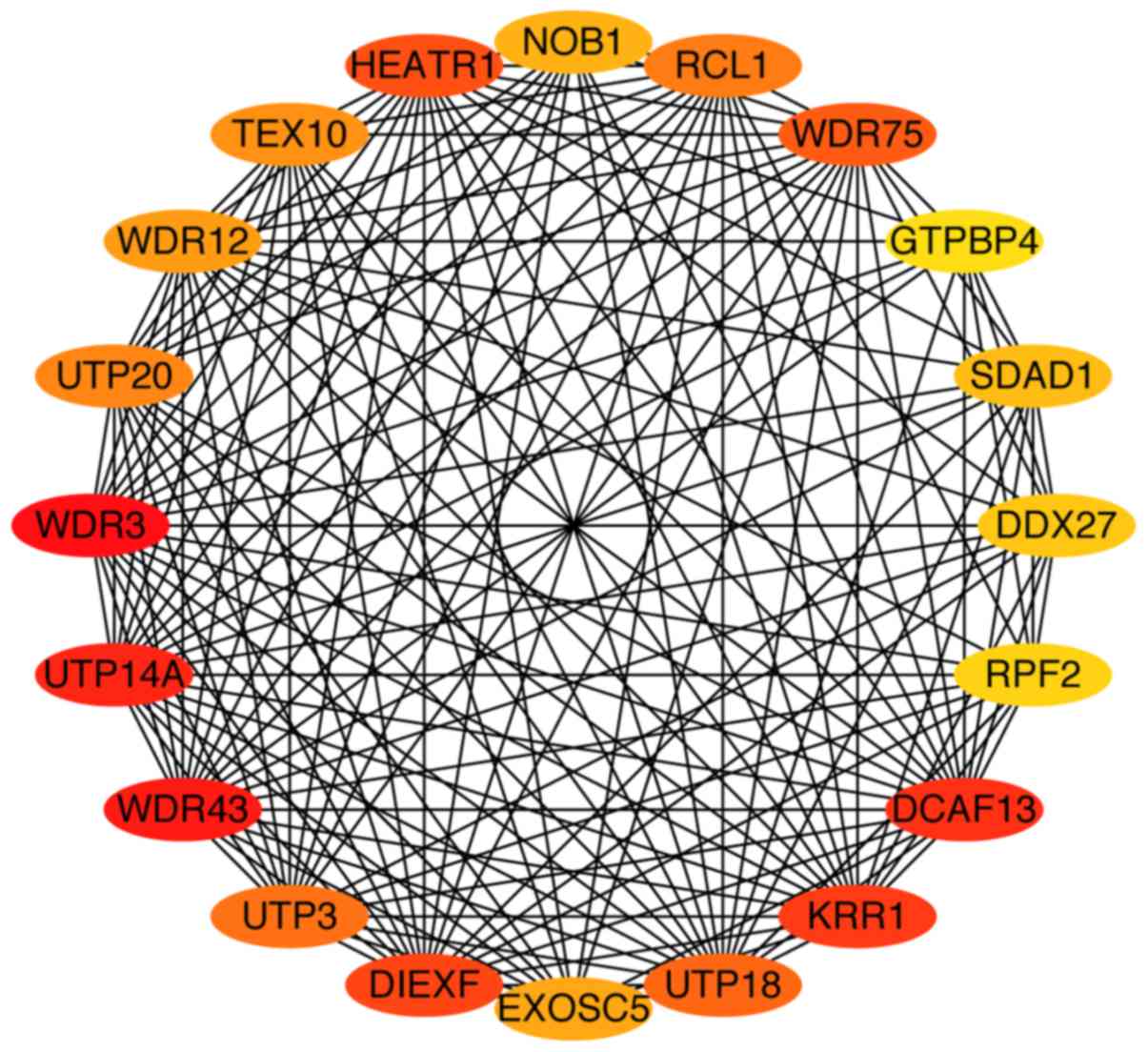
To identify potential hub genes among the 306 genes
previously identified, Matthew's correlation coefficient algorithms
were performed using the CytoHubba software plug-in. As presented
in Fig. 4, 20 potential hub genes
for CRC were identified, all of which were closely associated with
each other. The characteristics of the 20 hub genes are presented
in Table II. Four of the 20 hub
genes are genes that encode for small subunit processome components
(UTP3, UTP14A, UTP18 and UTP20) and four are genes that encode for
WD-repeat domains (WDR3, WDR12, WDR43, WDR75). The remaining 12
genes encode binding domains or proteins.
| Table II.List of 20 hub genes associated with
colorectal cancer. |
Table II.
List of 20 hub genes associated with
colorectal cancer.
| Rank | Gene | Location |
|---|
| 1 | WDR3 | 1p12 |
| 2 | WDR43 | 2p23.2 |
| 3 | UTP14A | Xq26.1 |
| 4 | DCAF13 | 8q22.3 |
| 5 | KRR1 | 12q21.2 |
| 6 | DIEXF | 1q32.2 |
| 7 | HEATR1 | 1q43 |
| 8 | WDR75 | 2q32.2 |
| 9 | UTP18 | 17q21.33 |
| 10 | UTP3 | 4q13.3 |
| 11 | RCL1 | 9p24.1 |
| 12 | UTP20 | 12q23.2 |
| 13 | TEX10 | 9q31.1 |
| 14 | WDR12 | 2q33.2 |
| 15 | EXOSC5 | 19q13.2 |
| 16 | NOB1 | 16q22.1 |
| 17 | SDAD1 | 4q21.1 |
| 18 | DDX27 | 20q13.13 |
| 19 | RPF2 | 6q21 |
| 20 | GTPBP4 | 10p15.3 |
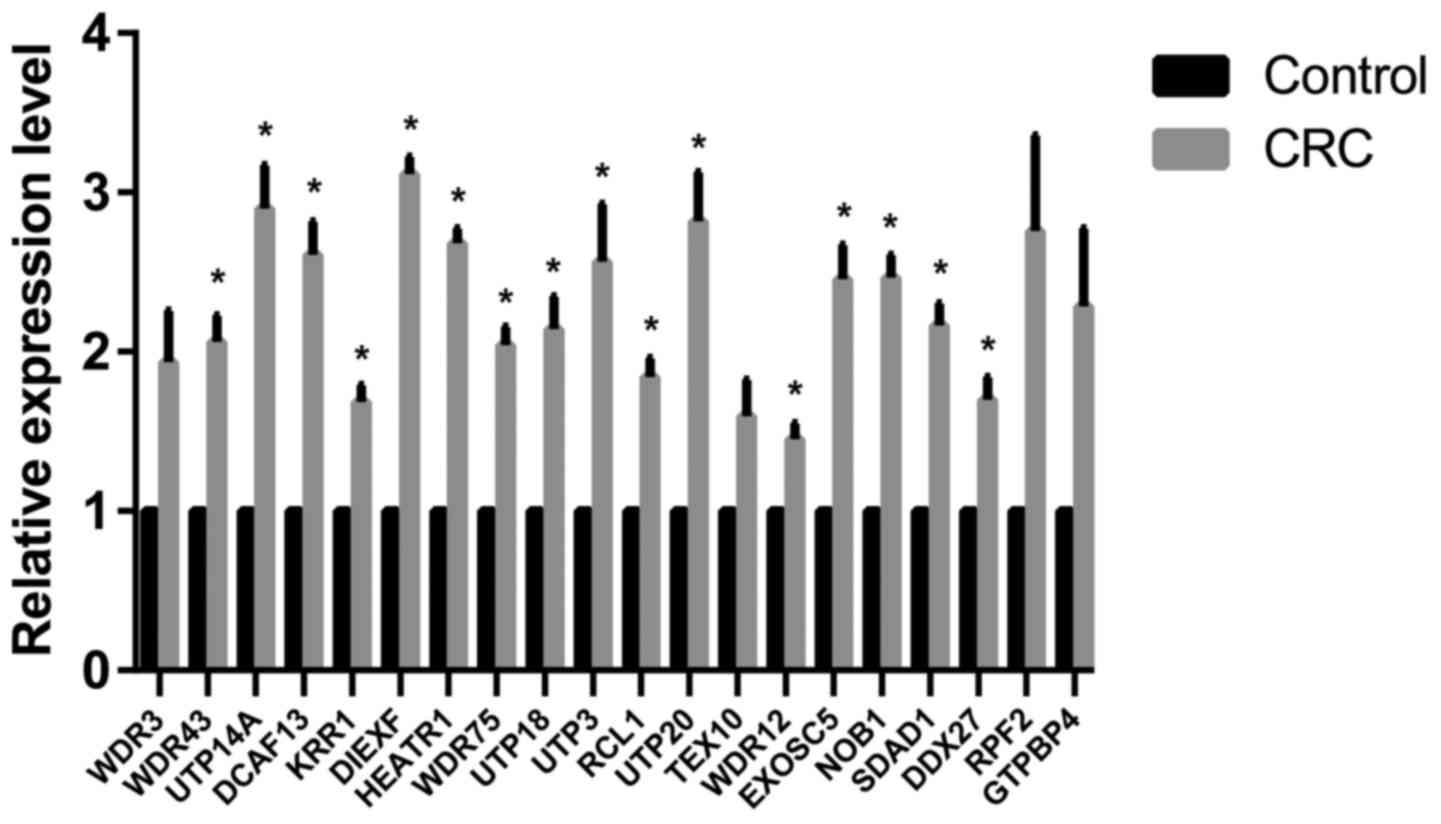
Verification of Affymetrix gene
expression data
To further investigate the gene expression profiles,
the expression levels of the 20 hub genes were determined via
RT-qPCR analysis. The results demonstrated that the majority of the
expression levels of the 20 hub genes were significantly
upregulated in CRC tissues compared with control tissues (Fig. 5), which is consistent with the
results of the microarray data analyses.
Discussion
CRC progression is characterized by the
transformation of the normal mucosa of the bowel into an adenoma
and then into a malignant tumor. This progression involves the
acquisition of gene mutations that enable the tumor cells to
proliferate and migrate within the tissues (20).
In the present study, a previously published
microarray dataset (11) was used
to identify significant genes in CRC tumor tissues via comparison
of gene expression profiles in CRC tissues with those in adjacent
paired tissue samples. A total of 1,594 genes were revealed to be
downregulated, and 1,670 genes were revealed to be upregulated, in
CRC tumor tissues. KEGG enrichment analysis of each of these
subgroups provided information regarding the function and other
biological characteristics of associated genes. The upregulated
genes were predominantly enriched in ‘cell cycle’ (hsa04110), ‘RNA
transport’ (hsa03013) and ‘DNA replication’ (hsa03030) pathways,
which is consistent with the findings of a previous study
investigating gene involvement in tumor pathogenesis (21). The mammalian cell cycle is highly
organized and regulated to ensure the correct functioning of cell
division and other biological activities (22). The four phases of the cell cycle
(G0/G1, S, G2 and M), are regulated by numerous cyclin-dependent
kinases (CDKs) (23). Aberrant
cell cycle activity, which may occur as a result of genetic lesion
within genes encoding cell cycle proteins, represents one of the
typical characteristics of cancer (24). Therefore, synthetic inhibitors of
CDKs are widely used as anticancer drugs in current cancer
treatment therapies (25,26).
Notably, in the present study, the upregulated genes
identified in CRC were also revealed to be enriched in ‘small cell
lung cancer’ (hsa05222), ‘bladder cancer’ (hsa05219) and ‘basal
cell carcinoma’ (hsa05217) KEGG pathways, suggesting that CRC may
exhibit similar gene regulation and molecular mechanisms with small
cell lung cancer, bladder cancer and basal cell carcinoma.
Similarly, the results from the enrichment analysis of the
downregulated genes revealed that there were 10 and 7 downregulated
DEGs enriched in the ‘melanoma’ (hsa05218) term and ‘bladder
cancer’ (hsa05219) term, respectively. Merlo et al (27) revealed that one of the
microsatellite alterations exhibited in small cell lung cancer is
also exhibited in CRC. Furthermore, a clinical study in Japan has
suggested that patients with bladder cancer have the potential to
develop colon cancer (1.44%) during anticancer therapy (28), and an immunotherapy study have
demonstrated that type I and II interferons can be used to treat
both CRC and melanoma due to their dual role in promoting
proliferation and inhibiting growth (29). However, the extent to which genes
that have been previously revealed to be implicated in these other
cancers are expressed in CRC remains largely unknown, and warrants
further research.
Song et al (30) downloaded CRC microarray data from
the GEO database (GSE17538) and screened for genes associated with
enhancer of zeste homolog 2 (EZH2). Song et al (30) revealed that EZH2 may represent a
potential prognostic marker of patients with CRC. By contrast, the
present study was performed using all DEGs in CRC, and aimed to
uncover the molecular mechanisms underlying the progression of CRC.
Liang et al (31) analyzed
141 samples (132 CRC and 9 normal colon epitheliums) and 3,500 DEGs
were identified. In addition, Liang et al (31) performed GO and KEGG pathway
enrichment analyses, and the top 10 hub genes from PPI network were
identified, including G protein subunit γ2, angiotensin precursor,
serum amyloid A1, adenylate cyclase 5, lysophosphatidic acid
receptor 1, neuromedin-U, interleukin 8, C-X-C motif chemokine
ligand 12, G protein subunit α1, and C-C motif chemokine receptor
2. There was no intersection between the 10 hub genes and 20 hub
genes detected in the present study, which suggested that the novel
CRC-associated genes were identified. Furthermore, Guo et al
(32) acquired overlapping DEGs in
CRC from four GEO datasets (GSE28000, GSE21815, GSE44076 and
GSE75970) (33–36) and performed GO enrichment analysis,
KEGG pathway analysis and PPI network analysis. Guo et al
(32) found also that cell cycle
term may serve an important role in CRC; 31 hub genes were acquired
in a PPI network. However, there was no intersection between these
31 hub genes and the results of the present study. A total of 17
pairs of cancer tissues and adjacent tissues were analyzed in the
present study, which made obtained DEGs more reliable and accurate
relative to unpaired samples. The follow-up analyses, including GO
enrichment analysis, KEGG pathway enrichment analysis, PPI network
analysis and MCODE software analysis, could be more reliable based
on these DEGs. In addition, CRC tissues and adjacent tissues from
patients with CRC were collected, and RT-qPCR was performed to
verify the expression levels of the identified 20 hub genes in CRC
tissue compared with normal tissue.
In the present study, DEGs in CRC and adjacent
tissues were identified and were separated into two subgroups
(upregulated and downregulated groups), but the accounts of DEGs
are still enormous. Furthermore, the present study aimed to
construct a protein interaction network based on the selected
genes, and following this, a total of 306 genes were revealed to be
involved in this network. From these 306 genes, 20 hub genes, which
represent genes exhibiting close interactions with each other, were
identified using the Cytohubba plug-in analysis. These hub genes
may serve important molecular roles in the pathogenesis of CRC.
GO analysis of the 20 hub genes revealed that there
are four subunit processome component encoded genes: UTP3, UTP14A,
UTP18 and UTP20 differentially expressed in CRC. The subunit
processome component is an essential part of the ribonucleoprotein
complex that once bound to the U3 small nuclear RNA, participates
in ribosome biogenesis and 18S ribosomal RNA synthesis (37). It has been demonstrated that
alternative splicing of said genes may result in multiple
transcript variants; which can promote cancer development (38). For example, UTP18 has been revealed
to be localized in the cytoplasm of cells, and serum withdrawal has
been revealed to increase cytoplasmic UTP18, which can associate
with the translation complex and Hsp90 to upregulate the
translation of HIF1a, Myc and VEGF, thus inducing a cellular stress
response (39). UTP18
overexpression promotes transformation and tumorigenesis; however,
UTP18 knockdown can inhibit these processes (23). In addition, the subunit processome
component is represented by a large gene family (uridine
triphosphate, UTP), which exhibits different mechanisms in cell
proliferation and cancer pathogenesis (40). Furthermore, the associations
between the four subunit processome component encoded genes and
CRC, to the best of our knowledge, have not previously been
investigated. The present study demonstrated that the
aforementioned four genes may have important roles in the
progression of CRC and represent potential biomarkers for CRC.
In the present study, members of the WD repeat
domain family (including WDR3, WDR12, WDR43, WDR75) was a further
group of genes revealed to be represented in the 20 hub genes. It
has been previously established that WD repeats are ~30–40 amino
acid domains in length, and contain conserved repeating units,
which are frequently terminated with Trp-Asp at the C-terminal
(41). Proteins belonging to the
WD repeat family are involved in numerous cellular processes, such
as cell proliferation, apoptosis, signal transduction, gene
regulation and human disease (42). In a recent study, Izumi et
al (43) concluded that c-Myc
expression alterations are regulated by upregulation of F-box/WD
repeat-containing protein 7 (FBXW7), and that knockdown of FBXW7
via application of small interfering RNA could enhance cell
sensitivity to anticancer agents.
Akdi et al (44) revealed that mRNA and protein levels
of WDR3 are dysregulated in human thyroid cancer cells. In
addition, a further study demonstrated that WDR3 can regulate
genome stability in patients with thyroid cancer (45). However, studies investigating the
associations between WD repeats and CRC are currently very limited.
In the present study, it was revealed that these WD repeats are hub
genes in progression of CRC. Therefore, further in vitro and
in vivo studies on this topic are required. He et al
(46) demonstrated that knockdown
of NOB1 can induce apoptosis of human colorectal cells. In
addition, Zeng et al (47)
demonstrated that knockdown of SDAD1 can suppress the
proliferation, migration and invasion of colon cancer cells
(47). The results of the previous
two studies are consistent with the results of the present study.
In addition, a previous study demonstrated that overexpression of
DCAF13 in hepatocellular carcinoma is correlated with poor survival
of patients (48). Dyachenko et
al (49) demonstrated that
KRR1 may represent a potential biomarker of particular histological
types (invasive ductal) of breast tumor. Furthermore, Liu et
al (50) revealed that HEATR1
can negatively regulate protein kinase B and further decrease
resistance to gemcitabine and other chemotherapeutics. Tsukamoto
et al (51) demonstrated
that DDX27 is upregulated in gastric cancer tissues and may
represent a potential therapeutic target for patients with gastric
cancer. In addition, Liu et al (52) revealed that GTPBP4 serves an
important role in hepatocellular carcinoma development, and that
increased GTPBP4 expression is correlated with poor survival of
patients with hepatocellular carcinoma. However, the functions of
aforementioned five genes (DCAF13, KRR1, HEATR1, DDX27 and GTPBP4)
have, to the best of our knowledge, not been investigated with
regards to CRC. The results of the present study demonstrated that
the aforementioned five genes and the remaining 15 genes in the top
20 hub genes (DIEXF, RCL1, TEX10, EXOSC5 and RPF2) may serve
important roles in CRC development and represent potential
biomarkers for CRC.
In conclusion, the present study identified
significant genes associated with the pathogenesis of CRC via
analysis of genome-wide expression profiles of CRC as well as
comparison of expression levels of significant genes in CRC tissues
compared with healthy adjacent tissue samples. Furthermore, 20 hub
genes were revealed via genetic analysis as validated by RT-qPCR.
GO analysis revealed that ‘small subunit processsome component’ and
‘WD repeat domains’ were two protein family subgroups encoded by
the 20 hub genes, and could represent novel molecular markers
associated with CRC. The patterns of the expression levels of the
20 hub genes in CRC were further verified by RT-qPCR. However, a
small sample size (three paired samples) represents a limitation of
the present study, and therefore this should be further
investigated using a greater sample size in future studies.
Therefore, additional studies are required to further investigate
the associations between identified hub genes and CRC.
Acknowledgements
We would like to express our thanks to the National
Library of Medicine (https://www.nlm.nih.gov/) for giving user the
privilege to freely download the raw data of various GEO
series.
Funding
No funding was received.
Availability of data and materials
The datasets used and analyzed during the current
study are available from GEO database (GSE32323, https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE32323).
Authors' contributions
All the research was conducted by SL. The design of
the present study was conceived by QH. All authors read and
approved the final manuscript.
Ethics approval and consent to
participate
Written informed consent was obtained from all
patients included in the present study. The Ethics Committee of
Renji Hospital Affiliated to Shanghai Jiaotong University School of
Medicine approved the present study and protocols.
Consent for publication
Written informed consent was obtained from all
patients for publication of reverse transcription-quantitative
polymerase chain reaction results in this study; personal
identifying information was not included in this article.
Competing interests
The authors declare that they have no competing
interests.
References
|
1
|
McWhirter JE and Hoffman-Goetz L: Coverage
of skin cancer and recreational tanning in North American magazines
before and after the landmark 2006 international agency for
research on cancer report. BMC Public Health. 15:1692015.
View Article : Google Scholar : PubMed/NCBI
|
|
2
|
Navarro M, Nicolas A, Ferrandez A and
Lanas A: Colorectal cancer population screening programs worldwide
in 2016: An update. World J Gastroenterol. 23:3632–3642. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
3
|
Marley AR and Nan H: Epidemiology of
colorectal cancer. Int J Mol Epidemiol Genet. 7:105–114.
2016.PubMed/NCBI
|
|
4
|
Siegel RL, Miller KD and Jemal A: Cancer
statistics, 2016. CA Cancer J Clin. 66:7–30. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
5
|
Ranzani M, Annunziato S, Adams DJ and
Montini E: Cancer gene discovery: Exploiting insertional
mutagenesis. Mol Cancer Res. 11:1141–1158. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
6
|
Li MH, Fu SB and Xiao HS: Genome-wide
analysis of microRNA and mRNA expression signatures in cancer. Acta
Pharmacol Sin. 36:1200–1211. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
7
|
Choi JY, Kim JG, Lee YJ, Chae YS, Sohn SK,
Moon JH, Kang BW, Jung MK, Jeon SW, Park JS and Choi GS: Prognostic
impact of polymorphisms in the CASPASE genes on survival of
patients with colorectal cancer. Cancer Res Treat. 44:32–36. 2012.
View Article : Google Scholar : PubMed/NCBI
|
|
8
|
Lascorz J, Chen B, Hemminki K and Försti
A: Consensus pathways implicated in prognosis of colorectal cancer
identified through systematic enrichment analysis of gene
expression profiling studies. PLoS One. 6:e188672011. View Article : Google Scholar : PubMed/NCBI
|
|
9
|
Karagiannis GS, Berk A, Dimitromanolakis A
and Diamandis EP: Enrichment map profiling of the cancer invasion
front suggests regulation of colorectal cancer progression by the
bone morphogenetic protein antagonist, gremlin-1. Mol Oncol.
7:826–839. 2013. View Article : Google Scholar : PubMed/NCBI
|
|
10
|
Lascorz J, Hemminki K and Försti A:
Systematic enrichment analysis of gene expression profiling studies
identifies consensus pathways implicated in colorectal cancer
development. J Carcinog. 10:72011. View Article : Google Scholar : PubMed/NCBI
|
|
11
|
Khamas A, Ishikawa T, Shimokawa K, Mogushi
K, Iida S, Ishiguro M, Mizushima H, Tanaka H, Uetake H and Sugihara
K: Screening for epigenetically masked genes in colorectal cancer
Using 5-Aza-2′-deoxycytidine, microarray and gene expression
profile. Cancer Genomics Proteomics. 9:67–75. 2012.PubMed/NCBI
|
|
12
|
Irizarry RA, Hobbs B, Collin F,
Beazer-Barclay YD, Antonellis KJ, Scherf U and Speed TP:
Exploration, normalization, and summaries of high density
oligonucleotide array probe level data. Biostatistics. 4:249–264.
2003. View Article : Google Scholar : PubMed/NCBI
|
|
13
|
Wu Z, Irizarry RA, Gentleman R,
Martinez-Murillo F and Spencer F: A model-based background
adjustment for oligonucleotide expression arrays. J Am Stat Assoc.
99:909–917. 2004. View Article : Google Scholar
|
|
14
|
Denoeux T: A k-nearest neighbor
classification rule based on Dempster-Shafer theory. Syst Man
Cybernetics IEEE Trans. 25:804–813. 1995. View Article : Google Scholar
|
|
15
|
Smyth GK: Limma: Linear models for
microarray data. Bioinformatics Comput Biol Solutions Using R
Bioconductor. 397–420. 2011.
|
|
16
|
Maere S, Heymans K and Kuiper M: BiNGO: A
Cytoscape plugin to assess overrepresentation of gene ontology
categories in biological networks. Bioinformatics. 21:3448–3449.
2005. View Article : Google Scholar : PubMed/NCBI
|
|
17
|
Jiao X, Sherman BT, da Huang W, Stephens
R, Baseler MW, Lane HC and Lempicki RA: DAVID-WS: A stateful web
service to facilitate gene/protein list analysis. Bioinformatics.
28:1805–1806. 2012. View Article : Google Scholar : PubMed/NCBI
|
|
18
|
Kanehisa M and Goto S: KEGG: Kyoto
encyclopedia of genes and genomes. Nucleic Acids Res. 28:27–30.
2000. View Article : Google Scholar : PubMed/NCBI
|
|
19
|
Livak KJ and Schmittgen TD: Analysis of
relative gene expression data using real-time quantitative PCR and
the 2(-Delta Delta C(T)) method. Methods. 25:402–408. 2001.
View Article : Google Scholar : PubMed/NCBI
|
|
20
|
Huang D, Sun W, Zhou Y, Li P, Chen F, Chen
H, Xia D, Xu E, Lai M, Wu Y and Zhang H: Mutations of key driver
genes in colorectal cancer progression and metastasis. Cancer
Metastasis Rev. 37:173–187. 2018. View Article : Google Scholar : PubMed/NCBI
|
|
21
|
Janin N: A simple model for carcinogenesis
of colorectal cancers with microsatellite instability. Adv Cancer
Res. 77:189–221. 2000. View Article : Google Scholar : PubMed/NCBI
|
|
22
|
Efe Yagdi E, Mazumder A, Lee JY, Gaigneaux
A, Radogna F, Nasim MJ, Christov C, Jacob C, Kim KW, Dicato M, et
al: Tubulin-binding anticancer polysulfides induce cell death via
mitotic arrest and autophagic interference in colorectal cancer.
Cancer Lett. 410:139–157. 2017. View Article : Google Scholar : PubMed/NCBI
|
|
23
|
Otto T and Sicinski P: Cell cycle proteins
as promising targets in cancer therapy. Nat Rev Cancer. 17:93–115.
2017. View Article : Google Scholar : PubMed/NCBI
|
|
24
|
Liu Z, Li J, Chen J, Shan Q, Dai H, Xie H,
Zhou L, Xu X and Zheng S: MCM family in HCC: MCM6 indicates adverse
tumor features and poor outcomes and promotes S/G2 cell cycle
progression. BMC Cancer. 18:2002018. View Article : Google Scholar : PubMed/NCBI
|
|
25
|
Owa T, Yoshino H, Yoshimatsu K and Nagasu
T: Cell cycle regulation in the G1 phase: A promising target for
the development of new chemotherapeutic anticancer agents. Curr Med
Chem. 8:1487–1503. 2001. View Article : Google Scholar : PubMed/NCBI
|
|
26
|
Roberge M, Berlinck RG, Xu L, Anderson HJ,
Lim LY, Curman D, Stringer CM, Friend SH, Davies P, Vincent I, et
al: High-throughput assay for G2 checkpoint inhibitors and
identification of the structurally novel compound isogranulatimide.
Cancer Res. 58:5701–5706. 1998.PubMed/NCBI
|
|
27
|
Merlo A, Mabry M, Gabrielson E, Vollmer R,
Baylin SB and Sidransky D: Frequent microsatellite instability in
primary small cell lung cancer. Cancer Res. 54:2098–2101.
1994.PubMed/NCBI
|
|
28
|
Tashiro K, Iwamuro S, Hatano T, Furuta A,
Takizawa A, Ohishi Y, Igarashi H, Hasegawa N, Asano K and Aoki H:
Double cancer observed from bladder cancer. Nihon Hinyokika Gakkai
Zasshi. 90:509–513. 1999.PubMed/NCBI
|
|
29
|
Di Franco S, Turdo A, Todaro M and Stassi
G: Role of type I and II interferons in colorectal cancer and
melanoma. Front Immunol. 8:8782017. View Article : Google Scholar : PubMed/NCBI
|
|
30
|
Song D, Huang R, Tang Q, et al:
Identification of EZH2-related key pathways and genes in colorectal
cancer using bioinformatics analysis. Chin J Colorectal Dis.
5:475–479. 2016.
|
|
31
|
Liang B, Li C and Zhao J: Identification
of key pathways and genes in colorectal cancer using bioinformatics
analysis. Med Oncol. 33:1112016. View Article : Google Scholar : PubMed/NCBI
|
|
32
|
Guo Y, Bao Y, Ma M and Yang W:
Identification of key candidate genes and pathways in colorectal
cancer by integrated bioinformatical analysis. Int J Mol Sci.
18:pii: E722. 2017. View Article : Google Scholar
|
|
33
|
Sanz-Pamplona R, Berenguer A, Cordero D,
Molleví DG, Crous-Bou M, Sole X, Paré-Brunet L, Guino E, Salazar R,
Santos C, et al: Aberrant gene expression in mucosa adjacent to
tumor reveals a molecular crosstalk in colon cancer. Mol Cancer.
13:462014. View Article : Google Scholar : PubMed/NCBI
|
|
34
|
Kogo R, Shimamura T, Mimori K, Kawahara K,
Imoto S, Sudo T, Tanaka F, Shibata K, Suzuki A, Komune S, et al:
Long noncoding RNA HOTAIR regulates polycomb-dependent chromatin
modification and is associated with poor prognosis in colorectal
cancers. Cancer Res. 71:6320–6326. 2011. View Article : Google Scholar : PubMed/NCBI
|
|
35
|
Jovov B, Araujo-Perez F, Sigel CS,
Stratford JK, McCoy AN, Yeh JJ and Keku T: Differential gene
expression between African American and European American
colorectal cancer patients. PLoS One. 7:e301682012. View Article : Google Scholar : PubMed/NCBI
|
|
36
|
Huang FT, Chen WY, Gu ZQ, Zhuang YY, Li
CQ, Wang LY, Peng JF, Zhu Z, Luo X, Li YH, et al: The novel long
intergenic noncoding RNA UCC promotes colorectal cancer progression
by sponging miR-143. Cell Death Dis. 8:e27782017. View Article : Google Scholar : PubMed/NCBI
|
|
37
|
Melnik S, Deng B, Papantonis A, Baboo S,
Carr IM and Cook PR: The proteomes of transcription factories
containing RNA polymerases I, II or III. Nat Methods. 8:963–968.
2011. View Article : Google Scholar : PubMed/NCBI
|
|
38
|
Narla A and Ebert BL: Ribosomopathies:
Human disorders of ribosome dysfunction. Blood. 115:3196–3205.
2010. View Article : Google Scholar : PubMed/NCBI
|
|
39
|
Yang HW, Kim TM, Song SS, Menon L, Jiang
X, Huang W, Black PM, Park PJ, Carroll RS and Johnson MD: A small
subunit processome protein promotes cancer by altering translation.
Oncogene. 34:4471–4481. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
40
|
Guan Y, Huang D, Chen F, Gao C, Tao T, Shi
H, Zhao S, Liao Z, Lo LJ, Wang Y, et al: Phosphorylation of Def
regulates Nucleolar p53 turnover and cell cycle progression through
Def recruitment of Calpain3. PLoS Biol. 14:e10025552016. View Article : Google Scholar : PubMed/NCBI
|
|
41
|
Neer EJ, Schmidt CJ, Nambudripad R and
Smith TF: The ancient regulatory-protein family of WD-repeat
proteins. Nature. 371:297–300. 1994. View Article : Google Scholar : PubMed/NCBI
|
|
42
|
Li D and Roberts R: WD-repeat proteins:
Structure characteristics, biological function, and their
involvement in human diseases. Cell Mol Life Sci. 58:2085–2097.
2001. View Article : Google Scholar : PubMed/NCBI
|
|
43
|
Izumi D, Ishimoto T, Miyake K, Eto T,
Arima K, Kiyozumi Y, Uchihara T, Kurashige J, Iwatsuki M, Baba Y,
et al: Colorectal cancer stem cells acquire chemoresistance through
the upregulation of F-Box/WD repeat-containing protein 7 and the
consequent degradation of c-Myc. Stem Cells. 35:2027–2036. 2017.
View Article : Google Scholar : PubMed/NCBI
|
|
44
|
Akdi A, Giménez EM, Garcia-Quispes W,
Pastor S, Castell J, Biarnés J, Marcos R and Velázquez A: WDR3 gene
haplotype is associated with thyroid cancer risk in a Spanish
population. Thyroid. 20:803–809. 2010. View Article : Google Scholar : PubMed/NCBI
|
|
45
|
Garcia-Quispes WA, Pastor S, Galofré P,
Biarnés J, Castell J, Velázquez A and Marcos R: Possible role of
the WDR3 gene on genome stability in thyroid cancer patients. PLoS
One. 7:e442882012. View Article : Google Scholar : PubMed/NCBI
|
|
46
|
He XW, Feng T, Yin QL, Jian YW and Liu T:
NOB1 is essential for the survival of RKO colorectal cancer cells.
World J Gastroenterol. 21:868–877. 2015. View Article : Google Scholar : PubMed/NCBI
|
|
47
|
Zeng M, Zhu L, Li L and Kang C: miR-378
suppresses the proliferation, migration and invasion of colon
cancer cells by inhibiting SDAD1. Cell Mol Biol Lett. 22:122017.
View Article : Google Scholar : PubMed/NCBI
|
|
48
|
Cao J, Hou P, Chen J, Wang P, Wang W, Liu
W, Liu C and He X: The overexpression and prognostic role of DCAF13
in hepatocellular carcinoma. Tumour Biol. 39:10104283177057532017.
View Article : Google Scholar : PubMed/NCBI
|
|
49
|
Dyachenko L, Havrysh K, Lytovchenko A,
Dosenko I, Antoniuk S, Filonenko V and Kiyamova R: Autoantibody
response to ZRF1 and KRR1 SEREX antigens in patients with breast
tumors of different histological types and grades. Dis Markers.
2016:51287202016. View Article : Google Scholar : PubMed/NCBI
|
|
50
|
Liu T, Fang Y, Zhang H, Deng M, Gao B, Niu
N, Yu J, Lee S, Kim J, Qin B, et al: HEATR1 negatively regulates
Akt to help sensitize pancreatic cancer cells to chemotherapy.
Cancer Res. 76:572–581. 2016. View Article : Google Scholar : PubMed/NCBI
|
|
51
|
Tsukamoto Y, Fumoto S, Noguchi T,
Yanagihara K, Hirashita Y, Nakada C, Hijiya N, Uchida T, Matsuura
K, Hamanaka R, et al: Expression of DDX27 contributes to
colony-forming ability of gastric cancer cells and correlates with
poor prognosis in gastric cancer. Am J Cancer Res. 5:2998–3014.
2015.PubMed/NCBI
|
|
52
|
Liu WB, Jia WD, Ma JL, Xu GL, Zhou HC,
Peng Y and Wang W: Knockdown of GTPBP4 inhibits cell growth and
survival in human hepatocellular carcinoma and its prognostic
significance. Oncotarget. 8:93984–93997. 2017.PubMed/NCBI
|